 ,, 刘华东, 王敬国, 刘化龙, 孙健, 杨洛淼, 贾琰, 吴文申, 郑洪亮,, 邹德堂,东北农业大学寒地粮食作物种质创新与生理生态教育部重点实验室,哈尔滨 150030
,, 刘华东, 王敬国, 刘化龙, 孙健, 杨洛淼, 贾琰, 吴文申, 郑洪亮,, 邹德堂,东北农业大学寒地粮食作物种质创新与生理生态教育部重点实验室,哈尔滨 150030Genome-Wide Association Study and Candidate Gene Mining of Tillering Number in Japonica Rice
ZHANG JiFeng,, LIU HuaDong, WANG JingGuo, LIU HuaLong, SUN Jian, YANG LuoMiao, JIA Yan, WU WenShen, ZHENG HongLiang,, ZOU DeTang,Key Laboratory of Germplasm Enhancement, Physiology and Ecology of Food Crops in Cold Region, Ministry of Education, Northeast Agricultural University, Harbin 150030通讯作者:
责任编辑: 李莉
收稿日期:2019-10-18接受日期:2020-01-13网络出版日期:2020-08-16
| 基金资助: |
Received:2019-10-18Accepted:2020-01-13Online:2020-08-16
作者简介 About authors
张继峰,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1054KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
张继峰, 刘华东, 王敬国, 刘化龙, 孙健, 杨洛淼, 贾琰, 吴文申, 郑洪亮, 邹德堂. 粳稻分蘖数全基因组关联分析及候选基因的挖掘[J]. 中国农业科学, 2020, 53(16): 3205-3213 doi:10.3864/j.issn.0578-1752.2020.16.001
ZHANG JiFeng, LIU HuaDong, WANG JingGuo, LIU HuaLong, SUN Jian, YANG LuoMiao, JIA Yan, WU WenShen, ZHENG HongLiang, ZOU DeTang.
0 引言
【研究意义】水稻是世界上主要粮食作物之一,随着全球人口的增长和耕地面积的日益减少,水稻每年需要达到1%的增产才能满足人类对食物的需求。在经历矮化育种和杂种优势2次突破性飞跃后,水稻增产达到了瓶颈。长期栽培实践证明,杂种优势与理想株型相互利用是实现水稻进一步增产的重要途径。分蘖数是水稻理想株型的重要组成部分之一,也是水稻产量形成的重要性状。分蘖数决定有效穗数,适量的分蘖数会有助于高产,分蘖数过多会导致营养物质的浪费,分蘖数过少会导致过少的有效穗数,因此,适量的分蘖数是决定水稻高产的重要条件。【前人研究进展】水稻分蘖由分蘖节上各叶腋下处的分生组织发育而来,水稻分蘖形成包括2个部分,分别是分蘖芽的形成和向外生长过程。植物遗传学家现在已经克隆了MOC1、MOC2、MOC3、LAX1、LAX2和DTE1等与分蘖芽形成相关的基因。MOC1编码一个GRAS家族的核蛋白,主要在腋芽和叶腋原基表达,在营养生长和生殖生长阶段控制叶腋分生组织形成,moc1突变体表现为少蘖,同时花序轴和小穗减少[1,2]。MOC2/FBP1编码果糖-1,6二磷酸酶,该基因缺失表现为单孽、矮化、叶色变淡、每穗实粒数减少等,moc2突变体与moc1突变体不同,可以生长分蘖芽,但是分蘖芽的生长受到阻碍,不能正常发育成分蘖,果糖-1,6二磷酸酶是一个胞质FBP酶,该酶失活会导致植株蔗糖供应不足,从而导致moc2缺失突变体分蘖芽受到抑制[3]。MOC3/OsWUS/TAB1编码一个WOX蛋白家族成员,是一个转录抑制因子,主要在地上部愈伤组织中表达[4],该基因正向调控腋芽发育,可能是通过促进水稻同源异型基因OSH1的表达,进而在茎端分生组织中发挥重要作用[5],moc3-1突变体和moc2突变体一样,在分蘖芽发育过程中遭受破坏[3,4],Tab1/wus突变体表现为不能形成腋芽,导致没有分蘖[5]。LAX1编码一个植物特有的bHLH转录因子,该转录子含有一个碱性螺旋-环-螺旋(bHLH)结构域,该基因在地上部分顶端分生组织与新形成的分生组织之间的边界区域表达,LAX1蛋白在叶腋分生组织中积累,被mRMA和随后的蛋白转运进行严格调控,该步骤对叶腋分生组织建立发挥重要作用[6,7]。LAX2/Gnp4编码一个核蛋白,和LAX1蛋白互作共同参与调控水稻腋生分生组织形成,进而调控水稻分蘖形态[8],Lax2与lax1突变体在营养阶段叶腋分生组织数目减少且大多数侧生小穗腋分生组织减少[8]。DTE1编码一个硼酸通道蛋白,在根的外层皮以及成熟叶片节区域表达,在硼饥饿处理下,dtel突变体表现出分蘖数增加、生长缓慢和育性下降等生长缺陷[9]。研究表明,大部分基因均通过双亲分离群体(RIL、NIL和BIL等)进行遗传位点挖掘,但鉴定的分蘖数遗传位点受到双亲遗传差异或分子标记密度的限制,所能挖掘鉴定的位点相对有限。全基因组关联分析以连锁不平衡(linkage disequilibrium,LD)为基础,通过群体基因型数据与表型数据检测有效关联位点,可以实现对同一复杂性状多数基因座以及一个基因座上多个复等位基因的检测[10,11]。同时,全基因组关联分析基于自然群体,不需要构建专门的作图群体,并且自然群体经过天然杂交多代,重组更加充分,定位精度更高。