Profiling and Regulation Network of Differentially Expressed Genes During the Development Process of Apis mellifera ligustica Worker’s Midgut
DU Yu, ZHOU DingDing, WAN JieQi, LU JiaXuan, FAN XiaoXue, FAN YuanChan, CHEN Heng, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, XU GuoJun, CHEN DaFu, GUO Rui,College of Animal Sciences (College of Bee Science), Fujian Agriculture and Forestry University, Fuzhou 350002
Received:2019-07-9Accepted:2019-09-2Online:2020-01-01 作者简介 About authors 杜宇,E-mail:m18505700830@163.com。
周丁丁,E-mail:ZDD03569981@163.com。
摘要 【目的】前期已对意大利蜜蜂(Apis mellifera ligustica,简称意蜂)7日龄工蜂中肠(Am7)、10日龄工蜂中肠(Am10)进行全转录组测序,本研究基于高质量的组学数据探究中肠发育过程中的差异基因表达谱及其调控网络,以期解析意蜂工蜂中肠发育的分子机理。【方法】根据FPKM(fragments per kilobase of transcript per million mapped reads)算法计算基因表达量,并以|log2 fold change|≥1且P≤0.05作为标准筛选得到差异表达基因(differentially expressed gene,DEG)。利用TargetFinder软件预测ame-miR-6001-3p的靶mRNA。利用相关生物信息学软件,对全部DEG进行GO和KEGG数据库注释。筛选出与AMPK、P13K-Akt、Wnt、cAMP、FoxO、Hippo、mTOR、Jak-STAT、Toll-like受体、TGF-beta、Notch、MAPK和NF-κB 13条信号通路存在富集关系的DEG,以及与ame-miR-6001-3p存在靶向结合关系的DEG,通过Cytoscape软件构建调控网络将其富集关系与调控关系可视化。利用茎环反转录PCR(Stem loop RT-PCR)和实时荧光定量PCR(RT-qPCR)验证ame-miR-6001-3p以及DEG在Am7和Am10中的差异表达情况。【结果】Am7 vs Am10比较组中共有1 038个DEG,包括515个上调基因和523个下调基因。这些DEG涉及细胞进程、代谢进程和催化活性等功能条目,并显著富集在氧化磷酸化、氨基糖与核苷酸糖代谢、脂肪酸代谢和嘌呤代谢等能量和物质代谢通路,表明工蜂中肠内存在旺盛细胞生命与新陈代谢活动。表达量聚类分析发现分别有20、18、15和14个DEG富集在AMPK信号通路、P13K-Akt信号通路、内吞作用和Hippo信号通路。57个DEG与P13K-Akt、Wnt、Jak-STAT等上述13条与生长发育和免疫防御相关的信号通路存在富集关系,且1个DEG可与多条信号通路存在富集关系。调控网络分析结果显示,分别有54个上调基因和44个下调基因可被ame-miR-6001-3p靶向结合;上调基因富集在磷酸肌醇代谢、胰岛素信号通路、Hippo信号通路和谷胱甘肽代谢等43条代谢通路,而下调基因富集在Hippo信号通路、新陈代谢途径、谷胱甘肽代谢和花生四烯酸代谢等20条代谢通路。RT-qPCR结果显示随机挑选的6个DEG的表达量变化趋势与测序数据一致,证实了本研究中基因差异表达真实可靠。此外ame-miR-6001-3p在Am7和Am10内均真实表达,并在Am10中的相对表达量显著较低。【结论】对意蜂工蜂中肠发育过程的DEG表达谱和DEG与ame-miR-6001-3p之间的调控关系,以及DEG的潜在作用进行深入分析和探讨,发现DEG可参与TGF-beta、Wnt、Hippo、Notch、PI3K-Akt、mTOR、AMPK和NF-κB等各类信号通路进而影响中肠的生长发育和免疫防御,DEG可通过与显著下调表达的ame-miR-6001-3p形成复杂的调控网络参与意蜂工蜂中肠发育过程中胰岛素信号通路等多条代谢途径。 关键词:意大利蜜蜂;中肠;发育;差异表达基因;竞争性内源RNA;调控网络
Abstract 【Objective】The whole transcriptome sequencing of Apis mellifera ligustica 7- and 10-day-old workers’ midguts (Am7 and Am10) was previously conducted. In this study, the differential expression profile and regulation network of genes were investigated to reveal the molecular mechanism underlying the midgut development.