 ,, 靳学慧,, 张晓玉, 姜军黑龙江八一农垦大学农学院/黑龙江省植物抗性研究中心,黑龙江大庆 163000
,, 靳学慧,, 张晓玉, 姜军黑龙江八一农垦大学农学院/黑龙江省植物抗性研究中心,黑龙江大庆 163000Detection and Analysis of Magnaporthe oryzae Avirulence Genes AVR-Pib, AVR-Pik and AvrPiz-t in Heilongjiang Province
MENG Feng, ZHANG YaLing,, JIN XueHui,, ZHANG XiaoYu, JIANG JunCollege of Agronomy, Heilongjiang Bayi Agricultural University/Heilongjiang Plant Resistance Research Center, Daqing 163000, Heilongjiang通讯作者:
责任编辑: 岳梅
收稿日期:2019-05-30接受日期:2019-08-1网络出版日期:2019-12-01
| 基金资助: |
Received:2019-05-30Accepted:2019-08-1Online:2019-12-01
作者简介 About authors
孟峰,E-mail:xmfengf@126.com

摘要
关键词:
Abstract
Keywords:
PDF (2746KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
孟峰, 张亚玲, 靳学慧, 张晓玉, 姜军. 黑龙江省稻瘟病菌无毒基因AVR-Pib、AVR-Pik和AvrPiz-t的检测与分析[J]. 中国农业科学, 2019, 52(23): 4262-4273 doi:10.3864/j.issn.0578-1752.2019.23.007
MENG Feng, ZHANG YaLing, JIN XueHui, ZHANG XiaoYu, JIANG Jun.
0 引言
【研究意义】稻瘟病菌(Magnaporthe oryzea)引起的稻瘟病是世界水稻生产中最具有毁灭性的病害之一[1,2],目前,控制该病害最为经济有效并且对环境安全的策略是种植抗病品种。但经常一个高抗水稻品种由于病原菌无毒基因的突变或者有毒群体的增长,导致抗病品种应用3—5年后便“丧失”抗性,变为高度的感病品种。因此,对水稻与稻瘟病菌的互作调控机制深入研究,了解病原物丧失无毒功能的机制并阐明无毒基因变异对水稻抗性的影响,从而进一步了解无毒基因和对应抗性基因之间的互作关系,对长久、有效地防治稻瘟病具有重要意义。【前人研究进展】稻瘟病系统是一个经典的基因对基因系统[3],其中致病菌中的无毒(AVR)基因在功能上与水稻中的特定抗病基因(R)相对应。只有在稻瘟病菌的无毒基因与寄主的抗性基因相互作用时,水稻品种才能表现出抗瘟性[4,5]。近年来在水稻抗瘟基因与稻瘟病菌无毒基因研究方面已经取得了非常重要的研究进展,据不完全统计,目前已鉴定的无毒基因有40多个[6],其中PWL1、PWL2、PWL2D、ACE1、AVR-Pita、AVR1-CO39、AvrPiz-t、AVR-Pia、AVR-Pii、AVR-Pik/kp/km、AVR-Pi9和AVR-Pib等被克隆[7,8,9,10,11,12,13,14,15,16]。无毒基因在病菌群体中的存在、缺失及变异会影响与寄主植物抗性基因的识别,对于病菌群体中无毒基因的研究有利于抗病品种的选育和利用。