 ,21
,21 2
Response and the Expression of Pi-Responsive Genes in Leymus chinensis Under Inorganic Phosphate Treatment
WAN DongLi1, HOU XiangYang1, DING Yong1, REN WeiBo1, WANG Kai1, LI XiLiang1, WAN YongQing,2通讯作者:
责任编辑: 李莉
收稿日期:2019-06-4接受日期:2019-09-19网络出版日期:2019-12-01
| 基金资助: |
Received:2019-06-4Accepted:2019-09-19Online:2019-12-01
作者简介 About authors
万东莉,E-mail:wandongli@caas.cn

摘要
关键词:
Abstract
Keywords:
PDF (1880KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
万东莉, 侯向阳, 丁勇, 任卫波, 王凯, 李西良, 万永青. 磷胁迫下羊草的响应及磷响应相关基因表达分析[J]. 中国农业科学, 2019, 52(23): 4215-4227 doi:10.3864/j.issn.0578-1752.2019.23.003
WAN DongLi, HOU XiangYang, DING Yong, REN WeiBo, WANG Kai, LI XiLiang, WAN YongQing.
0 引言
【研究意义】羊草(Leymus chinensis)隶属禾本科(Gramineae)赖草属,是欧亚大陆草原区东部草甸草原及干旱草原上的重要建群种之一。在中国内蒙古、东北三省、河北、山西和新疆等地广泛分布[1]。近半个世纪以来,由于过度放牧等原因,中国北方草原大面积退化,羊草等植物出现小型化,草原群落生产力锐减和优势种更替[2]。磷(phosphorus,P)是草原生产力的限制因子之一[3,4]。虽然通过施用磷肥可以提高退化草地土壤肥力、促进羊草等草原植物的生产力[4,5],但是磷是不可再生资源,并且过多施用磷肥会造成环境问题[6]。因此,通过分子改良获得羊草磷高效利用植物,是促进草原生产力恢复和提高的一个重要途径。【前人研究进展】磷是植物生长发育所必需的营养元素,不但是核酸、磷脂和ATP等生命大分子的重要组成成分,也是植物体内许多新陈代谢进程的关键调节子[7]。磷储量低和植物磷利用效率低是一个全球性的问题[6,8-9]。内蒙古天然草原土壤中全磷和有效磷含量的平均值分别为0.33 mg·g-1和3.53 mg·kg-1,其中,有效磷含量与全国土壤水平接近,但低于全球土壤水平[10]。中国草地植物磷的含量平均值为1.9 mg·g-1[11],内蒙古天然草地植物叶片磷的含量平均值为1.51 mg·g-1[10],低于英国草地植物的磷含量2.7 mg·g-1[12]。总体上,内蒙古草地和草地植物普遍处于缺磷状态[3]。过度放牧干扰下,典型草原严重退化样地(冷蒿+糙隐子草群落),与围封恢复样地(羊草+大针茅群落)相比,土壤全磷含量减少[13]。土壤中添加丛枝菌根真菌(arbuscular mycorrhizal fungi,AMF),能促进羊草根围土壤有效磷含量、对磷的吸收以及地上生物量的提高,有利于退化羊草草地的修复[14,15]。磷限制条件下植物会从形态、生理、生化和分子水平激发一系列的适应响应,例如减少植物生长,改变根系统体系结构,分泌有机酸、磷酸酶和和核酸酶以增加磷的获取[16]。基因表达模式的研究是现代分子生物学的基石之一。通过基因表达分析有助于深入了解复杂的生物过程,增加对环境响应信号和代谢途径的理解[17]。实时荧光定量PCR(quantitative real-time reverse transcription PCR,qRT-PCR)作为一个高效的基因表达检测工具,在生物学研究的各个领域广泛应用[18]。但是基因表达数据需要利用内参基因进行标准化处理[18]。内参基因的稳定表达是精确的标准化靶基因表达的关键,筛选不同条件下的合适内参基因是精确分析qRT-PCR结果的必要步骤[19]。在羊草中通过对不同组织中内参基因的稳定性分析,分别获得了根、茎、叶和穗中相对稳定的内参基因,可用作羊草不同组织qRT-PCR检测的内参基因[20]。【本研究切入点】目前,关于Pi处理条件下,羊草的内参基因筛选、以及Pi响应相关基因的表达研究尚缺乏。另外,在模式植物拟南芥、水稻和玉米等作物中对低磷响应的生理、分子适应机制研究较多[8-9,16,21-22]。在羊草中已有关于低磷响应的生理生化水平研究[23],但分子水平上的研究鲜见报道。【拟解决的关键问题】本研究通过对羊草进行不同浓度的Pi处理,从表型、生理水平上对羊草的响应情况进行分析。同时,对不同浓度Pi水平下羊草内参基因进行qRT-PCR检测,基于合适的内参基因筛选,对磷响应相关基因的表达进行标准化分析,为羊草Pi胁迫响应的分子机制研究提供理论基础和试验支持。1 材料与方法
1.1 植物培养和处理
表型鉴定和分子试验分别于2014—2015年和2018年在内蒙古自治区植物逆境生理与分子生物学重点实验室进行,所用材料为羊草吉生4号种子。将羊草种子播种于装有营养土和蛭石(V﹕V=1﹕2)的培养钵中,并置于植物温室培养,温度为23℃—25℃,光照为16 h光照/8 h黑暗。待种子发芽后,将幼苗移入Hoagland培养液中培养2 d,然后将幼苗移到分别含有0、0.01、0.33、5和30 mmol·L-1 KH2PO4的Hoagland培养液中进行Pi胁迫处理培养,以含1 mmol·L-1 KH2PO4作为对照,每隔4 d更换一次培养液并测量根长和株高(除25—31 d间隔6 d外),每种处理至少测量20株羊草。生长35 d后,分别取根和地上组织样品用于Pi含量检测。基因表达检测:羊草培养和处理方法同前面所述,共设5个Pi浓度梯度(0、0.01、1、5和30 mmol·L-1),生长35 d左右,每个处理取3株羊草混合作为RNA提取检测的样品,并迅速放于液氮速冻后于-80℃冰箱保存。3次独立生物学重复。
1.2 植物Pi含量检测
将经过不同浓度Pi处理的羊草不同组织烘干并研成粉末,利用钒钼黄比色法对Pi的含量进行检测。1.3 总RNA提取和cDNA合成
取0.05—0.1 g羊草样品,利用植物RNA提取试剂盒MiniBEST Plant RNA Extraction Kit(TaKaRa)提取总RNA,具体方法按照说明书进行操作。通过吸光率A260/280和琼脂糖凝胶电泳分别对RNA的纯度和完整性进行检测。取2 μg总RNA,利用反转录试剂盒PrimeScriptTM RT reagent Kit with gDNA Eraser(TaKaRa)进行cDNA合成,具体步骤按产品说明书进行。将反转录得到的cDNA原液稀释10倍作为基因表达检测的模板。1.4 内参基因选择和引物设计评价
试验共用到8个qRT-PCR候选内参基因,其中7个基因序列(包括LcTUA、LcTUB、Lc18SrRNA、LcCAP、LcGAPDH、LcEF1α和LcAPRT)来源于羊草转录组数据库,LcACT序列来自于NCBI(GenBank登录号HM623326.1)。采用生物软件Primer Premier 5设计qRT-PCR检测特异引物(表1),LcACT引物序列参考文献[20,24]。引物设计参数为:引物退火温度59℃—61℃,引物序列长度18—25 bp,扩增产物长度80—200 bp,GC含量40%—60%。Table 1
表1
表1基因和qRT-PCR引物特性
Table 1
| 基因名称 Gene name | 基因描述 Gene description | 引物序列 Primer sequences (5′-3′) | 引物扩增效率 Amplification efficiency (%) | 产物大小 Amplicon length (bp) | 溶解温度 Product melting temperature (℃) |
|---|---|---|---|---|---|