在人类和动物研究的基因挖掘中,关联分析是重要的手段,并且已在植物基因定位中得到广泛应用。2005年,Science杂志首次报道了人类年龄相关的黄斑性病症全基因组关联分析的研究[12]。受到人类遗传疾病研究的启示,植物遗传学家也将关联分析应用于作物遗传育种中,并取得了重要研究进展,为挖掘水稻有效等位基因提供了新方法。随着基因分型与测序技术的日益进步,目前,对于水稻的粒型、株高、抽穗期和剑叶形态等很多农艺性状开展了GWAS分析,同时也对病虫害抗性等生物胁迫和盐碱耐受性等非生物胁迫进行了全基因组关联分析,检测出许多重要的显著性SNP位点及候选基因,解析了许多重要性状的遗传基础[13,14,15,16,17,18,19,20,21,22]。【本研究切入点】尽管已有很多关于水稻分蘖数QTL/基因的研究报道,但大多是通过突变体基因定位或反向遗传学的方法进行研究,所挖掘到的QTL/基因有限。【拟解决的关键问题】本研究以来自不同国家和地区的295份温带粳稻品种为试验材料,结合高通量重测序获得的788 396个高质量多态性SNP,对粳稻分蘖数进行全基因组关联分析,鉴定影响粳稻分蘖数稳定存在的主效QTL,并对QTL区间内所有基因进行单倍型分析,根据基因注释筛选候选基因,为分子辅助育种提高产量提供理论基础。1 材料与方法
1.1 试验材料以及性状考察
试验材料由国内外广泛收集的295份粳稻品种构成,其中,国内材料主要来自黑龙江省、吉林省和辽宁省等,国外材料主要来自日本、韩国和俄罗斯等(电子附表1)。所有材料于2018—2019年种植在东北农业大学阿城水稻试验基地,每份材料种植8行,每行20株,行、株间距为30 cm×13.3 cm,单株插秧。田间管理依照常规大田生产的基本方法。为了减少边际效应对表型的影响,于水稻分蘖盛期,在每个品种的第4行,选取表型相似的连续5株,考察每株水稻分蘖数,并计算平均值。1.2 全基因组重测序
使用植物基因组DNA提取试剂盒提取295份粳稻品种的DNA,将检测合格的样品DNA采用Illumina HiSeq XTen进行高通量测序。利用GATK软件工具包进行SNP检测,使用Plink软件从初步获得的群体SNP中筛选出最小等位基因频率(minimum allele frequency,MAF)大于5%,缺失率(missing rate)小于20%的788 396个SNP用于后续分析[23]。1.3 群体结构与亲缘关系分析
利用ADMIXTURE软件计算每个品种的遗传成分,将K值设置为1—10,运行结束后,提取每个K值的交叉验证(cross validation,CV)值,最小CV值所对应的K值为最优亚群分组数,所用群体被划分为3个亚群[23]。另外使用GCTA软件[24]进行主成分分析(principal component analysis,PCA),利用R语言绘制三维PCA图。利用MEGA 7[25,26]的邻接法(neighbor-joining)构建系统进化树(NJ树)。使用TASSEl 5.0软件[27]对群体材料间的亲缘关系进行评估[23]。1.4 粳稻分蘖数全基因组关联分析
利用TASSEl 5.0软件的混合线性模型(mixed linear model,MLM),结合群体SNP基因型数据,进行粳稻分蘖数的全基因组关联分析,对GEC软件计算出的有效独立SNP数目进行阈值的确定,最终将P<5.46×10-6作为阈值,判定SNP标记与目标性状关联的显著性。利用R语言的“CMplot”包绘制曼哈顿图和Q-Q图。依据每条染色体LD衰减结果[23],以GWAS检测到的峰值SNP所在染色体上下游LD衰减距离区域界定为该QTL的染色体区间。1.5 候选基因单倍型分析
对2年环境下共同检测到的QTL内所有基因进行单倍型分析。具体操作过程如下:从3KRGP的Rice SNP-Seek Database网站[28]中提取位于QTL区间内所有基因外显子区非同义突变SNP。利用之前研究中筛选得到的788 396个高质量SNP在位于该QTL区间内的所有SNP与3KRGP中提取的所有非同义突变SNP进行比对,确定其是否为非同义突变。若基因存在非同义突变,则先利用DnaSP软件对该QTL区间内所有存在非同义突变的基因进行单倍型分析,对不同单倍型(≥15份材料)的表型值进行多重比较,筛选不同单倍型表型值之间存在显著性差异的基因。对于不同单倍型表型值之间差异不显著的基因,再用启动子区(ATG前1.5 kb)的SNP进行单倍型分析。结合基因注释预测可能的候选基因。2 结果
2.1 295份粳稻材料分蘖数表现
2018和2019年295份粳稻品种的分蘖数均具有较大的表型分布,分别为7.00—49.00个和6.00—33.80个(表1和图1),且2年总趋势基本一致,分蘖数平均值分别为18.35和15.75个。Table 1
表1
表12年环境下295份粳稻材料的分蘖数表型值
Table 1
| 年份 Year | 均值±标准差 Mean±standard | 变幅 Range | 变异系数 CV (%) | 偏度 Skewness | 峰度 Kurtosis |
|---|---|---|---|---|---|
| 2018 | 18.35±5.98 | 7.00—49.00 | 32.58 | 1.31 | 0.49 |
| 2019 | 15.75±4.65 | 6.00—33.80 | 29.55 | 3.09 | 0.05 |
新窗口打开|下载CSV
图1
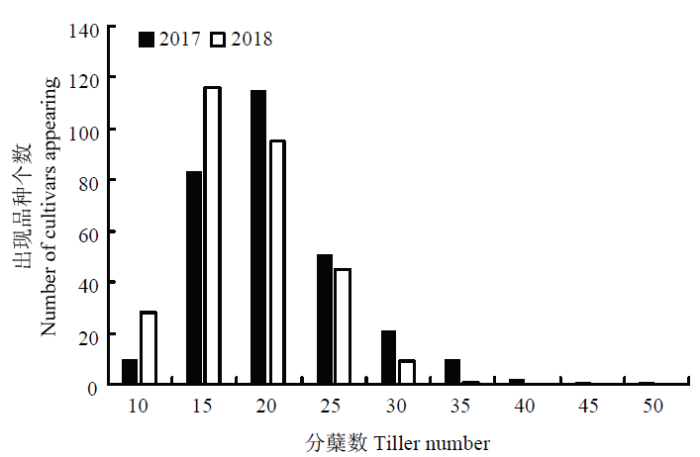 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图12年环境条件下分蘖数频次分布直方图