【Method】Gene expressions were calculated based on FPKM (fragments per kilobase of transcript per million mapped reads) algorithm, and differentially expressed genes (DEGs) were gained following the standard |log2 fold change|≥1 and P≤0.05. Target mRNAs of ame-miR-6001-3p were predicted utilizing TargetFinder. Annotations of all DEGs in GO and KEGG databases were performed using related software. In addition, DEGs enriched in 13 signaling pathways including AMPK, P13K-Akt, Wnt, cAMP, FoxO, Hippo, mTOR, Jak-STAT, Toll-like receptor, TGF-beta, Notch, MAPK and NF-κB, as well as DEGs targeted by ame-miR-6001-3p were screened out, followed by visualization of enrichment networks and regulation networks with Cytoscape. Finally, Stem loop RT-PCR and RT-qPCR were used to verify the expression and differential expression pattern of ame-miR-6001-3p and DEGs in Am7 and Am10.【Result】A total of 1 038 DEGs were identified in Am7 vs Am10 comparison group, including 515 up- and 523 down-regulated genes, respectively. These DEGs were associated with cellular process, metabolic process and catalytic activity, and significantly enriched in some material and energy metabolisms such as oxidative phosphorylation, amino sugar and nucleotide sugar metabolisms, fatty acid metabolism and purine metabolism, indicative of the active cellular and metabolic activities. Expression cluster analysis suggested that 20, 18, 15 and 14 DEGs were respectively enriched in AMPK signaling pathway, PI3K-Akt signaling pathway, endocytosis and Hippo signaling pathway. In addition, 57 DEGs were enriched in the aforementioned 13 signaling pathways associated with growth and development as well as immune defense, among them one DEG was enriched in several signaling pathways. Moreover, regulation network analysis showed that 54 up-regulated genes and 44 down-regulated genes were targets of ame-miR-6001-3p, respectively; these up-regulated genes were enriched in 43 pathways including inositol phosphate metabolism, Hippo signaling pathway, glutathione metabolism and insulin signaling pathway, while these down-regulated genes were enriched in 20 pathways including Hippo signaling pathway, metabolic pathways, glutathione metabolism and arachidonic acid metabolism. Moreover, RT-qPCR result showed that the variation trend of six randomly selected DEGs were consistent with that in sequencing data, confirming the reliability of DEGs. Finally, ame-miR-6001-3p was definitely expressed and significantly down-regulated in Am10.【Conclusion】In this work, the expression pattern of DEGs and the regulation network between DEGs and ame-miR-6001-3p as well as the potential role of DEGs during the developmental process of A. m. ligustica worker’s midgut were deeply analyzed. The results revealed that the DEGs may participate in various signaling pathways including TGF-beta, Wnt, Hippo, Notch, PI3K-Akt, mTOR, AMPK, NF-κB signaling pathways, thus affecting the growth and development as well as immune defense of the midgut; DEGs were likely to regulate several signaling pathways such as insulin signaling pathway during the midgut development via formation regulation networks with significantly down-regulated ame-miR-6001-3p. Keywords:Apis mellifera ligustica;midgut;development;differentially expressed gene (DEG);competitive endogenous RNA;regulation network