李祥晓等[17]对177个采自黑龙江省稻瘟病菌单孢菌株进行PCR扩增检测,并采用20个已知抗性基因单基因系对48个黑龙江省稻瘟病菌进行毒力分析,结果表明黑龙江省稻瘟病菌无毒基因具有明显的地理特征和品种的特异性,在不同地区应根据无毒基因组成选取有效的抗病基因用于水稻抗瘟育种;王世维等[18]选取辽宁省稻瘟病常发区的26株稻瘟病菌单孢菌株,对6个无毒基因的PCR产物进行碱基与氨基酸序列比较分析,结果表明不同地区稻瘟病菌菌株的无毒基因类型及发生频率差异较大,可为辽宁省不同稻区的品种种植提供参考依据;汪文娟等[19]采用抗稻瘟病单基因系对稻瘟病菌单孢分离菌株进行致病型测定,并根据已经克隆的且与稻瘟病菌致病性相关的8个无毒基因的功能性分子标记进行分离菌株的无毒基因型分析,结果表明在华南主栽品种广8 A杂交稻组合中分布较为广泛的无毒基因有AVR-Pik和AVR-Pi9。【本研究切入点】深入阐明稻瘟病菌无毒基因的变异特征,需要从等位基因型与病原菌致病型两方面开展研究,据前人报道抗性基因Pib、Pik和Piz-t在黑龙江省主栽水稻品种中存在较为广泛,并且具有较高的利用价值[20,21]。为准确探明黑龙江省稻瘟病菌无毒基因AVR-Pib、AVR-Pik和AvrPiz-t的分布与变异机制,本研究结合无毒基因的扩增与测序结果和抗病单基因系品种致病型鉴定,对采集自黑龙江省的稻瘟病菌进行分析,研究不同菌株中无毒基因的类型及变异情况。【拟解决的关键问题】通过基因扩增,首先判断不同地区的稻瘟病菌是否携带无毒基因或其等位基因,再通过测序比对分析菌株之间无毒基因序列的变化,最后通过对不同变异类型的稻瘟病菌单孢分离菌株进行致病型测定,从而明确黑龙江省稻瘟病菌无毒基因分布情况及变异类型,以期为黑龙江省抗瘟品种的合理布局与稻瘟病的有效防控提供依据。1 材料与方法
试验于2018年6月至2019年4月在黑龙江八一农垦大学植物抗性研究中心实验室完成。1.1 供试菌株
2016年和2017年在黑龙江省10个市20个县水稻种植区内采集稻瘟病穗颈瘟标样,经单孢分离获得单孢菌株335个(2016年菌株127个,2017年菌株208个),采用滤纸片保存法[22]保存备用。1.2 稻瘟病菌基因组DNA提取
将分离纯化的稻瘟病菌单孢菌株在PDA固体培养基上活化培养,挑取适量菌丝块接到酵母液体培养基中,于28℃摇床,120 r/min 振荡培养3—5 d,收集菌丝体,分装于1.5 mL离心管中,置于-20℃冰箱中冷冻保存。使用真菌DNA提取试剂盒(D3390-01 OmegaBio-Tek公司)提取稻瘟病菌基因组DNA,DNA提取之后使用微量分光光度计测定DNA浓度,并将DNA原液稀释成60 ng·μL-1的工作液备用。原液-20℃保存。1.3 引物设计
根据文献中已克隆的无毒基因于NCBI上查找基因序列,利用Primer premier 5.0在基因全长和CDS区设计6对特异性引物,所有引物均委托上海生工生物工程技术有限公司合成,引物序列见表1。Table 1
表1
表1用于扩增稻瘟病菌无毒基因的引物
Table 1
| 无毒基因 Avirulence gene | 引物序列 Primer sequence (5′-3′) | 目的片段长度 Length of targeted fragments (bp) | 目的 Purpose |
|---|---|---|---|
| AVR-Pib | F1: AAGTCCTTCCCATTACCCTA R1: GCAATAACCATCCAGCCATA | 484 | 检测AVR-Pib CDS Detection of AVR-Pib CDS |
| F2: GAAGTACCCACCCATAACCC R2: CAAGGGAGGCAAATCTAACC | 1147 | 检测AVR-Pib基因全长 Detection of AVR-Pib gene full length | |
| AVR-Pik | F1: AATTTATTCAACTGCCACTCTG R1: AACCTCGTCAAACCTCCCTA | 526 | 检测AVR-Pik CDS Detection of AVR-Pik CDS |