Fig. 1Histogram of tillering number frequency distribution under 2-year environmental conditions
2.2 全基因组关联分析
利用TASSLE 5.0软件的MLM模型分析,2018年在水稻第8、9和10染色体共检测到3个QTL(qTiller8、qTillerq9和qTiller10),2019年在水稻第9染色体检测到1个QTL(qTiller9),其中,qTiller9在2年中均被检测到(表2)。依据第9染色体LD衰减距离(52 kb)[23],选取峰值SNP所在染色体的上下游LD衰减距离区域,界定为该QTL的区间(表2),结果显示,qTiller9被定位于水稻第9染色体15.01—15.12 Mb,与已定位影响水稻分蘖数QTL(qNOT9-1)[29]位于相同区间。Table 2
表2
表2粳稻分蘖数显著关联位点
Table 2
| QTL | 年份 Year | 峰值SNP Peak SNP | 染色体 Chr. | P值 P value | 表型贡献率 R2(%) | 已定位QTL QTL located |
|---|---|---|---|---|---|---|
| qTiller8 | 2018 | 7527329 | 8 | 2.02E-06 | 9.36 | |
| qTiller9 | 2018 | 15065450 | 9 | 6.99E-08 | 11.86 | qNOT9-1[29] |
| qTiller10 | 2018 | 15406671 | 10 | 2.64E-06 | 9.10 | |
| qTiller9 | 2019 | 15060619 | 9 | 2.55E-07 | 10.61 | qNOT9-1[29] |
新窗口打开|下载CSV
图2
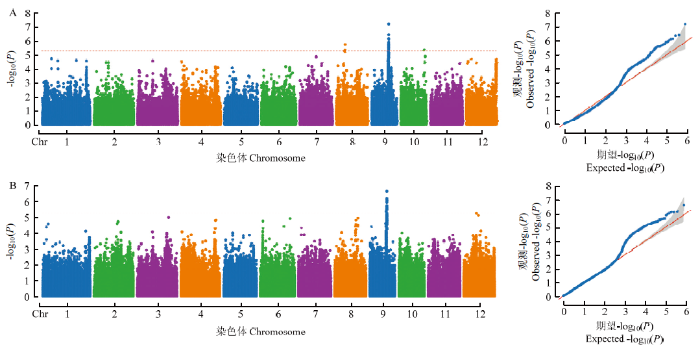 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2分蘖数GWAS结果的曼哈顿图和QQ散点图
A和B分别表示2018和2019年的分析结果
Fig. 2Manhattan plots and quantile-quantile (Q-Q) plots of genome-wide association studies for the tillering number
A and B indicate the result of 2018 and 2019, respective
2.3 候选基因分析
2018—2019年共检测到3个影响粳稻分蘖数的QTL,其中,2年中能够稳定表达的QTL——qTiller9位于已定位影响水稻分蘖数QTL(qNOT9-1)区间内。因此,对qTiller9区间内全部15个基因的外显子区非同义突变SNP和启动子区SNP进行单倍型分析。结果显示,共有6个基因(LOC_Os09g25090、LOC_ Os09g25100、LOC_Os09g25150、LOC_Os09g25190、LOC_Os09g25200和LOC_Os09g25220)的不同单倍型分蘖数之间均检测到极显著差异水平(图3)。LOC_Os09g25090被启动子区SNP分为2种单倍型,hap2(TAA)分蘖数极其显著大于hap1(AGG);LOC_Os09g25100被非同义突变SNP分为2种单倍型,hap2(GAGA)分蘖数极其显著大于hap1(AGCC);LOC_Os09g25150被非同义突变SNP分为2种单倍型,hap2(ATG)分蘖数极其显著大于hap1(GCC);LOC_Os09g25190被启动子区SNP分为2种单倍型,hap2(GCATCGCATCGACGCCGA)分蘖数极其显著大于hap1(ATGCTGATGAAGTCATCC);LOC_ Os09g25200被非同义突变SNP分为2种单倍型,hap2(TAG)分蘖数极其显著大于hap1(AGA);LOC_Os09g25220被非同义突变SNP分为2种单倍型,hap1(GG)分蘖数极其显著大于hap2(AA)(表3和图3)。其中,LOC_Os09g25090和LOC_Os09g25100编码钙调蛋白依赖性蛋白激酶,它是脱落酸(ABA)表达所必需Ca2+的传感器。因此,推测LOC_Os09g25090和LOC_Os09g25100可能是影响水稻分蘖数的候选基因。Table 3
表3
表3候选基因单倍型分组及每种单倍型的SNP组成
Table 3
| 基因 Gene | 单倍型1/品种数 hap1/Number | 单倍型2/品种数 hap2/Number | 单倍型3/品种数 hap3/Number |
|---|---|---|---|
| LOC_Os09g25090 | AGG/182 | TAA/81 | |
| LOC_Os09g25100 | AGCC/188 | GAGA/89 | |
| LOC_Os09g25150 | ATG/190 | GCC/69 | GTC/17 |
| LOC_Os09g25190 | ATGCTGATGAAGTCATCC/126 | GCATCGCATCGACGCCGA/76 | |
| LOC_Os09g25200 | AGA/181 | TAG/89 | |
| LOC_Os09g25220 | GG/97 | AA/187 |
新窗口打开|下载CSV
图3
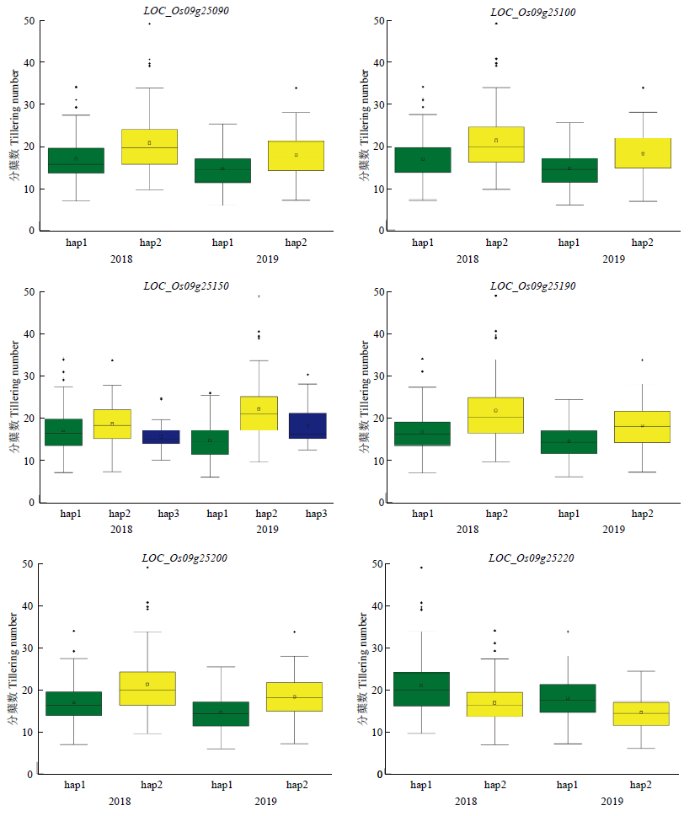 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3候选基因不同单倍型之间分蘖数的箱线图
绿色、黄色和蓝色分别代表hap1、hap2和hap3的表型结果