| LcTUA | α-微管蛋白Alpha-tubulin | F: ACGAGGCCATCTACGACATCT | 95.0 | 135 | 87.6 |
| R: CCACGTTCAGGGCACCAT | |||||
| LcTUB | β-微管蛋白Beta-tubulin | F: TCCCTCGCCTCCACTTCTT | 107.2 | 197 | 88.8 |
| R: CTCCTTTGTGCTCATCTTCCC | |||||
| LcGAPDH | 甘油醛3-磷酸脱氢酶Glyceraldehyde-3-phosphate dehydrogenase | F: AAACGCAACGAAGCAACTACA | 97.7 | 105 | 82.2 |
| R: AGAGCACCAAGCCGCAAG | |||||
| LcEF1α | 延伸因子1-α Elongation factor 1-alpha | F: CCGCCATCAAGAAGAAATAGG | 103.9 | 98 | 82.6 |
| R: CGCCGAAGCAGGAATAAAA | |||||
| Lc18SrRNA | 18S核糖体RNA 18S ribosomal RNA | F: TTGAAGCTGATAGGAGGGAGG | 96.7 | 98 | 82.8 |
| R: CTGAAGGTTGGGCGGAATA | |||||
| LcCAP | 腺苷酰环化酶相关蛋白 Adenylyl cyclase-associated protein | F: AGGACGGGCAGTTCACCA | 103.6 | 114 | 83.1 |
| R: ACAAGGCAAAGGCAGCATC | |||||
| LcAPRT | 腺嘌呤磷酸核糖转化酶 Adenine phosphoribosyl transferase | F: GAATCCTCTGGCTTCAACACC | 101.4 | 118 | 83.7 |
| R: GGACATCACAACCTTGCTTCTT | |||||
| LcACT | 肌动蛋白Actin | F: ATTGTGCTCAGTGGTGGGTCA | 106.5 | 137 | 85.5 |
| R: CCAATCCAAACACTGTACTTCCTC | |||||
| LcPHO1-2 | 磷酸盐1-2 Phosphate1-2 | F: CTCGCCTACTGGATTTCTCCC | - | 97 | 82.5 |
| R: CCAACATTGTTCAAGTGTTCGTTC | |||||
| LcPAP2 | 紫色酸性磷酸酶2 Purple acid phosphatase2 | F: CTCGCGTCCGCACACATAAT | - | 83 | 82.9 |
| R: TCCGCTTCCAGCCACTTATA | |||||
| LcPAP27 | 紫色酸性磷酸酶27 Purple acid phosphatase27 | F: CGACTCCTTCACCATCCACA | - | 172 | 88.3 |
| R: TTCACGCACACGAGATTCTTT |
新窗口打开|下载CSV
引物扩增效率:取不同Pi处理下的cDNA样品等量混合,按照10倍的梯度稀释5个浓度梯度,并以此为模板,按照1.5方法进行qRT-PCR反应后计算扩增效率。
1.5 实时荧光定量PCR
利用TaKaRa的SYBR Premix Ex Taq II对Pi处理下内参基因进行qRT-PCR检测。qRT-PCR反应总体系为20 μL,包括cDNA模板5 μL、SYBR Premix Ex Taq II 10 μL、正和反向引物各0.8 μL(10 μmol·L-1)和DEPC水3.4 μL。qRT-PCR反应程序为95℃ 30 s;95℃ 5 s,60℃ 30 s,72℃ 15 s,40个循环。溶解曲线:以0.5℃逐渐增加,从70℃加热至95℃,然后95℃恒温15 s。每个样品3个技术重复,每个试验进行3次独立生物学重复。1.6 内参基因稳定性分析
利用软件geNorm(version 3.5)[25]、NormFinder(version 0.953)[26]和BestKeeper(version 1)[27]对内参基因的稳定性进行分析评估。具体方法参考文献[28]。geNorm基于对数转换表达率的标准差,确定每个内参基因与其他所有内参基因配对变异的平均值,作为内参基因的表达稳定性M值(average expression stability values),通过逐步排除M值最高的基因,对被测基因的表达稳定性进行排名,M值越低稳定性越高,M值小于1.5则默认为可用作内参基因[25]。NormFinder基于方差分析计算每个基因的表达稳定值(stability value),对候选内参基因的稳定性进行排名,稳定值越低则稳定性越高[26]。BestKeeper通过比较CT(cycle threshold)值的标准偏差(standard deviation,SD)和变异系数(coefficient of variance,CV)来评估内参基因的稳定性,SD值越高基因表达越不稳定[27]。geNorm和NormFinder将qRT-PCR检测获得的CT值换算为2-ΔCT值作为分析的输入数据,其中,ΔCT=相应样品CT值-最小CT值。BestKeeper直接将CT值作为输入数据进行分析。1.7 磷响应相关基因表达分析
LcPHO1-2、LcPAP2和LcPAP27来源于羊草转录组数据库,利用qRT-PCR对不同Pi处理下相关基因的表达进行检测。选用获得的较稳定和不稳定的内参基因,利用2-ΔΔCT法[29]对磷响应相关基因的相对表达量进行分析。每个样品3个技术重复。qRT-PCR引物序列列于表1。2 结果
2.1 羊草对不同浓度Pi处理的响应
通过对羊草进行不同浓度Pi处理,结果显示,对照(1 mmol·L-1 Pi)条件下,羊草的株高和根长均随着处理时间的增加而增长;与对照相比,低Pi(0和0.01 mmol·L-1)和高Pi(5和30 mmol·L-1)处理都使得羊草的生长受阻(图1)。在低Pi或者缺Pi条件下,株高在处理17d时显著低于对照,根长在处理的25d显著低于对照,并且均随着处理时间的延长差异显著增强,与根长相比,株高对低Pi或缺Pi较为敏感。在高Pi处理条件下,株高分别在5和30 mmol·L-1 Pi处理的25 d和13 d显著低于对照,并且随着处理时间的延长差异显著增强;根长分别从5和30 mmol·L-1 Pi处理的13 d和9 d开始显著低于对照,并且一直持续至处理结束(除5 mmol·L-1 Pi处理21 d外),与株高相比,根长对高Pi更为敏感。在地上组织中,与高Pi处理相比,株高对低Pi较为敏感;而在地下组织中,与低Pi相比,根长对高Pi(30 mmol·L-1)较为敏感。表明羊草地上、地下组织对Pi处理响应的模式不同。0.33 mmol·L-1 Pi处理条件下,与对照相比,株高和根长都没有显著性差异,表明0.33—1 mmol·L-1 Pi浓度范围是羊草正常生长的合适浓度。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1不同浓度Pi处理下羊草的表型分析
A:表型;B:株高检测;C:根长检测。*、**和***分别代表在P<0.05、P<0.01和P<0.001水平差异显著。下同
Fig. 1Phenotype analysis of Leymus chinensis under different concentration of Pi treatments
A: Phenotype; B: Shoot height measurement; C: Root length measurement. *, ** and *** represent significantly different at levels of P<0.05, P<0.01 and P<0.001 compared with control, respectively. The same as below