| F2: GACAAACAGGATGGGATT R2: AGGTCGTAGGTCGGAAAC | 1929 | 检测AVR-Pik基因全长 Detection of AVR-Pik gene full length | |
| AvrPiz-t | F1: ATCAAATGAACACCAGGAA R1: CCAGCCGAAGATACAAAA | 450 | 检测AvrPiz-t CDS Detection of AvrPiz-t CDS |
| F2: AATCTCCCAATGGTTCGC R2: AAAGTGGCTCGTTCCTAA | 1965 | 检测AvrPiz-t基因全长 Detection of AvrPiz-t gene full length |
新窗口打开|下载CSV
1.4 PCR扩增及电泳检测
PCR反应体系(20 μL):rTaq酶(5 U·μL-1)0.1 μL,10×Buffer(Mg+)2.0 μL,dNTP Mixture 1.6 μL(2.5 m Meach),正反向引物(10 μmol·L-1)各0.3 μL,DNA模板(20—30 ng·μL-1)1 μL,加ddH2O补足20 μL。扩增程序:94℃预变性4 min,94℃变性45 s,55—60℃退火45 s(具体温度根据引物的GC含量设定),72℃延伸30—90 s(具体时间根据扩增片段大小确定),30个循环,72℃延伸10 min,4℃保存待检测。扩增产物在1%的琼脂糖凝胶中电泳,150 V,20 min,电泳液为0.5×TBE,在凝胶电泳成像系统下观察并拍照,统计无毒基因扩增频率。1.5 部分无毒基因序列测序分析
在黑龙江省2016年和2017年的稻瘟病菌菌株中挑选不同带型与不同地区的部分菌株送去上海生工生物工程技术有限公司测序,测序结果采用Lasergene 7.0的SeqMan软件进行比对与拼接;并采用DNAMAN软件对有差异的核苷酸序列进行比较分析。1.6 致病型测定
植物材料:2个抗稻瘟病单基因系分别为BL1(Pib)和IRBLzt-T(Piz-t),均为国际水稻研究所选育,感病对照:丽江新团黑谷(LTH)。菌株活化及产孢培养:在无菌条件下,将之前保存的滤纸片置入PDA培养基内培养,5 d后转置米糠培养基内进行产孢培养。当培养皿内长满菌丝后,将气生菌丝用涂布棒刮掉,将其放置在恒温恒湿环境中,用蓝紫光灯照射3 d,进行产孢培养。
植物材料种植:水稻品种采用室内水培方式进行培养。挑选饱满的种子用1.25‰的多菌灵溶液室温(20℃)浸泡24 h后,用清水冲洗干净,放入培养皿内并用滤纸保湿。于30℃的恒温培养箱内催芽,播种于固定在塑料水培盒(尺寸:225 mm×155 mm×55 mm)内的定植篮(上端内径34 mm,下端内径28 mm,高45 mm)中,每个定植篮内播种10粒,自来水培养3 d以后,换营养液培养,营养液采用经典的霍格兰(Hoagland)配方[23],每4 d换一次营养液,待苗长至3叶1心期进行人工喷雾接种。
接种及调查:每个培养皿用5 mL无菌水洗下孢子,用双层纱布过滤,经显微镜检测当孢子量为1×105个/mL时,将菌悬液装入喷壶中,并加入5 mL明胶溶液摇匀后均匀地喷洒在秧苗表面,接种后将其移入25℃的保湿棚内进行遮光保湿培养24 h,然后返回室温自然正常培养,接种后7 d调查,调查标准参考靳学慧等[24]。
2 结果
2.1 稻瘟病菌无毒基因AVR-Pib的基因型与致病表型分析