Fig. 3Boxplots for the tillering number based on the haplotypes(hap) for candidate gene
Green, yellow and blue indicate phenotypic result for hap1, hap2 and hap3, respectively
3 讨论
水稻分蘖数是由多基因控制的数量性状,挖掘水稻分蘖数相关QTL和基因对水稻增产具有重要意义。连锁分析已经被证明可以有效地挖掘水稻分蘖数QTL,但是由于亲本数目较少,后代群体重组机会有限,因此,当等位基因在双亲中无差异时,QTL无法通过连锁分析检测出来。与连锁分析相比,全基因组关联分析以数百个品种组成的自然群体为研究材料,自然群体已经过天然杂交多代,重组更加充分,定位精度更高,并且能够同时检测同一基因座上的多个等位基因。本研究利用全基因组关联分析方法,于2018和2019年在水稻第8、9和10染色体共检测到3个QTL,分别为qTiller8、qTiller9和qTiller10。HEMAMALINI等[29]利用IR64和Azucena杂交得到的56个随机DH系在水稻第9染色体11.80—15.55 Mb区间内定位到一个影响水稻分蘖数的QTL(qNOT9-1),与本研究qTiller9位于相同区间,这验证了本研究结果可靠性的同时进一步缩小了QTL区间,为后续候选基因分析提供了坚实的理论依据;qTiller8和qTiller10在前人研究中未见报道,且2个QTL仅在2018年检测到,需要通过遗传群体进一步验证。本研究进一步通过QTL所在的物理位置与前人已克隆基因进行比较,未发现QTL区间内有已知基因的报道。Table 4
表4
表4候选基因的基因注释
Table 4
| 基因 Gene | 基因功能注释 Gene annotation |
|---|---|
| LOC_Os09g25090 | 钙调蛋白依赖性蛋白激酶 Calmodulin depedent protein kinases |
| LOC_Os09g25100 | 钙调蛋白依赖性蛋白激酶 Calmodulin depedent protein kinases |
| LOC_Os09g25150 | 肉桂酰辅酶A还原酶 Cinnamoyl-CoA reductase |
| LOC_Os09g25190 | 蛋白结合蛋白Protein binding protein |
| LOC_Os09g25200 | 蛋白结合蛋白Protein binding protein |
| LOC_Os09g25220 | 蛋白结合蛋白Protein binding protein |
新窗口打开|下载CSV
以往的全基因组关联分析挖掘相关性状候选基因,大多依据显著性峰值SNP位点上下游固定物理距离确定候选基因区域,然后通过基因功能注释进行候选基因的预测,然而水稻每条染色体的LD衰减距离各不相同,仅仅依靠固定距离确定候选基因区域,降低了该区域的精准性。本研究的检测到的QTL区域是依据每条染色体LD衰减距离及显著性峰值SNP位点确定的,这大大增加候选基因区域的精准性,同时本研究对共定位QTL区域内所有基因进行单倍型分析,再通过基因注释筛选单倍型表型值之间存在显著性差异的基因,这大大增加了候选基因筛选的效率及可靠性。
本研究通过对qTiller9区间内全部15个基因进行单倍型分析,发现有6个基因不同单倍型的分蘖数存在极显著差异。进一步通过基因功能注释筛选候选基因,发现6个候选基因分别属于钙调蛋白依赖性蛋白激酶、肉桂酰辅酶A还原酶和蛋白结合蛋白。其中LOC_Os09g2509和LOC_Os09g25100均预测编码钙调蛋白依赖性蛋白激酶,其结构包括CaM结合域、丝氨酸/苏氨酸激酶域和3个EF手基序,是多个Ca2+传感器中的一员,可以检测到Ca2+的瞬时浓度[30,31],同时,Ca2+对脱落酸(ABA)的表达是必需的[37,38,39]。ZHU等[32]研究发现,9顺式-环氧类胡萝卜素双加氧酶OsNCED2是控制ABA合成代谢的关键酶,过表达OsNCED2的水稻转基因品系中,其分蘖数减少[33];另外,拟南芥基因MAX3和MAX4均编码类胡萝卜素裂解双加氧酶,是ABA生物合成中的关键酶[34],拟南芥突变体max4存在一种侧芽抑制信号,该信号由根部向上运输,并与生长素互作进而抑制植株侧芽发生,这种信号的前体是类胡萝卜素,而植物中ABA的前体同样为类胡萝卜素[35];STEVEN等[36]进一步研究发现,拟南芥中的max3和max4突变体是由MAX3和MAX4损伤引起的,导致2种突变体侧向分支急剧增加。以上研究表明,ABA在植物侧芽发生中发挥重要作用。本研究在水稻表达数据库(Rice Expression Profile Database)中查询LOC_Os09g25090和LOC_Os09g25100在田间整个生长生育时期各组织的时空表达量,发现2个候选基因在水稻茎部均有较高强度的表达。因此,推测2个候选基因可能在水稻分蘖处表达,通过调节Ca2+浓度介导ABA合成代谢进而影响水稻分蘖数。
4 结论
2018和2019年共检测到3个与粳稻分蘖数相关QTL(qTiller8、qTiller9和qTiller10),其中,qTiller9在2年中均稳定表达,并与已定位影响水稻分蘖数QTL(qNOT9-1)重合。LOC_Os09g25090和LOC_ Os09g25100可能为影响qTiller9的候选基因。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/nature01518URLPMID:12687001 [本文引用: 1]