不同浓度Pi处理条件下,羊草根和叶中Pi含量随着处理浓度的增加均呈上升趋势(图2)。与对照相比,低Pi和高Pi处理下,除30 mmol·L-1 Pi处理的根外,根和叶片中Pi的积累达到差异显著水平。与对照相比,0.33 mmol·L-1 Pi处理差异不显著。表明不同浓度Pi处理下羊草的表型与Pi的含量相关。
图2
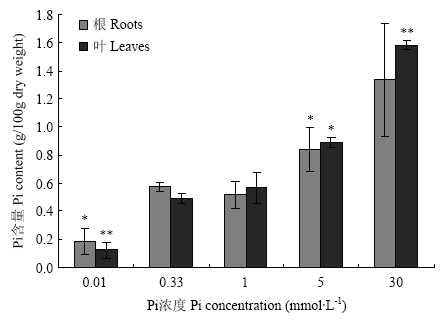 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2不同浓度Pi处理下羊草组织中Pi的含量
Fig. 2Pi contents in tissues of Leymus chinensis under different concentration of Pi treatments
2.2 候选内参基因的引物特异性和扩增效率分析
根据前期转录组数据,筛选出7个表达稳定的基因(表达水平变化小于2倍),同时从NCBI获得1个基因作为候选内参基因。利用Primer Premier 5软件设计基因特异引物,以不同浓度Pi胁迫下羊草组织cDNA为模板进行qRT-PCR,对8个候选内参基因的引物扩增效率进行分析,其范围在95.0%—107.2%(表1)。qRT-PCR溶解曲线分析显示,8个候选内参基因均具有单一的溶解峰(图3),表明候选基因引物特异性较好。图3
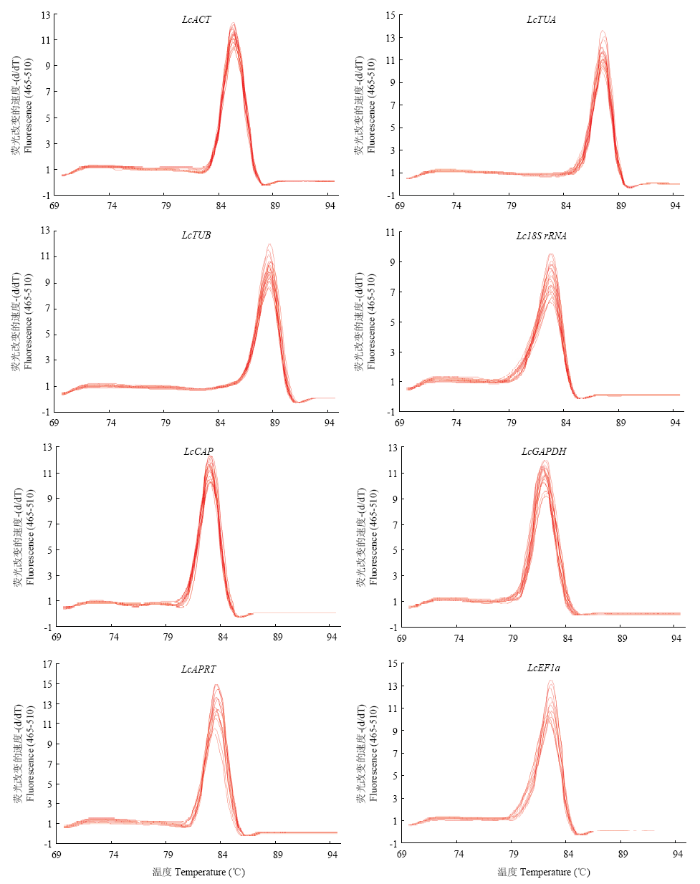 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3候选内参基因的溶解曲线
Fig. 3Melting curves of candidate reference genes
2.3 Pi胁迫条件下候选内参基因的表达谱分析
根据qRT-PCR结果中的CT值,对候选内参基因的表达谱进行分析。结果(图4)表明,在所有样品中,8个候选内参基因的CT值范围是17.16—26.61,其中,大多数基因的CT值范围在18.84—23.95。在所有基因中,LcGAPDH的表达丰度最高,为17.16—20.22,其次为LcACT,其CT值范围为18.84—21.78;Lc18SrRNA的表达丰度最低,为23.28—26.61,其次为LcARPT和LcTUA,CT值范围分别为22.26—23.95和20.35—26.30。通过对候选内参基因在所有样品中的CT值进行平均数(mean)、标准偏差SD和变异系数CV分析(表2),结果显示,LcTUA和LcTUB变异系数最大,分别为6.8%和6.6%,稳定性较差;LcAPRT的变异系数最小,为2.09%,稳定性较好;其他基因的变异系数居中。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4候选内参基因在所有测试样品中的CT值分布情况
方框代表CT值的第一和第三四分位值;红线代表中位数;短横线代表最大值和最小值
Fig. 4Distribution of CT values of candidate reference genes across all samples
Boxes represent the first and third quartiles of the data; Red lines across the boxes indicate the median CT values; Short horizontal lines show the maximum and minimum values
Table2
表2
表2候选内参基因的CT值
Table2
| 基因 Gene | 平均数 Mean | 标准差 SD | 变异系数 CV(%) |
|---|---|---|---|
| LcACT | 20.10 | 0.97 | 4.82 |
| LcTUA | 23.15 | 1.58 | 6.80 |
| LcTUB | 20.75 | 1.37 | 6.60 |
| LcAPRT | 23.32 | 0.49 | 2.09 |
| LcEF1α | 21.11 | 0.92 | 4.35 |
| Lc18SrRNA | 25.27 | 0.93 | 3.68 |
| LcCAP | 22.56 | 0.89 | 3.95 |
| LcGAPDH | 18.61 | 0.92 | 4.96 |
新窗口打开|下载CSV
2.4 Pi胁迫条件下候选内参基因的稳定性分析
利用geNorm、NormFinder和Bestkeeper软件,对候选内参基因的表达稳定性进行分析。geNorm分析结果显示(表3),在不同浓度Pi处理条件下,所有基因的M值均小于1.5,Lc18SrRNA和LcCAP的M值最低(0.42),稳定性最高,然后依次为LcEF1α(0.49)、LcACTIN(0.52)、LcGAPDH(0.54)和LcAPRT(0.60),LcTUB(0.68)和LcTUA(0.78)的M值最高,稳定性最差。geNorm通过分析配对变异系数(pairwise variation,V)评估标准化过程中合适的内参基因数目,Vn/n+1的值小于0.15作为阈值,判断是否需要再增加一个内参基因。V值分析结果显示,V2/3的值(0.16)大于0.15(图5),表明2个内参基因不足以进行标准化过程分析,需要引进第三个内参基因,V3/4及其他V值均小于0.15,即3个内参基因可满足对靶基因精准的标准化分析。Table 3
表3
表3候选内参基因的稳定性分析
Table 3
| 排名 Rank | geNorm | NormFinder | Bestkeeper | |||
|---|---|---|---|---|---|---|
| 基因Gene | M值M value | 基因Gene | 稳定值Stability value | 基因Gene | 标准差Std dev (± CP) | |
| 1 | LcCAP | 0.42 | Lc18SrRNA | 0.138 | LcAPRT | 0.40 |
| 2 | Lc18SrRNA | 0.42 | LcCAP | 0.231 | Lc18SrRNA | 0.73 |
| 3 | LcEF1α | 0.49 | LcEF1α | 0.255 | LcCAP | 0.75 |
| 4 | LcACT | 0.52 | LcACT | 0.342 | LcEF1α | 0.76 |
| 5 | LcGAPDH | 0.54 | LcGAPDH | 0.390 | LcGAPDH | 0.76 |
| 6 | LcAPRT | 0.60 | LcTUB | 0.457 | LcACT | 0.81 |
| 7 | LcTUB | 0.68 | LcAPRT | 0.470 | LcTUB | 1.04 |
| 8 | LcTUA | 0.78 | LcTUA | 0.692 | LcTUA | 1.16 |
新窗口打开|下载CSV
图5
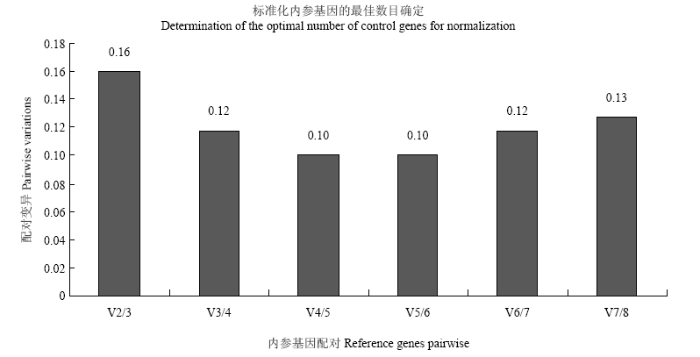 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5候选内参基因配对变异系数分析
Fig. 5Pairwise variations (V) analysis of the candidate reference genes
NormFinder分析结果显示(表3),排名最靠前的基因为Lc18SrRNA,紧随其后的3个基因为LcCAP、LcEF1α和LcACT,最不稳定的2个基因为LcAPRT和LcTUA。
根据BestKeeper分析结果(表3),稳定性较好的3个基因为LcAPRT、Lc18SrRNA和LcCAP,其次为LcEF1α和LcGAPDH,LcTUB和LcTUA的SD值最高最不稳定。
综上所述,通过计算几何平均数对8个候选内参基因排名进行综合评价(表4),其中,排名靠前的3个基因为Lc18SrRNA、LcCAP和LcEF1α,并且在3个软件稳定性分析结果中排名均在前四名,表明稳定较好,最适合作为内参基因。排名最后的2个基因为LcTUA和LcTUB,这与稳定性分析结果基本相符,表明最不适合作为内参基因。
Table 4
表4
表4候选内参基因稳定性整体排名