PDF (6040KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 杜宇, 周丁丁, 万洁琦, 卢家轩, 范小雪, 范元婵, 陈恒, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 陈大福, 郭睿. 意大利蜜蜂工蜂中肠发育过程中的差异基因 表达谱及调控网络[J]. 中国农业科学, 2020, 53(1): 201-212 doi:10.3864/j.issn.0578-1752.2020.01.019 DU Yu, ZHOU DingDing, WAN JieQi, LU JiaXuan, FAN XiaoXue, FAN YuanChan, CHEN Heng, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, XU GuoJun, CHEN DaFu, GUO Rui. Profiling and Regulation Network of Differentially Expressed Genes During the Development Process of Apis mellifera ligustica Worker’s Midgut[J]. Scientia Acricultura Sinica, 2020, 53(1): 201-212 doi:10.3864/j.issn.0578-1752.2020.01.019
利用Perl脚本剔除测序数据原始读段中未知核苷酸含量>5%以及含量>50%的质量低的读段(reads),获得有效读段(clean reads)。过滤可比对上核糖体RNA数据库的reads,进而通过TopHat2将unmapped reads比对到西方蜜蜂参考基因组(Amel_4.5)(https://www.ncbi.nlm.nih.gov/assembly/GCF_000002195.4)。校准参数如下:(1)最大读取不匹配为2;(2)配对读取目标区域为50 bp;(3)配对读取目标区域误差为±80 bp。利用Cufflinks将对比到转录本上的reads进行组装。根据FPKM(fragments per kilobase of transcript per million mapped reads)算法计算基因表达量,并利用R软件计算Pearson相关性系数来评估同组样品的生物学重复性。
A:AMPK信号通路AMPK signaling pathway;B:内吞作用Endocytosis;C:P13K-Akt信号通路PI3K-Akt signaling pathway;D:Hippo信号通路Hippo signaling pathway Fig. 3Expression clustering of DEGs enriched in AMPK, PI3K-Akt, Hippo signaling pathway and endocytosis
57个DEG在13条信号通路(AMPK、P13K-Akt、Wnt、cAMP、FoxO、Hippo、mTOR、Jak-STAT、Toll-like受体、TGF-beta、Notch、MAPK和NF-κB)的富集网络关系,利用Cytoscape软件进行可视化,结果显示一个基因可同时富集在多条信号通路(图4),例如磷脂酰肌醇3-激酶调节亚基α-类编码基因(XM_016917733.1和XM_392119.6)富集在7条信号通路(AMPK、PI3-Akt、cAMP、FoxO、mTOR、Jak-STAT和Toll-like受体);Ras相关蛋白rac1亚型编码基因(TCONS_00021690和TCONS_00038549)富集在5条信号通路(P13K-Akt、cAMP(NF-κB、AMPK)、Toll-like受体、Wnt、MAPK)(图4);Smad(mothers against decapentaplegic homolog)3亚型编码基因(TCONS_00001946)与4条信号通路(Wnt、FoxO、Hippo和TGF-beta)存在富集关系;类胰岛素样受体编码基因(XM_016918001.1)、mTOR的调节相关蛋白编码基因(TCONS_00038733)、磷酸烯醇丙酮酸羧激酶编码基因(XM_006560464.2)等8个DEG均与3条信号通路存在富集关系。
六边形代表信号通路Hexagons indicate signaling pathways;红色圆形代表上调基因Red circles indicate up-regulated genes;绿色圆形代表下调基因 Green circles indicate down-regulated genes;六边形和圆形的大小代表连接DEG或信号通路的数量 The size of hexagons and circles indicates the number of DEGs or signaling pathways connected Fig. 4Relationship networks between 13 signaling pathways and related DEGs in A. m. ligustica worker’s midgut