Tillering in rice (Oryza sativa L.) is an important agronomic trait for grain production, and also a model system for the study of branching in monocotyledonous plants. Rice tiller is a specialized grain-bearing branch that is formed on the unelongated basal internode and grows independently of the mother stem (culm) by means of its own adventitious roots. Rice tillering occurs in a two-stage process: the formation of an axillary bud at each leaf axil and its subsequent outgrowth. Although the morphology and histology and some mutants of rice tillering have been well described, the molecular mechanism of rice tillering remains to be elucidated. Here we report the isolation and characterization of MONOCULM 1 (MOC1), a gene that is important in the control of rice tillering. The moc1 mutant plants have only a main culm without any tillers owing to a defect in the formation of tiller buds. MOC1 encodes a putative GRAS family nuclear protein that is expressed mainly in the axillary buds and functions to initiate axillary buds and to promote their outgrowth.
DOI:10.1016/j.molp.2019.04.008URLPMID:31048024 [本文引用: 1]
Tillering in rice is one of the most important agronomic traits. Rice tiller development can be divided into two main processes: the formation of the axillary bud and its subsequent outgrowth. Several genes critical for bud formation in rice have been identified by genetic studies; however, their molecular functions and relationships are still largely unknown. Here, we report that MONOCULM 1 (MOC1) and MONOCULM 3/TILLERS ABSENT 1/STERILE AND REDUCED TILLERING 1 (MOC3/TAB1/SRT1), two vital regulators for tiller formation in rice, physically interact to regulate tiller bud outgrowth through upregulating the expression of FLORAL ORGAN NUMBER1 (FON1), the homolog of CLAVATA1 in rice. We found that MOC3 is able to directly bind the promoter of FON1 and subsequently activate FON1 expression. MOC1 functions as a co-activator of MOC3, whereas it could not directly bind the FON1 promoter, and further activated FON1 expression in the presence of MOC3. Accordingly, FON1 is highly expressed at axillary meristems and shows remarkably decreased expression levels in moc1 and moc3 mutants. Loss-of-function mutants of FON1 exhibit normal bud formation but defective bud outgrowth and reduced tiller number. Collectively, these results shed light on a joint transcriptional regulatory mechanim by MOC1 and MOC3, and establish a new framework for the control of tiller bud formation and outgrowth.