以稻瘟病菌335个菌株的DNA为模板,根据无毒基因AVR-Pib外显子区域与全序列设计的引物(图1)进行PCR扩增,电泳结果显示有4种不同类型的条带(图2),分别为无带、高带、中高带与低带,用0、1、2、3表示,带型0含有82个菌株,带型1含有8个菌株,带型2为2个,带型3的菌株有243个。将包含带型1、2、3的稻瘟病菌菌株共40株的PCR产物进行测序(其中带型1的菌株4个;带型2的菌株2个;带型3的菌株34个)。测序结果发现4个新的基因型,即带型1的菌株一个基因型是在基因阅读框上游-37 bp处有转座子Pot2的插入,另一个基因型是阅读框的5′端26 bp处有转座子Pot2的插入,将其分别命名为AVR-Pib-1-1和AVR-Pib-1-2。带型2的菌株碱基序列在基因上游-129 bp处有500 bp的碱基序列插入,将其命名为AVR-Pib-2;带型3的菌株YL17077序列比对AVR-Pib有4处差异,即32(C/G)35(T/A)36(T/A)38(T/A)(图3),导致翻译提前终止,将其命名为AVR-Pib-3-1,其他菌株的序列均与AVR-Pib序列完全一致,且出现频率最高,命名为AVR-Pib-3。图1
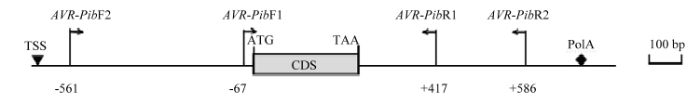 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1无毒基因AVR-Pib两个引物AVR-Pib F1/R1和AVR-Pib F2/R2的位置
Fig. 1Location of AVR-Pib F1/R1 and AVR-Pib F2/R2 primers in avirulence gene AVR-Pib
图2
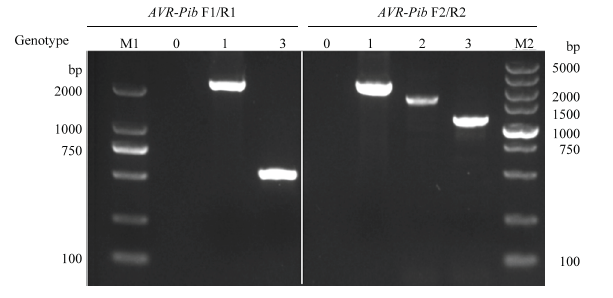 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2供试菌株AVR-Pib等位基因型扩增的琼脂糖凝胶电泳条带
M1:Marker,DL2000;M2:Marker,DL5000;0:未扩增出条带No band;1:高带型基因 High band alleles;2:中高带型基因 Mid to high band alleles;3:低带型基因(正常基因)Lower band alleles (general gene)
Fig. 2Agarose gel electrophoresis bands of AVR-Pib amplification of tested strains
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3AVR-Pib等位基因变异的特征
Fig. 3Characterization of allelic variation at AVR-Pib
采用AVR-Pib-3及4种变异类型AVR-Pib-1-1、AVR-Pib-1-2、AVR-Pib-2和AVR-Pib-3-1的菌株接种水稻单基因系BL1(Pib),以丽江新团黑谷(LTH)为对照。结果显示正常AVR-Pib基因型菌株能够诱导Pib产生无毒性(A),而4种新变异类型均不能被Pib识别而表现为有毒性(V),表明此变异类型造成无毒功能丧失(图4)。
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4AVR-Pib-3、AVR-Pib-1-1、AVR-Pib-1-2、AVR-Pib-2和AVR-Pib-3-1菌株的致病表型
A:无毒性Avirulent;V:有毒性Virulent
Fig. 4Disease phenotype of the AVR-Pib-3, AVR-Pib-1-1, AVR-Pib-1-2, AVR-Pib-2 and AVR-Pib-3-1 strains
2.2 稻瘟病菌无毒基因AVR-Pik的扩增结果与序列分析
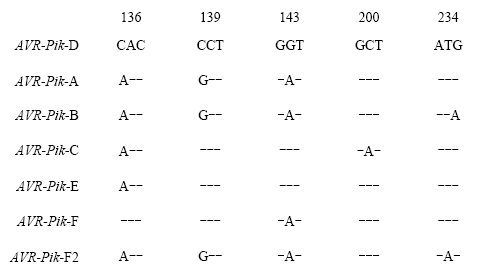