肠道是蜜蜂吸收消化以及免疫防御的主要场所[16,17],肠道的发育情况及内环境稳态共同影响着不同虫态蜜蜂的正常生命活动。前期研究中,笔者团队对意蜂幼虫肠道发育及响应蜜蜂球囊菌(Ascosphaera apis)胁迫的mRNA和miRNA应答进行了全面细致的组学研究[7,15,18-19];也分别在lncRNA和circRNA组学层面探究了意蜂工蜂中肠的发育机理[10,11]。基于前期已获得的意蜂工蜂中肠的全转录组数据,本研究对意蜂工蜂中肠发育过程的DEG及其潜在作用,以及融合DEG与ame-miR-6001-3p的调控网络进行深入分析,发现Am7 vs Am10比较组的上调基因和下调基因数分别为515和523个,其中分别有285、273和258个DEG涉及细胞进程、代谢进程和催化活性等功能条目,分别有13、8、8、6、6和5个DEG富集在氧化磷酸化、磷酸肌醇代谢、碳代谢、氨基糖与核苷酸糖代谢、脂肪酸代谢和嘌呤代谢等能量和物质代谢通路,说明意蜂工蜂中肠发育过程伴随着较为旺盛细胞生命与新陈代谢活动。
COON KL, VOGEL KJ, BROWN MR, STRAND MR . Mosquitoes rely on their gut microbiota for development Molecular Ecology, 2014,23(11):2727-2739. [本文引用: 1]
CHOUAIAB, ROSSIP, MONTAGNAM, RICCII, CROTTIE, DAMIANIC, EPISS, FAYEI, SAGNONN, ALMAA, FAVIAG, DAFFONCHIOD, BANDIC . Molecular evidence for multiple infections as revealed by typing of Asaia bacterial symbionts of four mosquito species Applied and Environmental Microbiology, 2010,76(22):7444-7450. [本文引用: 1]
STORELLIG, DEFAYEA, ERKOSARB, HOLSP, ROYETJ, LEULIERF . Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing Cell Metabolism, 2011,14(3):403-414. [本文引用: 1]
ZHENGH, POWELL JE, STEELE MI, DIETRICHC, MORAN NA . Honeybee gut microbiota promotes host weight gain via bacterial metabolism and hormonal signaling Proceedings of the National Academy of Sciences of the United States of America, 2017,114(18):4775-4780. [本文引用: 1]
FENG QQ . Optimal level of VA and VE in pollen substitutes of Apis mellifera ligustica during the period of spring multiplication [D]. Taian: Shandong Agricultural University, 2012. (in Chinese) [本文引用: 1]
GUOR, XIE YL, XIONG CL, YIN WX, ZHENG YZ, FU ZM, CHEN DF . Trend analysis for differentially expressed genes in the developmental process of 4-, 5- and 6-day-old larval guts of Apis mellifera ligustica Journal of Shanghai Jiaotong University (Agricultural Science), 2018,36(4):14-21, 29. (in Chinese) [本文引用: 2]
GUOR, DUY, XIONG CL, ZHENG YZ, FU ZM, XU GJ, WANG HP, CHEN HZ, GENG SH, ZHOU DD, SHI CY, ZHAO HX, CHEN DF . Differentially expressed microRNA and their regulation networks during the developmental process of Apis mellifera ligustica larval gut Scientia Agricultura Sinica, 2018,51(21):4197-4209. (in Chinese) [本文引用: 4]
MELLO T RP, ALEIXO AC, PINHEIRO DG, NUNES F MF, BITONDI M MG, HARTFELDERK, BARCHUK AR, SIM?ES Z LP . Developmental regulation of ecdysone receptor (EcR) and EcR-controlled gene expression during pharate-adult development of honeybees ( Apis mellifera) Frontiers in Genetics, 2014,5:445. [本文引用: 2]
COLLINS DH, MOHORIANUI, BECKERSM, MOULTONV, DALMAYT, BOURKE AF . MicroRNAs associated with caste determination and differentiation in a primitively eusocial insect Scientific Reports, 2017,7:45674. [本文引用: 2]