Table 4
| 排名Rank | 基因Gene | 几何平均数Geomean |
|---|---|---|
| 1 | Lc18SrRNA | 1.260 |
| 2 | LcCAP | 1.817 |
| 3 | LcEF1α | 3.302 |
| 4 | LcAPRT | 3.476 |
| 5 | LcACT | 4.579 |
| 6 | LcGAPDH | 4.642 |
| 7 | LcTUB | 6.649 |
| 8 | LcTUA | 8.000 |
新窗口打开|下载CSV
2.5 Pi处理条件下磷响应相关基因表达分析
选用稳定性较好的Lc18SrRNA、LcCAP、LcEF1α以及Lc18SrRNA+LcCAP+LcEF1α分别作为内参基因,对不同Pi处理下LcPHO1-2、LcPAP2和LcPAP27进行qRT-PCR分析(图6)。结果显示,利用不同内参基因分析得到的表达模式类似。与对照相比,LcPHO1-2的表达受低Pi或者缺Pi诱导;LcPAP2的表达受高Pi诱导;而LcPAP27的表达同时受低Pi和高Pi下调。而以稳定性较差的LcTUA作为内参基因时,LcPHO1-2、LcPAP2和LcPAP27的表达模式与以稳定性较好的内参基因不同。表明筛选的内参基因比较可靠。图6
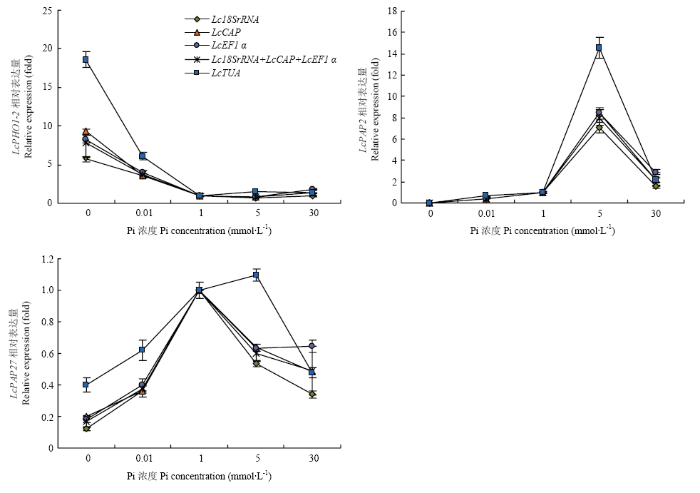 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6不同浓度Pi处理下磷响应基因表达分析
Fig. 6Relative expression of Pi-responsive genes under different concentration of Pi treatments
3 讨论
3.1 不同浓度Pi处理对羊草生长的影响
磷是植物生长发育所必需的大量无机营养元素之一,Pi缺乏条件下植物会从形态、生理和分子水平产生一系列反应以保持磷稳态[7,16]。LI等[23]对缺Pi条件下羊草的生理特性分析结果表明,Pi缺乏致使叶片的生长、生物量积累和净光合速率显著降低。本研究中通过进行不同浓度的Pi胁迫处理,对羊草的响应和候选内参基因进行了分析。研究结果表明,低Pi(或缺Pi)条件下,羊草的茎和根生长均受到明显的抑制,这与LI等[23]研究结果一致。同时,高浓度Pi条件下,羊草的生长也受到了抑制,表明外源施加较高浓度的Pi使得羊草体内Pi过量积累,产生P毒性症状(P-toxicity symptoms)。P毒性症状在不同植物物种间的差异不是很大,表现为生长减缓、叶片失绿坏死、细胞器损伤和细胞活力丧失等特征,但是在不同物种间出现P毒性症状的Pi浓度不同[30,31,32,33],本研究中Pi浓度达到5 mmol·L-1时羊草的生长减缓,表明产生了P毒性症状,当Pi浓度达到30 mmol·L-1时少数羊草植株出现了死亡的现象,表明P毒性症状加重。但是羊草中低Pi响应及P毒性症状的分子机制如何?是否与其他物种类似?值得进一步研究验证。3.2 不同浓度Pi处理条件下羊草内参基因稳定性分析
内参基因筛选是qRT-PCR结果准确分析的关键步骤。本研究利用软件geNorm、NormFinder和Bestkeeper,对8个候选内参基因在不同浓度Pi处理下的表达稳定性进行了分析。geNorm分析结果显示,8个候选内参基因的M值均小于默认阈值1.5[25],表明均可以作为内参基因;配对变异系数V2/3的值大于0.15,V3/4的值小于0.15,表明理想的内参基因数目为3个。由于每个软件评估的策略不同,因此分析的结果稍有不同。通过计算不同软件排名的几何平均数,对内参基因进行整体排名,排名靠前的3个基因为Lc18SrRNA、LcCAP和LcEF1α,其在geNorm和NormFinder中的排名均为前三名之内,在Bestkeeper排名为前四名之内。LcTUB在所有软件排名中均为最后一名,表明其稳定性最差,不适合作为内参基因。作为传统的内参基因,18SrRNA和EF1α在不同的植物中被广泛应用,但是在不同条件下的表达不同[34]。在桂花(Osmanthus fragrans Lour.)不同品种、发育阶段和低温胁迫材料中,18SrRNA不适合作为内参基因[34];在草莓(Fragaria×ananassa)的不同组织和发育阶段,18SrRNA的表达稳定性较低[35];而在水稻中,不同环境胁迫下,18SrRNA的表达稳定性最高[36]。在高羊茅(Festuca arundinacea Schreb.)中,不同的逆境胁迫条件下EF1α是最不稳定的内参基因[37];而在板蓝根(Isatis indigotica)中,EF1α是在所有试验样品中最稳定的内参基因之一[38]。胡宁宁等[20]对羊草不同组织部位内参基因筛选结果显示,18SrRNA和EF1α分别是根和茎中较为稳定的内参基因。作为新的内参基因,CAP编码的是环化酶相关蛋白,在不同条件下其表达稳定性不同,例如在柠条锦鸡儿(Caragana korshinskii)中,CAP在干旱、冷和高pH胁迫下稳定性较好,而在NaCl胁迫下稳定性较差[19];在大针茅(Stipa grandis)中,JA处理下CAP的表达稳定性较差[28]。本研究通过综合分析显示,Lc18SrRNA、LcCAP和LcEF1α稳定性最好,但是geNorm、NormFinder和Bestkeeper软件在分析过程均没有考虑到基因的表达丰度,根据2.3中分析结果,Lc18SrRNA的CT值较高、表达丰度较低,因此,综合考虑可优先选用LcCAP和LcEF1α作为内参基因。
3.3 不同浓度Pi处理下磷响应基因表达分析
WU等[7]通过拟南芥基因芯片分析显示,在Pi饥饿72 h以内,6 172个基因中的1 800多个基因有响应;通过RNA印记杂交进一步证实,一个PAP在叶片中的表达受Pi饥饿诱导。MISSION等[21]利用Affymetrix基因芯片技术,对低Pi胁迫下拟南芥的22 810个基因表达模式进行了分析,共发现612和254个基因的表达分别受低Pi诱导和抑制,其中包括11个受低Pi诱导表达的PAPs。与拟南芥中部分PAPs的表达模式不同,本研究检测的2个LcPAPs中,LcPAP2的表达受高Pi诱导,LcPAP27的表达同时受低Pi和高Pi下调,表明这两个基因参与了羊草对Pi胁迫响应的进程,并且LcPAP2主要与高Pi胁迫应答响应有关。PHO1编码的是SPX-EXS蛋白,主要负责将Pi从根运输到地上组织,其表达受低Pi诱导[6,39]。本研究LcPHO1-2的表达特异的受低Pi诱导,表明其参与了羊草对低Pi的应答响应,但是是否参与Pi从根到地上组织的运输还不清楚,有待于进一步试验验证。4 结论
低Pi和高Pi胁迫均使羊草生长受阻,但地上组织与地下组织对Pi胁迫响应的模式不同,随着处理浓度的增加羊草中Pi的含量增加。筛选出3个表达较为稳定的基因Lc18SrRNA、LcCAP和LcEF1α,可作为Pi处理下羊草基因表达分析的内参基因。LcPHO1-2和LcPAP2分别参与了羊草对低Pi和高Pi的应答响应,LcPAP27同时参与了羊草对低Pi和高Pi的响应进程。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[D].
[本文引用: 2]
[D].
[本文引用: 2]
DOI:10.11733/j.issn.1007-0435.2014.06.013URL [本文引用: 1]

以中科2号羊草(Leymus chinensis ‘Zhongke No.2’)为材料,系统分析了氮磷肥配施对干草产量、品质及养分吸收的影响.结果表明:增施氮肥羊草产草量显著增加(P<0.05),以120 kg·hm-2 P2O5为肥底、施用120 kg·hm-2 N时产草量最高,较对照增产1.5倍;氮、磷、钾吸收量随氮磷肥施用量的增加均呈现先增加后减少趋势,适当增施磷肥促进氮素的吸收,增施氮肥促进磷素的吸收,钾吸收量对氮肥响应显著;氮磷肥合理配施有效增加羊草粗蛋白含量,降低粗纤维和无氮浸出物含量,以120 kg·hm-2 P2O5为肥底、施用90 kg·hm-2 N时羊草品质最优,且经济效益较对照增收1.48倍,建议同类地区推广施用.