DOI:10.5511/plantbiotechnology.12.1210aURL [本文引用: 2]
We characterized a rice monoculm mutant moc2, which showed significantly reduced tiller numbers, pale-green leaves, a reduced growth rate, and a consequent dwarf phenotype. The monoculm feature was attributed to a deficiency in the efficient outgrowth of tiller buds, although the moc2 mutant produced tiller buds. Inconsistent change was observed in the expression of genes involved in tiller bud outgrowth, suggesting that the moc2 mutant has a defective function necessary for the tiller bud outgrowth. The gene responsible for the moc2 mutant was mapped to a locus encoding cytosolic fructose-1,6-bisphosphatase 1 (FBP1), in which a Tos17 retrotransposon was inserted in exon 4. Reverse-transcription PCR for the FBP1 gene amplified a shorter transcript from the moc2 mutant than from the wild-type plant. The sequence of the shorter transcript revealed a deletion of exon 4 by abnormal splicing, and the resulting frameshift generated a new translation termination signal. The moc2 mutant showed a very low level of FBPase activity, suggesting that it involves a loss-of-function mutation of FBP1. Cytosolic FBPase is considered a key enzyme in the sucrose biosynthesis pathway. Defective FBPase activity is anticipated to lead a shortage of sucrose supply, which probably causes the inhibition of tiller bud outgrowth in the moc2 mutant. The monoculm phenotype of the moc2 mutant supports the idea that sucrose supply may be an important cue to outgrow tiller buds.
DOI:10.1016/j.jgg.2014.12.005URLPMID:25697101 [本文引用: 2]
WUSCHEL (WUS) plays an essential role for the maintenance of meristem activity in dicots, but its function is still elusive in monocots. We isolated a new monoculm mutant, monoculm 3 (moc3), in which a point mutation causes the premature termination of rice O. sativa WUS (OsWUS). Morphological observation revealed that the formation of tiller buds was disrupted in moc3. MOC3 was localized in the nuclear and could interact with TOPLESS-RELATED PROTEINS (TPRs). The expression of MOC3 was induced by cytokinins and defection of MOC3 affected the expression of several two-component cytokinin response regulators, OsRRs and ORRs. Our results suggest that MOC3 is required for the formation of axillary buds and has a complex relationship with cytokinins.
URLPMID:25841039 [本文引用: 2]
URLPMID:19346465 [本文引用: 1]
DOI:10.1073/pnas.1932414100URLPMID:13130077 [本文引用: 1]
The aerial architecture of plants is determined primarily by the pattern of shoot branching. Although shoot apical meristem initiation during embryogenesis has been extensively studied by molecular genetic approaches using Arabidopsis, little is known about the genetic mechanisms controlling axillary meristem initiation, mainly because of the insufficient number of mutants that specifically alter it. We identified the LAX PANICLE (LAX) and SMALL PANICLE (SPA) genes as the main regulators of axillary meristem formation in rice. LAX encodes a basic helix-loop-helix transcription factor and is expressed in the boundary between the shoot apical meristem and the region of new meristem formation. This pattern of LAX expression was repeatedly observed in every axillary meristem, consistent with our observation that LAX is involved in the formation of all types of axillary meristems throughout the ontogeny of a rice plant. Ectopic LAX expression in rice caused pleiotropic effects, including dwarfing, an altered pattern of stem elongation, darker color, bending of the lamina joint, absence of the midribs of leaves, and severe sterility.
URLPMID:21963665 [本文引用: 2]
URLPMID:26025517 [本文引用: 1]
DOI:10.1016/j.tplants.2006.12.001URLPMID:17224302 [本文引用: 1]
Linkage disequilibrium (LD) mapping in plants detects and locates quantitative trait loci (QTL) by the strength of the correlation between a trait and a marker. It offers greater precision in QTL location than family-based linkage analysis and should therefore lead to more efficient marker-assisted selection, facilitate gene discovery and help to meet the challenge of connecting sequence diversity with heritable phenotypic differences. Unlike family-based linkage analysis, LD mapping does not require family or pedigree information and can be applied to a range of experimental and non-experimental populations. However, care must be taken during analysis to control for the increased rate of false positive results arising from population structure and variety interrelationships. In this review, we discuss how suitable the recently developed alternative methods of LD mapping are for crops.
URLPMID:19584810 [本文引用: 1]
DOI:10.1126/science.1109557URLPMID:15761122 [本文引用: 1]
Age-related macular degeneration (AMD) is a major cause of blindness in the elderly. We report a genome-wide screen of 96 cases and 50 controls for polymorphisms associated with AMD. Among 116,204 single-nucleotide polymorphisms genotyped, an intronic and common variant in the complement factor H gene (CFH) is strongly associated with AMD (nominal P value <10(-7)). In individuals homozygous for the risk allele, the likelihood of AMD is increased by a factor of 7.4 (95% confidence interval 2.9 to 19). Resequencing revealed a polymorphism in linkage disequilibrium with the risk allele representing a tyrosine-histidine change at amino acid 402. This polymorphism is in a region of CFH that binds heparin and C-reactive protein. The CFH gene is located on chromosome 1 in a region repeatedly linked to AMD in family-based studies.
URLPMID:27898819 [本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12863-016-0340-2URL [本文引用: 1]
DOI:10.1007/s12041-016-0679-1URLPMID:27659335 [本文引用: 1]
Awn is one of the most important domesticated traits in rice (Oryza sativa). Understanding the genetic basis of awn length is important for grain harvest and production, because long awn length is disadvantageous for both grain harvest and milling. We investigated the awn length of 529 rice cultivars and performed a Genomewide association studies (GWAS) in the indica and japonica subpopulations, and the whole population. In total, we found 17 loci associated with awn length. Of these loci, seven were linked to previously reported quantitative trait loci, and one was linked to the awn gene An-1. Nine novel loci were repeatedly identified in different environments. One of the nine associations was identified in both the whole and japonica populations. Special interest was the detection of the most significant association SNP, sf0136352825, which was less than 95 kb from the seed shattering gene qSH1. These results may provide potentially favourable haplotypes for molecular breeding in rice.