GUOR, GENG SH, XIONG CL, ZHENG YZ, FU ZM, WANG HP, DUY, TONG XY, ZHAO HX, CHEN DF . Differential expression analysis of long non-coding RNAs during the developmental process of Apis mellifera ligustica worker’s midgut Scientia Agricultura Sinica, 2018,51(18):3600-3613. (in Chinese) [本文引用: 7]
GUOR, CHEN HZ, XIONG CL, ZHENG YZ, FU ZM, XU GJ, DUY, WANG HP, GENG SH, ZHOU DD, LIU SY, CHEN DF . Analysis of differentially expressed circular RNAs and their regulation networks during the developmental process of Apis mellifera ligustica worker’s midgut Scientia Agricultura Sinica, 2018,51(23):4575-4590. (in Chinese) [本文引用: 5]
HUANGQ, CHEN YP, WANG RW, CHENGS, EVANS JD . Host-parasite interactions and purifying selection in a microsporidian parasite of honey bees PLoS ONE, 2016,11(2):e0147549. [本文引用: 1]
HUANG WF, SOLTER LF . Comparative development and tissue tropism of Nosema apis and Nosema ceranae Journal of Invertebrate Pathology, 2013,113(1):35-41. [本文引用: 1]
SMOOT ME, ONOK, RUSCHEINSKIJ, WANG PL, IDEKERT . Cytoscape 2.8: new features for data integration and network visualization Bioinformatics (Oxford, England), 2011,27(3):431-432. [本文引用: 1]
GUOR, DUY, TONG XY, XIONG CL, ZHENG YZ, XU GJ, WANG HP, GENG SH, ZHOU DD, GUO YL, WU SZ, CHEN DF . Differentially expressed microRNAs and their regulation networks in Apis mellifera ligustica larval gut during the early stage of Ascosphaera apis infection Scientia Agricultura Sinica, 2019,52(1):166-180. (in Chinese) [本文引用: 2]
MARTINSON VG, MOYJ, MORAN NA . Establishment of characteristic gut bacteria during development of the honeybee worker Applied and Environmental Microbiology, 2012,78(8):2830-2840. [本文引用: 1]
KWONG WK, MANCENIDO AL, MORAN NA . Immune system stimulation by the native gut microbiota of honey bees Royal Society Open Science, 2017,4(2):170003. [本文引用: 1]
CHEND, GUOR, XUX, XIONGC, LIANGQ, ZHENGY, LUOQ, ZHANGZ, HUANGZ, KUMARD, XIW, ZOUX, LIUM . Uncovering the immune responses of Apis mellifera ligustica larval gut to Ascosphaera apis infection utilizing transcriptome sequencing Gene, 2017,621:40-50. [本文引用: 1]
GUOR, DUY, ZHOU NH, LIU SY, XIONG CL, ZHENG YZ, FU ZM, XU GJ, WANG HP, GENG SH, ZHOU DD, CHEN DF . Comprehensive analysis of differentially expressed microRNAs and their target genes in the larval gut of Apis mellifera ligustica during the late stage of Ascosphaera apis stress Acta Entomologica Sinica, 2019,62(1):49-60. (in Chinese) [本文引用: 1]
BARRY ER, CAMARGO FD . The Hippo superhighway: signaling crossroads converging on the Hippo/Yap pathway in stem cells and development Current Opinion in Cell Biology, 2013,25(2):247-253. [本文引用: 1]
CHENX . Identification of differentially expressed coding and noncoding RNAs during ovary activation and oviposition in honey bees [D]. Beijing: Chinese Academy of Agricultural Sciences, 2017. (in Chinese) [本文引用: 1]