DOI:10.11733/j.issn.1007-0435.2014.06.013URL [本文引用: 1]
以中科2号羊草(Leymus chinensis ‘Zhongke No.2’)为材料,系统分析了氮磷肥配施对干草产量、品质及养分吸收的影响.结果表明:增施氮肥羊草产草量显著增加(P<0.05),以120 kg·hm-2 P2O5为肥底、施用120 kg·hm-2 N时产草量最高,较对照增产1.5倍;氮、磷、钾吸收量随氮磷肥施用量的增加均呈现先增加后减少趋势,适当增施磷肥促进氮素的吸收,增施氮肥促进磷素的吸收,钾吸收量对氮肥响应显著;氮磷肥合理配施有效增加羊草粗蛋白含量,降低粗纤维和无氮浸出物含量,以120 kg·hm-2 P2O5为肥底、施用90 kg·hm-2 N时羊草品质最优,且经济效益较对照增收1.48倍,建议同类地区推广施用.
DOI:10.1177/0145561319879245URLPMID:31619067 [本文引用: 3]
Despite its effectiveness, radiochemotherapy treatment in the head and neck region is accompanied by acute oral complications such as oral mucositis, dysphagia, xerostomia, and dysgeusia. The aim of this study was to analyze and prospectively assess the frequency and evolution of acute oral complications during radiochemotherapy in patients diagnosed with squamous cell carcinoma in the head and neck region. We have analyzed oral complications of 20 patients during 6 weeks of radiochemotherapy treatment for squamous cell carcinoma. Oral mucositis was evaluated according to the World Health Organization criteria, dysphagia, and dysgeusia according to the National Cancer Institute Common Toxicity Criteria, and xerostomia according to parameters set by the Seminars in Radiation Oncology. Mucositis was first observed in the second week and all patients presented some degree of mucositis in the fourth week of radiotherapy. Xerostomia and dysphagia were initially reported already in the first week of radiotherapy. All patients presented xerostomia in the fourth week; however, dysphagia was observed in all patients, only in the sixth week. Dysgeusia was first observed in the second week, becoming more severe in the third week. Acute oral complications can be observed throughout the treatment, but the third week of radiotherapy seems to represent a critical week, regardless of the grade of the complication. The sixth week presents the worst grades of these complications. Knowledge about the natural course of oral complications during radiotherapy is important to develop better strategies for treatment and improve the patients' quality of life.
DOI:10.1104/pp.103.021022URLPMID:12857808 [本文引用: 3]
Arabidopsis genome expression pattern changes in response to phosphate (Pi) starvation were examined during a 3-d period after removal of Pi from the growth medium. Available Pi concentration was decreased after the first 24 h of Pi starvation in roots by about 22%, followed by a slow recovery during the 2nd and 3rd d after Pi starvation, but no significant change was observed in leaves within the 3 d of Pi starvation. Microarray analysis revealed that more than 1,800 of the 6,172 genes present in the array were regulated by 2-fold or more within 72 h from the onset of Pi starvation. Analysis of these Pi starvation-responsive genes shows that they belong to wide range of functional categories. Many genes for photosynthesis and nitrogen assimilation were down-regulated. A complex set of metabolic adaptations appears to occur during Pi starvation. More than 100 genes each for transcription factors and cell-signaling proteins were regulated in response to Pi starvation, implying major regulatory changes in cellular growth and development. A significant fraction of those regulatory genes exhibited distinct or even contrasting expression in leaves and roots in response to Pi starvation, supporting the idea that distinct Pi starvation response strategies are used for different plant organs in response to a shortage of Pi in the growth medium.
DOI:10.1199/tab.0024URLPMID:22303200 [本文引用: 2]
DOI:10.1038/s41477-018-0334-3URLPMID:30626920 [本文引用: 2]
Inorganic phosphate (Pi) is an essential component of all life forms. Land plants acquire Pi from the soil through roots and associated symbioses, and it is then transported throughout the plant. When sufficient, excess Pi is stored in vacuoles for remobilization following Pi deficiency. Although Pi release from the vacuoles to the cytoplasm serves as a critical mechanism for plants to adapt to low-Pi stress, the transporters responsible for vacuolar Pi efflux have not been identified. Here, we identified a pair of Oryza sativa vacuolar Pi efflux transporters (OsVPE1 and OsVPE2) that were more abundant in plants grown under Pi-deficient conditions. These OsVPE proteins can transport Pi into yeast cells and Xenopus laevis oocytes. Vacuolar Pi content was higher in the loss-of-function Osvpe1?Osvpe2 double mutant than in wild type, particularly under low-Pi stress. Overexpression of either OsVPE1 or OsVPE2 in transgenic plants reduced vacuolar Pi content, consistent with a role in vacuolar Pi efflux. We demonstrate that these VPE proteins evolved from an ancient plasma membrane glycerol-3-phosphate transporter protein. Together, these data indicate that this transporter was recruited to the vacuolar membrane to catalyse Pi efflux during the course of land plant evolution.
DOI:10.3724/SP.J.1258.2011.00001URL [本文引用: 2]
Aims Phosphorus status and N/P stoichiometry in plant leaves have been studied intensively with recent focus on large-scale patterns and driving factors. Studies of Chinese terrestrial plants found that leaf P was considerably lower than the global average, resulting in a higher N/P, probably due to the low soil total P content at the national scale. Inner Mongolia grassland offers a diverse array of taxa and soil conditions to examine the correlation between leaf and soil P concentrations. Our objective was to determine how and to what extent soil total and available P modify leaf P across the study region. Methods Leaf samples of 57 species were collected at 36 sites across Inner Mongolia grassland during July and August 2007. We determined leaf P concentration, N/P, soil total and available P concentrations and tested pairwise relationships between leaf and soil variables at species-by-site, inter-specific and inter-site levels. Important findings Findings of relatively low leaf P and high N/P across Inner Mongolia grassland were consistent with previous findings. Neither soil total nor available P appeared to be related with leaf P concentration, although soil available P had a stronger explanatory power than soil total P content. Moreover, Inner Mongolian grassland did not show a great shortage of soil available P compared with USA, Australia and the global average. The hypothesis that low leaf P and high N/P of plants are caused by low soil P content do not hold in Inner Mongolian grassland. Instead, neither soil total nor available P shapes the pattern of leaf P and N/P across this grassland.
DOI:10.3724/SP.J.1258.2011.00001URL [本文引用: 2]
Aims Phosphorus status and N/P stoichiometry in plant leaves have been studied intensively with recent focus on large-scale patterns and driving factors. Studies of Chinese terrestrial plants found that leaf P was considerably lower than the global average, resulting in a higher N/P, probably due to the low soil total P content at the national scale. Inner Mongolia grassland offers a diverse array of taxa and soil conditions to examine the correlation between leaf and soil P concentrations. Our objective was to determine how and to what extent soil total and available P modify leaf P across the study region. Methods Leaf samples of 57 species were collected at 36 sites across Inner Mongolia grassland during July and August 2007. We determined leaf P concentration, N/P, soil total and available P concentrations and tested pairwise relationships between leaf and soil variables at species-by-site, inter-specific and inter-site levels. Important findings Findings of relatively low leaf P and high N/P across Inner Mongolia grassland were consistent with previous findings. Neither soil total nor available P appeared to be related with leaf P concentration, although soil available P had a stronger explanatory power than soil total P content. Moreover, Inner Mongolian grassland did not show a great shortage of soil available P compared with USA, Australia and the global average. The hypothesis that low leaf P and high N/P of plants are caused by low soil P content do not hold in Inner Mongolian grassland. Instead, neither soil total nor available P shapes the pattern of leaf P and N/P across this grassland.