URLPMID:28110091 [本文引用: 1]
DOI:10.1146/annurev-arplant-050213-035715URL [本文引用: 1]
DOI:10.1038/ng.1018URLPMID:22138690 [本文引用: 1]
A high-density haplotype map recently enabled a genome-wide association study (GWAS) in a population of indica subspecies of Chinese rice landraces. Here we extend this methodology to a larger and more diverse sample of 950 worldwide rice varieties, including the Oryza sativa indica and Oryza sativa japonica subspecies, to perform an additional GWAS. We identified a total of 32 new loci associated with flowering time and with ten grain-related traits, indicating that the larger sample increased the power to detect trait-associated variants using GWAS. To characterize various alleles and complex genetic variation, we developed an analytical framework for haplotype-based de novo assembly of the low-coverage sequencing data in rice. We identified candidate genes for 18 associated loci through detailed annotation. This study shows that the integrated approach of sequence-based GWAS and functional genome annotation has the potential to match complex traits to their causal polymorphisms in rice.
DOI:10.1038/ng.695URLPMID:20972439 [本文引用: 1]
Uncovering the genetic basis of agronomic traits in crop landraces that have adapted to various agro-climatic conditions is important to world food security. Here we have identified approximately 3.6 million SNPs by sequencing 517 rice landraces and constructed a high-density haplotype map of the rice genome using a novel data-imputation method. We performed genome-wide association studies (GWAS) for 14 agronomic traits in the population of Oryza sativa indica subspecies. The loci identified through GWAS explained approximately 36% of the phenotypic variance, on average. The peak signals at six loci were tied closely to previously identified genes. This study provides a fundamental resource for rice genetics research and breeding, and demonstrates that an approach integrating second-generation genome sequencing and GWAS can be used as a powerful complementary strategy to classical biparental cross-mapping for dissecting complex traits in rice.
DOI:10.1186/s13059-017-1289-9URLPMID:28838319 [本文引用: 1]
BACKGROUND: Soybean (Glycine max [L.] Merr.) is one of the most important oil and protein crops. Ever-increasing soybean consumption necessitates the improvement of varieties for more efficient production. However, both correlations among different traits and genetic interactions among genes that affect a single trait pose a challenge to soybean breeding. RESULTS: To understand the genetic networks underlying phenotypic correlations, we collected 809 soybean accessions worldwide and phenotyped them for two years at three locations for 84 agronomic traits. Genome-wide association studies identified 245 significant genetic loci, among which 95 genetically interacted with other loci. We determined that 14 oil synthesis-related genes are responsible for fatty acid accumulation in soybean and function in line with an additive model. Network analyses demonstrated that 51 traits could be linked through the linkage disequilibrium of 115 associated loci and these links reflect phenotypic correlations. We revealed that 23 loci, including the known Dt1, E2, E1, Ln, Dt2, Fan, and Fap loci, as well as 16 undefined associated loci, have pleiotropic effects on different traits. CONCLUSIONS: This study provides insights into the genetic correlation among complex traits and will facilitate future soybean functional studies and breeding through molecular design.
DOI:10.1023/B:PLAN.0000038256.89809.57URLPMID:15316287 [本文引用: 1]
Members of the GRAS gene family encode transcriptional regulators that have diverse functions in plant growth and development such as gibberellin signal transduction, root radial patterning, axillary meristem formation, phytochrome A signal transduction, and gametogenesis. Bioinformatic analysis identified 57 and 32 GRAS genes in rice and Arabidopsis, respectively. Here, we provide a complete overview of this gene family, describing the gene structure, gene expression, chromosome localization, protein motif organization, phylogenetic analysis, and comparative analysis between rice and Arabidopsis. Phylogenetic analysis divides the GRAS gene family into eight subfamilies, which have distinct conserved domains and functions. Both genome/segmental duplication and tandem duplication contributed to the expansion of the GRAS gene family in the rice and Arabidopsis genomes. The existence of GRAS-like genes in bryophytes suggests that GRAS is an ancient family of transcription factors, which arose before the appearance of land plants over 400 million years ago.
DOI:10.1186/s12284-019-0285-yURLPMID:30976929 [本文引用: 5]
BACKGROUND: Salinity-alkalinity stress is one of the major factors limiting rice production. The damage caused by alkaline salt stress to rice growth is more severe than that caused by neutral salt stress. At present, the genetic resources (quantitative trait loci (QTLs) and genes) that can be used by rice breeders to improve alkalinity tolerance are limited. Here, we assessed the alkalinity tolerance of rice at the seedling stage and performed a genome-wide association study (GWAS) based on genotypic data including 788,396 single-nucleotide polymorphisms (SNPs) developed by re-sequencing 295 japonica rice varieties. RESULTS: We used the score of alkalinity tolerance (SAT), the concentrations of Na(+) and K(+) in the shoots (SNC and SKC, respectively) and the Na(+)/K(+) ratio of shoots (SNK) as indices to assess alkalinity tolerance at the seedling stage in rice. Based on population structure analysis, the japonica rice panel was divided into three subgroups. Linkage disequilibrium (LD) analysis showed that LD decay occurred at 109.77 kb for the whole genome and varied between 13.79 kb and 415.77 kb across the 12 chromosomes, at which point the pairwise squared correlation coefficient (r(2)) decreased to half of its maximum value. A total of eight QTLs significantly associated with the SAT, SNC and SNK were identified by genome-wide association mapping. A common QTL associated with the SAT, SNC and SNK on chromosome 3 at the position of 15.0 Mb, which explaining 13.36~13.64% of phenotypic variation, was selected for further analysis. The candidate genes were filtered based on LD decay, Gene Ontology (GO) enrichment, RNA sequencing data, and quantitative real-time PCR (qRT-PCR) analysis. Moreover, sequence analysis revealed one 7-bp insertion/deletion (indel) difference in LOC_Os03g26210 (OsIRO3) between the alkalinity-tolerant and alkalinity-sensitive rice varieties. OsIRO3 encodes a bHLH-type transcription factor and has been shown to be a negative regulator of the Fe-deficiency response in rice. CONCLUSION: Based on these results, OsIRO3 maybe a novel functional gene associated with alkalinity tolerance in japonica rice. This study provides resources for improving alkalinity tolerance in rice, and the functional molecular marker could be verified to breed new rice varieties with alkalinity tolerance via marker-assisted selection (MAS).