以黑龙江省稻瘟病菌335个菌株的菌丝DNA为模板,根据无毒基因AVR-Pik-D序列设计引物扩增,电泳结果显示有292株可扩增出特异性片段。从292株菌株中挑选不同地区与不同年份的菌株40株进行测序,与NCBI中AVR-Pik-D的CDS序列比对分析,结果发现7种等位基因型,且均有文献报道[13,18,25]。其中有6个菌株序列与报道的AVR-Pik-D序列完全一致;其余菌株的PCR产物序列与已经克隆的AVR-Pik-D序列有着不同程度的差异(图5)。根据差异可分为6种类型:第1类,与YOSHIDA等[13]文中报道的AVR-Pik-A类型序列一致,与AVR-Pik-D相比有3处差异,即136(C/A)、139(C/G)和143(G/A);第2类,与YOSHIDA等[13]文中报道的AVR-Pik-B类型序列一致,与AVR-Pik-D相比有4处差异,即136(C/A)、139(C/G)、143(G/A)和234(G/A);第3类,与YOSHIDA等[13]文中报道的AVR-Pik-C类型序列一致,与AVR-Pik-D相比有2处差异,即136(C/A)和200(C/A);第4类,与YOSHIDA等[13]文中报道的AVR-Pik-E类型序列一致,与AVR-Pik-D相比有1处差异,即136(C/A);第5类,与王世维等[18]文中新增的AVR-Pik-F类型序列完全一致,与AVR-Pik-D相比有1处差异,即143(G/A);第6类,与LONGYA等[25]文中报道的新基因型AVR-Pik-F序列一致,与AVR-Pik-D相比有4处差异136(C/A)、139(C/G)、143(G/A)和234(T/A),本试验将其命名为AVR-Pik-F2。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5供试菌株的AVR-Pik 碱基序列比对
Fig. 5Base sequence alignment of AVR-Pik of tested strains
6类基因型碱基序列翻译的氨基酸序列与AVR-Pik基因组(D型)的氨基酸序列比对结果如图6。AVR-Pik-A的氨基酸序列存在3处错义突变,分别为46(H/N)、47(P/A)和48(G/D);AVR-Pik-B的氨基酸序列存在4处错义突变,分别为46(H/N)、47(P/A)、48(G/D)和78(M/I);AVR-Pik-C的氨基酸序列存在2处错义突变,分别为46(H/N)和67(A/D);AVR-Pik-E的136(C/A)SNP造成了氨基酸突变46(H/N);AVR-Pik-F的143(G/A)SNP造成了氨基酸突变48(G/D)。AVR-Pik-F2的氨基酸序列存在3处错义突变46(H/N)、47(P/A)、48(G/D)和78(M/K)。
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6AVR-Pik氨基酸序列对比分析
Fig. 6Comparison analysis of amino acid sequence of AVR-Pik genotypes
2.3 稻瘟病菌无毒基因AvrPiz-t的基因型与致病表型分析
以黑龙江省稻瘟病菌335个菌株的菌丝DNA为模板,根据无毒基因AvrPiz-t的外显子区域与基因全长设计引物扩增,电泳结果显示有287株可扩增出特异性片段(图7),有4个菌株扩增出高带型。从中挑选40个不同带型、不同地区有特异性扩增片段的稻瘟病菌菌株PCR产物进行测序,结果发现4种基因型:第1类,菌株序列与基因原序列相同将其命名为AvrPizt-A;第2类,在CDS的191 bp处有碱基A的插入,本试验将此变异类型命名为AvrPiz-t-B;第3类,与AvrPiz-t-A存在2处差异,即CDS的17 bp处碱基C的替换和19 bp处碱基C的插入,此变异类型命名为AvrPiz-t-C(图8);第4类,在启动子区域-462 bp处有转座子Pot3插入,此变异类型已有报道且无毒功能已丧失[12],本试验将其命名为AvrPiz-t-D(图9)。对推测的氨基酸序列进行比较分析发现,AvrPizt-B在64位发生移码突变,导致氨基酸序列至89位时提前终止;AvrPizt-C在第6位发生移码突变,并在46位处提前终止(图10)。图7