NINGY, MIX, CHEN XY, SHAO JH, ZAN LS . Silencing and overexpressing SMAD family member 1 (SMAD1) gene and its effect on myogenesis in primary myoblast of Qinchuan cattle(Bos taurus) Scientia Agricultura Sinica, 2019,52(10):1818-1829. (in Chinese) [本文引用: 1]
BRUMMELT, ABDOLLAHS, HAERRY TE, SHIMELL MJ, MERRIAMJ, RAFTERYL, WRANA JL, O’CONNOR MB . The Drosophila activin receptor baboon signals through dSmad2 and controls cell proliferation but not patterning during larval development Genes and Development, 1999,13(1):98-111. [本文引用: 1]
ZHANGZ, LU ZY . Research progress on insulin signaling pathway and its function in silkworm Newsletter of Sericultural Science, 2014,34(2):22-26. (in Chinese) [本文引用: 1]
GAUTAMS, ISHRATN, YADAVP, SINGHR, NARENDERT, SRIVASTAVA AK . 4-Hydroxyisoleucine attenuates the inflammation- mediated insulin resistance by the activation of AMPK and suppression of SOCS-3 coimmunoprecipitation with both the IR-β subunit as well as IRS-1. Molecular and Cellular Biochemistry, 2016,414(1/2):95-104. [本文引用: 1]
NEUFELD TP . Body building: regulation of shape and size by PI3K/TOR signaling during development Mechanisms of Development, 2003,120(11):1283-1296. [本文引用: 1]
KAURS, SASSANOA, DOLNIAKB, JOSHIS, MAJCHRZAK- KITAB, BAKER DP, HAYN, FISH EN, PLATANIAS LC . Role of the Akt pathway in mRNA translation of interferon-stimulated genes Proceedings of the National Academy of Sciences of the United States of America, 2008,105(12):4808-4813. [本文引用: 1]
LIUS, LUCAS KJ, ROYS, HAJ, RAIKHEL AS . Mosquito-specific microRNA-1174 targets serine hydroxymethyltransferase to control key functions in the gut Proceedings of the National Academy of Sciences of the United States of America, 2014,111(40):14460-14465. [本文引用: 1]
XIONG CL, DUY, CHEN DF, ZHENG YZ, FU ZM, WANG HP, GENG SH, CHEN HZ, ZHOU DD, WU SZ, SHI CY, GUOR . Bioinformatic prediction and analysis of miRNAs in the Apis mellifera ligustica larval gut Chinese Journal of Applied Entomology, 2018,55(6):1023-1033. (in Chinese) [本文引用: 1]
KARRENTH FA, TAYY, PERNAD, ALAU, TAN SM, RUST AG, DENICOLAG, WEBSTER KA, WEISSDPEREZ-MANCERAP A, KRAUTHAMMERM, HALABANR, PROVEROP, ADAMSD J, TUVESOND A, PANDOLFIP P . In vivo identification of tumor-suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma . Cell, 2011,147(2):382-395. [本文引用: 1]
 ,福建农林大学动物科学学院(蜂学学院),福州 350002
,福建农林大学动物科学学院(蜂学学院),福州 350002
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT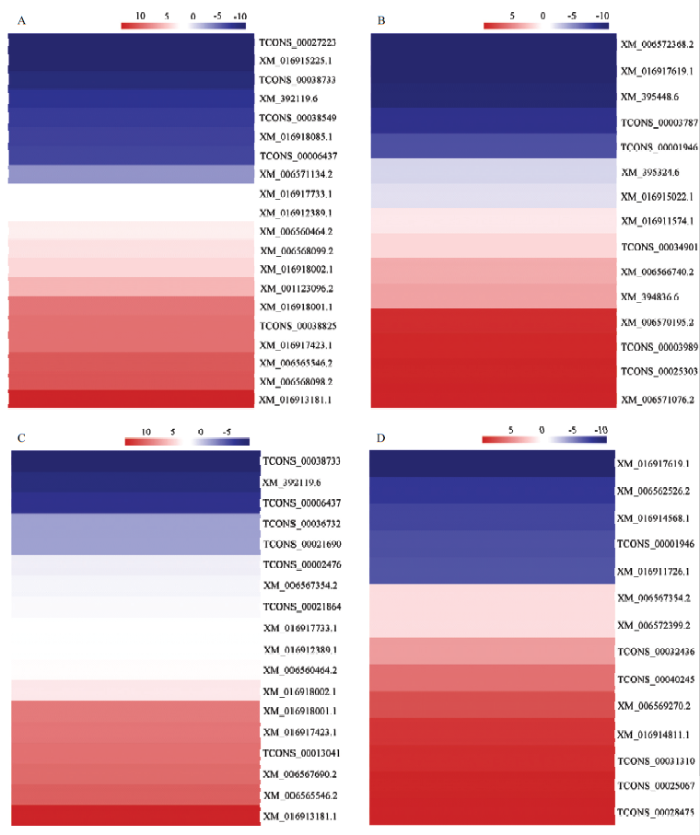 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT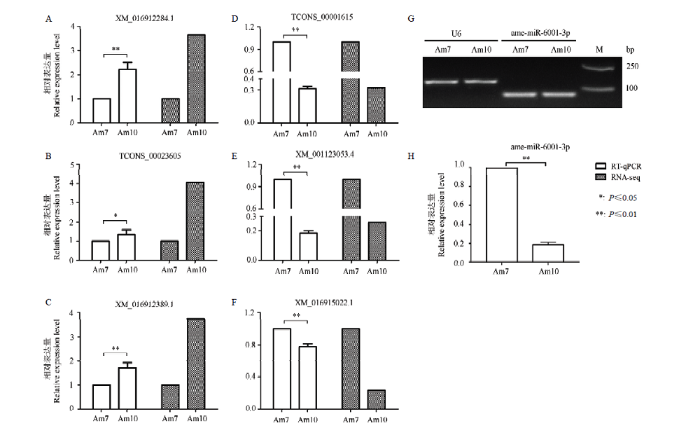 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}