DOI:10.1016/j.ajhg.2010.11.011URLPMID:21167468 [本文引用: 1]
For most human complex diseases and traits, SNPs identified by genome-wide association studies (GWAS) explain only a small fraction of the heritability. Here we report a user-friendly software tool called genome-wide complex trait analysis (GCTA), which was developed based on a method we recently developed to address the
DOI:10.1093/oxfordjournals.molbev.a040454URLPMID:3447015 [本文引用: 1]
A new method called the neighbor-joining method is proposed for reconstructing phylogenetic trees from evolutionary distance data. The principle of this method is to find pairs of operational taxonomic units (OTUs [= neighbors]) that minimize the total branch length at each stage of clustering of OTUs starting with a starlike tree. The branch lengths as well as the topology of a parsimonious tree can quickly be obtained by using this method. Using computer simulation, we studied the efficiency of this method in obtaining the correct unrooted tree in comparison with that of five other tree-making methods: the unweighted pair group method of analysis, Farris's method, Sattath and Tversky's method, Li's method, and Tateno et al.'s modified Farris method. The new, neighbor-joining method and Sattath and Tversky's method are shown to be generally better than the other methods.
URLPMID:27004904 [本文引用: 1]
DOI:10.1093/bioinformatics/btm308URLPMID:17586829 [本文引用: 1]
Association analyses that exploit the natural diversity of a genome to map at very high resolutions are becoming increasingly important. In most studies, however, researchers must contend with the confounding effects of both population and family structure. TASSEL (Trait Analysis by aSSociation, Evolution and Linkage) implements general linear model and mixed linear model approaches for controlling population and family structure. For result interpretation, the program allows for linkage disequilibrium statistics to be calculated and visualized graphically. Database browsing and data importation is facilitated by integrated middleware. Other features include analyzing insertions/deletions, calculating diversity statistics, integration of phenotypic and genotypic data, imputing missing data and calculating principal components.
[本文引用: 1]
[本文引用: 1]
[本文引用: 4]
URLPMID:7761420 [本文引用: 1]
DOI:10.1074/jbc.271.14.8126URLPMID:8626500 [本文引用: 1]
A chimeric Ca2+/calmodulin-dependent protein kinase (CCaMK) gene characterized by a catalytic domain, a calmodulin-binding domain, and a neural visinin-like Ca2+-binding domain was recently cloned from plants (Patil, S., Takezawa, D., and Poovaiah, B. W. (1995) Proc. Natl. Acad. Sci. U. S. A. 92, 4797-4801). The Escherichia coli-expressed CCaMK phosphorylates various protein and peptide substrates in a Ca2+/calmodulin-dependent manner. The calmodulin-binding region of CCaMK has similarity to the calmodulin-binding region of the alpha-subunit of multifunctional Ca2+/calmodulin-dependent protein kinase (CaMKII). CCaMK exhibits basal autophosphorylation at the threonine residue(s) (0.098 mol of 32P/mol) that is stimulated 3.4-fold by Ca2+ (0.339 mol of 32P/mol), while calmodulin inhibits Ca2+-stimulated autophosphorylation to the basal level. A deletion mutant lacking the visinin-like domain did not show Ca2+-stimulated autophosphorylation activity but retained Ca2+/calmodulin-dependent protein kinase activity at a reduced level. Ca2+-dependent mobility shift assays using E. coli-expressed protein from residues 358 520 revealed that Ca2+ binds to the visinin-like domain. Studies with site-directed mutants of the visinin-like domain indicated that EF-hands II and III are crucial for Ca2+-induced conformational changes in the visinin-like domain. Autophosphorylation of CCaMK increases Ca2+/calmodulin-dependent protein kinase activity by about 5-fold, whereas it did not affect its Ca2+-independent activity. This report provides evidence for the existence of a protein kinase in plants that is modulated by Ca2+ and Ca2+/calmodulin. The presence of a visinin-like Ca2+-binding domain in CCaMK adds an additional Ca2+-sensing mechanism not previously known to exist in the Ca2+/calmodulin-mediated signaling cascade in plants.
URLPMID:19208695 [本文引用: 1]
URLPMID:30740793 [本文引用: 1]
[本文引用: 1]
DOI:10.1101/gad.256603URLPMID:12815068 [本文引用: 1]
Shoot branching is inhibited by auxin transported down the stem from the shoot apex. Auxin does not accumulate in inhibited buds and so must act indirectly. We show that mutations in the MAX4 gene of Arabidopsis result in increased and auxin-resistant bud growth. Increased branching in max4 shoots is restored to wild type by grafting to wild-type rootstocks, suggesting that MAX4 is required to produce a mobile branch-inhibiting signal, acting downstream of auxin. A similar role has been proposed for the pea gene, RMS1. Accordingly, MAX4 and RMS1 were found to encode orthologous, auxin-inducible members of the polyene dioxygenase family.
URLPMID:15342640 [本文引用: 1]
URLPMID:17547665 [本文引用: 1]
URLPMID:16531486 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}