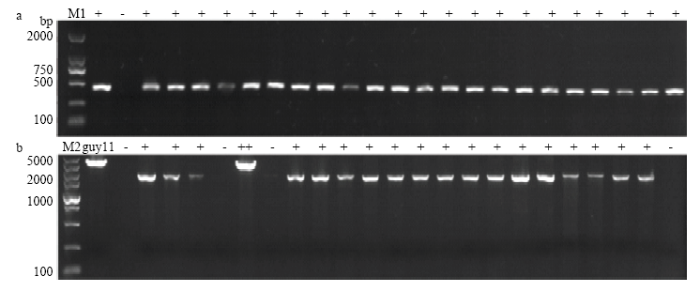 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7供试菌株无毒基因AvrPiz-t的扩增琼脂糖凝胶电泳条带
a:AvrPiz-t F1/R1的扩增结果Amplification result of AvrPiz-t F1/R1;b:AvrPiz-t F2/R2的扩增结果Amplification result of AvrPiz-t F2/R2。M1:Marker,DL2000;M2:Marker,DL5000;“+”:正常基因型Normal genotype;“++”:高带型High band type;“-”:未扩增出条带 No amplification band;“guy11”:标准菌株Standard strain
Fig. 7Agarose gel electrophoresis bands of AvrPiz-t amplification of tested strains
图8
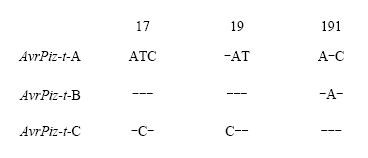 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8供试菌株AvrPiz-t碱基序列比对
Fig. 8Base sequence alignment of AvrPiz-t of tested strains
图9
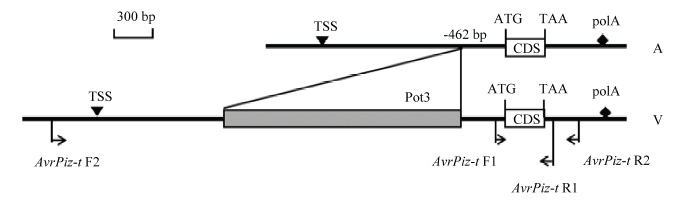 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9AvrPiz-t特点及引物设计
Fig. 9AvrPiz-t characteristics and primer design
图10
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10AvrPiz-t 氨基酸序列对比分析
Fig. 10Comparison analysis of amino acid sequence of AvrPiz-t genotypes
对无毒基因型AvrPiz-t 突变类型的致病型进行了分析,如图11所示,以丽江新团黑谷(LTH)为对照,结果显示,AvrPiz-t-A可以被Piz-t识别表现无毒性,AvrPiz-t-B、AvrPiz-t-C和AvrPiz-t-D均不能被Piz-t识别而表现为有毒性。
图11
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图11供试菌株的AvrPiz-t-A、AvrPiz-t-B、AvrPiz-t-C和AvrPiz-t-D致病表型
A:无毒性Avirulent;V:有毒性Virulent
Fig. 11Disease phenotype of the AvrPiz-t-A, AvrPiz-t-B, AvrPiz-t-C and AvrPiz-t-D strains
2.4 3种无毒基因在黑龙江省的分布情况
对2016年和2017年黑龙江省335个稻瘟病菌单孢菌株3个无毒基因AVR-Pib、AVR-Pik和AvrPiz-t的检测结果显示,这3个无毒基因在黑龙江省各稻区以不同分布频率与不同等位基因类型出现,连续两年出现频率较高且稳定遗传,平均出现频率均超过70%,分别为75.52%、87.16%和85.67%(表2)。表明这3个无毒基因为黑龙江省稻瘟病菌携带的重要无毒基因。Table 2
表2
表22016年和2017年黑龙江省稻瘟病菌无毒基因的扩增频率
Table 2