DOI:10.1007/s00442-007-0912-yURLPMID:18278518 [本文引用: 1]
Leaf N and P stoichiometry covaries with many aspects of plant biology, yet the drivers of this trait at biogeographic scales remain uncertain. Recently we reported the patterns of leaf C and N based on systematic census of 213 species over 199 research sites in the grassland biomes of China. With the expanded analysis of leaf P, here we report patterns of leaf P and N:P ratios, and analyze the relative contribution of climatic variables and phylogeny in structuring patterns of leaf N:P stoichiometry. Average values of leaf P and N:P ratio were 1.9 mg g(-1) and 15.3 (mass ratio), respectively, consistent with the previous observation of a higher N:P ratio in China's flora than the global averages (ca. 13.8), resulting from a lower leaf P. Climatic variables had very little direct correlation with leaf P and N:P ratios, with growing season precipitation and temperature together explaining less than 2% of the variation, while inter-site differences and within-site phylogenetic variation explained 55 and 26% of the total variation in leaf P and N:P ratios. Across all sites and species, leaf N and P were highly positively correlated at all levels. However, the within-site, within-species covariations of leaf N and P were weaker than those across sites and across species. Leaf N and P relationships are driven by both variation between sites at the landscape scale (explaining 58% of the variance) and within sites at the local scale (explaining 24%), while the climatic factors exerted limited influence (explaining less than 3%). In addition, leaf N:P ratios in two dominant genera Kobresia and Stipa had different responses to precipitation. This study suggests that geographic variation and between-species variation, rather than climatic variation, are the major determinants of grassland foliar stoichiometry at the biome level.
DOI:10.1046/j.1469-8137.1997.00787.xURL [本文引用: 1]
DOI:10.7717/peerj.7047URLPMID:31218124 [本文引用: 1]
Vegetation succession is one of the major driving processes of grassland degradation. Stoichiometry significantly contributes to vegetation dynamics. However, a knowledge gap exists in how soil nutrients and root enzymes influence the stoichiometric ratio to affect vegetation dynamics.
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1105/tpc.113.117325URLPMID:24249833 [本文引用: 3]
Using rice (Oryza sativa) as a model crop species, we performed an in-depth temporal transcriptome analysis, covering the early and late stages of Pi deprivation as well as Pi recovery in roots and shoots, using next-generation sequencing. Analyses of 126 paired-end RNA sequencing libraries, spanning nine time points, provided a comprehensive overview of the dynamic responses of rice to Pi stress. Differentially expressed genes were grouped into eight sets based on their responses to Pi starvation and recovery, enabling the complex signaling pathways involved in Pi homeostasis to be untangled. A reference annotation-based transcript assembly was also generated, identifying 438 unannotated loci that were differentially expressed under Pi starvation. Several genes also showed induction of unannotated splice isoforms under Pi starvation. Among these, PHOSPHATE2 (PHO2), a key regulator of Pi homeostasis, displayed a Pi starvation-induced isoform, which was associated with increased translation activity. In addition, microRNA (miRNA) expression profiles after long-term Pi starvation in roots and shoots were assessed, identifying 20 miRNA families that were not previously associated with Pi starvation, such as miR6250. In this article, we present a comprehensive spatio-temporal transcriptome analysis of plant responses to Pi stress, revealing a large number of potential key regulators of Pi homeostasis in plants.
DOI:10.1186/1471-2229-8-131URLPMID:19102748 [本文引用: 1]
The elucidation of gene expression patterns leads to a better understanding of biological processes. Real-time quantitative RT-PCR has become the standard method for in-depth studies of gene expression. A biologically meaningful reporting of target mRNA quantities requires accurate and reliable normalization in order to identify real gene-specific variation. The purpose of normalization is to control several variables such as different amounts and quality of starting material, variable enzymatic efficiencies of retrotranscription from RNA to cDNA, or differences between tissues or cells in overall transcriptional activity. The validity of a housekeeping gene as endogenous control relies on the stability of its expression level across the sample panel being analysed. In the present report we describe the first systematic evaluation of potential internal controls during tomato development process to identify which are the most reliable for transcript quantification by real-time RT-PCR.
DOI:10.1105/tpc.108.061143URLPMID:18664613 [本文引用: 2]
DOI:10.1007/s11033-014-3086-9URL [本文引用: 2]
Caragana korshinskii Kom., which is widely distributed in the northwest China and Mongolia, is an important forage bush belonging to the legume family with high economic and ecological value. Strong tolerance ability to various stresses makes C. korshinskii Kom. a valuable species for plant stress research. In this study, suitable reference genes for quantitative real-time reverse transcription PCR (qRT-PCR) were screened from 11 candidate reference genes, including ACT, GAPDH, EF1 alpha, UBQ, TUA, CAP, TUB, TUB3, SKIP1, SKIP5-1 and SKIP5-2. A total of 129 samples under drought, heat, cold, salt, ABA and high pH treatment were profiled, and software such as geNORM, NormFinder and BestKeeper were used for reference gene evaluation and selection. Different suitable reference genes were selected under different stresses. Across all 129 samples, GAPDH, EF1 alpha and SKIP5-1 were found to be the most stable reference genes, and EF1 alpha+SKIP5-1 is the most stable reference gene combination. Conversely, TUA, TUB and SKIP1 were not suitable for using as reference genes owing to their great expression variation under some stress conditions. The relative expression levels of CkWRKY1 were detected using the stable and unstable reference genes and their applicability was confirmed. These results provide some stable reference genes and reference gene combinations for qRT-PCR under different stresses in C. korshinskii Kom. for future research work, and indicate that CkWRKY1 plays essential roles in response to stresses in C. korshinskii.
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
DOI:10.1016/j.plaphy.2016.10.017URLPMID:27825075 [本文引用: 1]
Maize (Zea mays L.) is an important food and energy crop, and low phosphate (Pi) availability is one of the major constraints in maize production worldwide. Plants adapt suitably to acclimate to low Pi stress. However, the underlying molecular mechanism of Pi deficiency response is still unclear. In this study, comparative transcriptomic analyses were conducted to investigate the differences of transcriptional responses in two maize genotypes with different tolerances to low phosphorus (LP) stress. LP-tolerant genotype QXN233 maintained higher P and Pi levels in shoots than LP-sensitive genotype QXH0121 suffering from Pi deficiency at seedling stage. Moreover, the transcriptomic analysis identified a total of 1391 Pi-responsive genes differentially expressed between QXN233 and QXH0121 under LP stress. Among these genes, 468 (321 up- and 147 down-regulated) were identified in leaves, and 923 (626 up- and 297 down-regulated) were identified in roots. These Pi-responsive genes were involved in various metabolic pathways, the biosynthesis of secondary metabolites, ion transport, phytohormone regulation, and other adverse stress responses. Consistent with the differential tolerance to LP stress, five maize inorganic Pi transporter genes were more highly up-regulated in QXN233 than in QXH0121. Results provide important information to further study the changes in global gene expression between LP-tolerant and LP-sensitive maize genotypes and to understand the molecular mechanisms underlying maize's long-term response to Pi deficiency.
DOI:10.3389/fpls.2019.00170URLPMID:30873190 [本文引用: 3]
Sheepgrass [Leymus chinensis (Trin.) Tzvel] is a valuable forage plant highly significant to the grassland productivity of Euro-Asia steppes. Growth of above-ground tissues of L. chinensis is the major component contributing to the grass yield. Although it is generally known that this species is sensitive to ecosystem disturbance and adverse environments, detailed information of how L. chinensis coping with various nutrient deficiency especially phosphate deprivation (-Pi) is still limited. Here, we investigated impact of Pi-deprivation on shoot growth and biomass accumulation as well as photosynthetic properties of L. chinensis. Growth inhibition of Pi-deprived seedlings was most obvious and reduction of biomass accumulation and net photosynthetic rate (Pn) was 55.3 and 63.3%, respectively, compared to the control plants grown under Pi-repleted condition. Also, we compared these characters with seedlings subjected to low-Pi stress condition. Pi-deprivation caused 18.5 and 12.3% more reduction of biomass and Pn relative to low-Pi-stressed seedlings, respectively. Further analysis of in vivo chlorophyll fluorescence and thylakoid membrane protein complexes using 2D-BN/SDS-PAGE combined with immunoblot detection demonstrated that among the measured photosynthetic parameters, decrease of ATP synthase activity was most pronounced in Pi-deprived plants. Together with less extent of lipid peroxidation of the thylakoid membranes and increased ROS scavenger enzyme activities in the leaves of Pi-deprived seedlings, we suggest that the decreased activity of ATP synthase in their thylakoids is the major cause of the greater reduction of photosynthetic efficiency than that of low-Pi stressed plants, leading to the least shoot growth and biomass production in L. chinensis.
DOI:10.1186/gb-2002-3-7-research0034URLPMID:12184808 [本文引用: 3]
Gene-expression analysis is increasingly important in biological research, with real-time reverse transcription PCR (RT-PCR) becoming the method of choice for high-throughput and accurate expression profiling of selected genes. Given the increased sensitivity, reproducibility and large dynamic range of this methodology, the requirements for a proper internal control gene for normalization have become increasingly stringent. Although housekeeping gene expression has been reported to vary considerably, no systematic survey has properly determined the errors related to the common practice of using only one control gene, nor presented an adequate way of working around this problem.
DOI:10.1158/0008-5472.CAN-04-0496URLPMID:15289330 [本文引用: 2]
Accurate normalization is an absolute prerequisite for correct measurement of gene expression. For quantitative real-time reverse transcription-PCR (RT-PCR), the most commonly used normalization strategy involves standardization to a single constitutively expressed control gene. However, in recent years, it has become clear that no single gene is constitutively expressed in all cell types and under all experimental conditions, implying that the expression stability of the intended control gene has to be verified before each experiment. We outline a novel, innovative, and robust strategy to identify stably expressed genes among a set of candidate normalization genes. The strategy is rooted in a mathematical model of gene expression that enables estimation not only of the overall variation of the candidate normalization genes but also of the variation between sample subgroups of the sample set. Notably, the strategy provides a direct measure for the estimated expression variation, enabling the user to evaluate the systematic error introduced when using the gene. In a side-by-side comparison with a previously published strategy, our model-based approach performed in a more robust manner and showed less sensitivity toward coregulation of the candidate normalization genes. We used the model-based strategy to identify genes suited to normalize quantitative RT-PCR data from colon cancer and bladder cancer. These genes are UBC, GAPD, and TPT1 for the colon and HSPCB, TEGT, and ATP5B for the bladder. The presented strategy can be applied to evaluate the suitability of any normalization gene candidate in any kind of experimental design and should allow more reliable normalization of RT-PCR data.
DOI:10.1023/B:BILE.0000019559.84305.47URL [本文引用: 2]
The stability of standard gene expression is an elementary prerequisite for internal standardisation of target gene expression data and many so called housekeeping genes with assumed stable expression can exhibit either up- or down-regulation under some experimental conditions. The developed, and herein presented, software called BestKeeper determines the best suited standards, out of ten candidates, and combines them into an index. The index can be compared with further ten target genes to decide, whether they are differentially expressed under an applied treatment. All data processing is based on crossing points. The BestKeeper software tool was validated on four housekeeping genes and 10 members of the somatotropic axis differentially expressed in bovine corpora lutea total RNA. The BestKeeper application and necessary information about data processing and handling can be downloaded on http://www.wzw.tum.de/gene-quantification/bestkeeper.html
DOI:10.1371/journal.pone.0169465URLPMID:28056110 [本文引用: 2]
Stipa grandis P. Smirn. is a dominant plant species in the typical steppe of the Xilingole Plateau of Inner Mongolia. Selection of suitable reference genes for the quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) is important for gene expression analysis and research into the molecular mechanisms underlying the stress responses of S. grandis. In the present study, 15 candidate reference genes (EF1 beta, ACT, GAPDH, SamDC, CUL4, CAP, SNF2, SKIP1, SKIP5, SKIP11, UBC2, UBC15, UBC17, UCH, and HERC2) were evaluated for their stability as potential reference genes for qRT-PCR under different stresses. Four algorithms were used: GeNorm, NormFinder, BestKeeper, and RefFinder. The results showed that the most stable reference genes were different under different stress conditions: EF1beta and UBC15 during drought and salt stresses; ACT and GAPDH under heat stress; SKIP5 and UBC17 under cold stress; UBC15 and HERC2 under high pH stress; UBC2 and UBC15 under wounding stress; EF1beta and UBC17 under jasmonic acid treatment; UBC15 and CUL4 under abscisic acid treatment; and HERC2 and UBC17 under salicylic acid treatment. EF1beta and HERC2 were the most suitable genes for the global analysis of all samples. Furthermore, six target genes, SgPOD, SgPAL, SgLEA, SgLOX, SgHSP90 and SgPR1, were selected to validate the most and least stable reference genes under different treatments. Our results provide guidelines for reference gene selection for more accurate qRT-PCR quantification and will promote studies of gene expression in S. grandis subjected to environmental stress.
DOI:10.1006/meth.2001.1262URLPMID:11846609 [本文引用: 1]
The two most commonly used methods to analyze data from real-time, quantitative PCR experiments are absolute quantification and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control. The 2(-Delta Delta C(T)) method is a convenient way to analyze the relative changes in gene expression from real-time quantitative PCR experiments. The purpose of this report is to present the derivation, assumptions, and applications of the 2(-Delta Delta C(T)) method. In addition, we present the derivation and applications of two variations of the 2(-Delta Delta C(T)) method that may be useful in the analysis of real-time, quantitative PCR data.
DOI:10.1093/jxb/erj004URLPMID:16356944 [本文引用: 1]
Grevillea crithmifolia R. Br. is a species of Proteaceae that is resistant to developing P-toxicity symptoms at phosphorus supplies in the root environment that induce P-toxicity symptoms in the closely related Hakea prostrata (Proteaceae). It was discovered previously that development of P-toxicity symptoms in H. prostrata is related to its low capacity to down-regulate net P-uptake rates (i.e. its low plasticity). The plasticity of net P-uptake rates and whole-plant growth responses in G. crithmifolia has now been assessed in two separate experiments: (i) a range of P, from 0 to 200 micromol P d-1, was supplied to whole root systems; (ii) using a split-root design, one root half was supplied with 0, 3, 75, or 225 micromol P d-1, while the other root half invariably received 3 micromol P d-1. Fresh mass was significantly greater in G. crithmifolia plants that had received a greater daily P supply during the pretreatments, but symptoms of P toxicity were never observed. Cluster-root growth decreased from about half the total root fresh mass when the leaf [P] was lowest (c. 0.1 mg P g-1 DM) to complete suppression of cluster-root growth when leaf [P] was 1-2 mg P g-1 DM. Split-root studies revealed that cluster-root initiation and growth, and net P-uptake rates by roots were regulated systemically, possibly by shoot P concentration. It is concluded that, in response to higher P supply, G. crithmifolia does not develop symptoms of P toxicity because of (i) greater plasticity of its net P-uptake capacity, and (ii) its greater plasticity for allocating P to growth and P storage in roots. This ecologically important difference in plasticity is most probably related to a slightly higher nutrient availability in the natural habitat of G. crithmifolia when compared with that of H. prostrata.
DOI:10.1093/jxb/erh111URLPMID:15047760 [本文引用: 1]
Storage of phosphorus (P) in stem tissue is important in Mediterranean Proteaceae, because proteoid root growth and P uptake is greatest during winter, whereas shoot growth occurs mostly in summer. This has prompted the present investigation of the P distribution amongst roots, stems, and leaves of Hakea prostrata R.Br. (Proteaceae) when grown in nutrient solutions at ten P-supply rates. Glasshouse experiments were carried out during both winter and summer months. For plants grown in the low-P range (0, 0.3, 1.2, 3.0, or 6.0 micromol d(-1)) the root [P] was &gt; stem and leaf [P]. In contrast, leaf [P] &gt; stem and root [P] for plants grown in the high-P range (6.0, 30, 60, 150, or 300 micromol P d(-1)). At the highest P-supply rates, the capacity for P storage in stems and roots appears to have been exceeded, and leaf [P] thereafter increased dramatically to approximately 10 mg P g(-1) dry mass. This high leaf [P] was coincident with foliar symptoms of P toxicity which were similar to those described for many other species, including non-Proteaceae. The published values (tissue [P]) at which P toxicity occurs in a range of species are summarized. X-ray microanalysis of frozen, full-hydrated leaves revealed that the [P] in vacuoles of epidermal, palisade and bundle-sheath cells were in the mM range when plants were grown at low P-supply, even though very low leaf [P] was measured in bulk leaf samples. At higher P-supply rates, P accumulated in vacuoles of palisade cells which were associated with decreased photosynthetic rates.
DOI:10.1111/pce.13155URLPMID:29380389 [本文引用: 1]
Plants exhibit respiratory bypasses (e.g., the alternative oxidase [AOX]) and increase the synthesis of carboxylates in their organs (leaves and roots) in response to phosphorus (P) deficiency, which increases P uptake capacity. They also show differential expression of high-affinity inorganic phosphorus (Pi) transporters, thus avoiding P toxicity at a high P availability. The association between AOX and carboxylate synthesis was tested in Solanum lycopersicum plants grown at different soil P availability, by using plants grown under P-sufficient and P-limiting conditions and by applying a short-term (24?hr) P-sufficient pulse to plants grown under P limitation. Tests were also performed with plants colonized with arbuscular mycorrhizal fungi, which increased plant P concentration under reduced P availability. The in vivo activities of AOX and cytochrome oxidase were measured together with the concentration of carboxylates and the P concentration in plant organs. Gene transcription of Pi transporters (LePT1 and LePT2) was also studied. A coordinated response between plant P concentration with these traits was observed, indicating that a sufficient P availability in soil led to a suppression of both AOX activity and synthesis of citrate and a downregulation of the transcription of genes encoding high-affinity Pi transporters, presumably to avoid P toxicity.
DOI:10.1016/j.biortech.2018.07.148URLPMID:30081286 [本文引用: 1]
A high phosphorus concentration is widely accepted as favorable for enhancing both microalgae growth and lipid accumulation; however, excessively high P could be counter-productive. In this study, we investigated the effects of increasing P levels (5.4, 25, 45, 150, and 250?mg-P?L-1) on the heterotrophic cultivation of Chlorella regularis. Microalgae growth was inhibited and cells were severely damaged in response to highly excessive P levels (≥150?mg-P?L-1). In particular, 250?mg-P?L-1 resulted in a ~40% decrease in cell density and a ~70% loss of cell viability. Microalgae damage induced by excessive phosphorus included enlarged cell size, deformation of cell walls, and disorganization of organelles. These negative effects were associated with the over-accumulation of polyphosphates within cells, which may further cause binding of P to intracellular components. Although P is an essential nutrient, excessive P lowers cell growth and viability.
DOI:10.1371/journal.pone.0136355URLPMID:26302211 [本文引用: 2]
Quantitative real-time PCR (RT-qPCR), a sensitive technique for quantifying gene expression, depends on the stability of the reference gene(s) used for data normalization. Several studies examining the selection of reference genes have been performed in ornamental plants but none in sweet osmanthus (Osmanthus fragrans Lour.). Based on transcriptomic sequencing data from O. fragrans buds at four developmental stages, six reference genes (OfACT, OfEF1α, OfIDH, OfRAN1, OfTUB, and OfUBC2) with stable expression (0.5 to 2 fold change in expression levels between any two developmental stages), as well as the commonly used reference gene Of18S, were selected as candidates for gene expression normalization in the RT-qPCR analysis of O. fragrans. For the normalization of RT-qPCR with two dyes, SYBR Green and EvaGreen, the expressional stability of seven candidate reference genes in 43 O. fragrans samples was analyzed using geNorm, NormFinder and BestKeeper. For RT-qPCR using SYBR Green, OfRAN1 and OfUBC2 were the optimal reference genes for all samples and different cultivars, OfACT and OfEF1α were suitable for different floral developmental stages, and OfACT was the optimal reference gene for different temperature treatments. The geometric mean values of the optimal reference gene pairs for the normalization of RT-qPCR are recommended to be used for all samples, different cultivars and different floral developmental stages in O. fragrans. For RT-qPCR using EvaGreen, OfUBC2 was the optimal reference gene for all samples and different cultivars, and OfACT was the optimal reference gene for different floral developmental stages and different temperature treatments. As the worst reference gene, Of18S should not be used as a reference gene in O. fragrans in the future. Our results provide a reference gene application guideline for O. fragrans gene expression characterization using RT-qPCR.
DOI:10.1186/s12867-018-0109-4URLPMID:29933763 [本文引用: 1]
Strawberry has received much attention due to its nutritional value, unique flavor, and attractive appearance. The availability of the whole genome sequence and multiple transcriptome databases allows the great possibility to explore gene functions, comprehensively. Gene expression profiles of a target gene can provide clues towards the understanding of its biological function. Quantitative real-time PCR (qRT-PCR) is a preferred method for rapid quantification of gene expression. The accuracy of the results obtained by this method requires the reference genes with consistently stable expression to normalize its data.
DOI:10.1016/j.bbrc.2006.04.140URLPMID:16690022 [本文引用: 1]
For accurate and reliable gene expression results, normalization of real-time PCR data is required against a control gene, which displays highly uniform expression in living organisms during various phases of development and under different environmental conditions. We assessed the gene expression of 10 frequently used housekeeping genes, including 18S rRNA, 25S rRNA, UBC, UBQ5, UBQ10, ACT11, GAPDH, eEF-1alpha, eIF-4a, and beta-TUB, in a diverse set of 25 rice samples. Their expression varied considerably in different tissue samples analyzed. The expression of UBQ5 and eEF-1alpha was most stable across all the tissue samples examined. However, 18S and 25S rRNA exhibited most stable expression in plants grown under various environmental conditions. Also, a set of two genes was found to be better as control for normalization of the data. The expression of these genes (with more uniform expression) can be used for normalization of real-time PCR results for gene expression studies in a wide variety of samples in rice.
DOI:10.1371/journal.pone.0119569URLPMID:25786207 [本文引用: 1]
Tall fescue (Festuca arundinacea Schreb.) is widely utilized as a major forage and turfgrass species in the temperate regions of the world and is a valuable plant material for studying molecular mechanisms of grass stress tolerance due to its superior drought and heat tolerance among cool-season species. Selection of suitable reference genes for quantification of target gene expression is important for the discovery of molecular mechanisms underlying improved growth traits and stress tolerance. The stability of nine potential reference genes (ACT, TUB, EF1a, GAPDH, SAND, CACS, F-box, PEPKR1 and TIP41) was evaluated using four programs, GeNorm, NormFinder, BestKeeper, and RefFinder. The combinations of SAND and TUB or TIP41 and TUB were most stably expressed in salt-treated roots or leaves. The combinations of GAPDH with TIP41 or TUB were stable in roots and leaves under drought stress. TIP41 and PEPKR1 exhibited stable expression in cold-treated roots, and the combination of F-box, TIP41 and TUB was also stable in cold-treated leaves. CACS and TUB were the two most stable reference genes in heat-stressed roots. TIP41 combined with TUB and ACT was stably expressed in heat-stressed leaves. Finally, quantitative real-time polymerase chain reaction (qRT-PCR) assays of the target gene FaWRKY1 using the identified most stable reference genes confirmed the reliability of selected reference genes. The selection of suitable reference genes in tall fescue will allow for more accurate identification of stress-tolerance genes and molecular mechanisms conferring stress tolerance in this stress-tolerant species.
DOI:10.1186/s12867-019-0126-yURLPMID:30909859 [本文引用: 1]
Isatis indigotica, a traditional Chinese medicine, produces a variety of active ingredients. However, little is known about the key genes and corresponding expression profiling involved in the biosynthesis pathways of these ingredients. Quantitative real-time polymerase chain reaction (qRT-PCR) is a powerful, commonly-used method for gene expression analysis, but the accuracy of the quantitative data produced depends on the appropriate selection of reference genes.
DOI:10.1111/j.1365-313X.2007.03108.xURLPMID:17461783 [本文引用: 1]
The PHO1 family comprises 11 members in Arabidopsis thaliana. In order to decipher the role of these genes in inorganic phosphate (Pi) transport and homeostasis, complementation of the pho1 mutant, deficient in loading Pi to the root xylem, was determined by the expression of the PHO1 homologous genes under the control of the PHO1 promoter. Only PHO1 and the homologue PHO1;H1 could complement pho1. The PHO1;H1 promoter was active in the vascular cylinder of roots and shoots. Expression of PHO1;H1 was very low in Pi-sufficient plants, but was strongly induced under Pi-deficient conditions. T-DNA knock-out mutants of PHO1;H1 neither showed growth defects nor alteration in Pi transport dynamics, or Pi content, compared with wild type. However, the double mutant pho1/pho1;h1 showed a strong reduction in growth and in the capacity to transfer Pi from the root to the shoot compared with pho1. Grafting experiments revealed that phenotypes associated with the pho1 and pho1/pho1;h1 mutants were linked to the lack of gene expression in the root. The increased expression of PHO1;H1 under Pi deficiency was largely controlled by the transcription factor PHR1 and was suppressed by the phosphate analogue phosphite, whereas the increase of PHO1 expression was independent of PHR1 and was not influenced by phosphite. Together, these data reveal that although transfer of Pi to the root xylem vessel is primarily mediated by PHO1, the homologue PHO1;H1 also contributes to Pi loading to the xylem, and that the two corresponding genes are regulated by Pi deficiency by distinct signal transduction pathways.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}