| 无毒基因 Avirulence gene | 2016(127 a) | 2017(208 b) | 总计Total(335 c) | |||
|---|---|---|---|---|---|---|
| 出现个数 Number of occurrences | 出现频率 Frequency of occurrence (%) | 出现个数 Number of occurrences | 出现频率 Frequency of occurrence (%) | 出现个数 Number of occurrences | 出现频率 Frequency of occurrence (%) | |
| AVR-Pib | 104 | 81.89 | 149 | 71.63 | 253 | 75.52 |
| AVR-Pik | 105 | 82.68 | 187 | 89.90 | 292 | 87.16 |
| AvrPiz-t | 111 | 87.40 | 176 | 84.62 | 287 | 85.67 |
新窗口打开|下载CSV
3 讨论
3.1 黑龙江省稻瘟病菌的无毒基因型与品种的合理布局
黑龙江省是中国北方粳稻主产区,种植面积占东北地区总面积的58.1%[26],由于抗性品种抗性“丧失”使稻瘟病危害日益加重而造成粮食的大面积减产甚至绝收。稻瘟病菌的无毒基因可与相应的抗性基因互作,导致水稻植株发生非亲和反应而产生抗性,因此,监测黑龙江省主要水稻种植区稻瘟病菌的无毒基因型,合理利用抗病品种可以很大程度上降低稻瘟病发生。本试验检测到AVR-Pib、AVR-Pik和AvrPiz-t这3种无毒基因在黑龙江省稳定存在,无毒基因型AVR-Pib-3和AvrPiz-t-A出现频率最高且在黑龙江省各稻区均有分布,根据本试验的致病型测定来看,AVR-Pib-3对抗性品种Pib表现为无毒性。据时克等[27]报道在中国水稻品种中锦辉501等9个籼稻品种和粳稻品种辽粳454、武育粳7号携带抗性基因Pib,刘华招等[20]在黑龙江省种植品种中检测到垦稻13、垦稻14、松粳9、龙粳2这4份材料含有Pib。所以在品种选育时可以种植以上携带Pib的水稻品种。AvrPiz-t-A对Piz-t表现出无毒性,据于连鹏[21]报道黑龙江省48个主栽水稻品种中含有Piz-t的占77.08%且分布较广,因此携带抗性基因Piz-t的品种在黑龙江省抗病育种和品种合理布局时可以选用。在AVR-Pik的等位基因中,AVR-Pik-F2与AVR-Pik-C出现频率最高,据文献报道AVR-Pik-F2和AVR-Pik-C均不能被抗性基因Pik、Pikm和Pikp所识别[25,28],因此在生产上要避免利用携带Pik、Pikm或Pikp的品种。3.2 无毒基因的变异机制与功能验证
病原菌无毒基因不稳定,常常引发变异,无毒基因的变异导致其逃脱寄主抗病基因的识别使抗病品种丧失抗病性。因此,了解无毒基因变异机制对于培育抗病品种是至关重要的。无毒基因的变异机制主要包括点突变、缺失、插入、复制、移码突变等[29]。AVR-Pib变异能力较强,据ZHANG等[30]报道AVR-Pib检测出5种带型(无带、高带、中高带、低带与双带)和11种变异类型,且13位处的异亮氨酸为热点可变位置。在本研究中,黑龙江省稻瘟病菌检测出4种带型(无带、高带、中高带与低带)和5种基因型,未检测到双带型,变异类型AVR-Pib-3-1在热点可变位置翻译提前终止,由于此变异类型会导致抗性基因Pib的功能丧失,所以应引起研究者的关注。
AVR-Pik与Pik等位基因间呈现出了竞争性阶梯型共进化模式[31],据KANZAKI等[28]研究报道,AVR-Pik与Pik具有高度变异性,AVR-Pik主要突变类型是编码区单碱基的突变从而引起氨基酸的变异,5个氨基酸突变活性位点为46、47、48、67和78[13]。本试验检测出7种等位基因型,且无毒基因序列变异会导致其毒性发生变化。因此如何使用抗谱更广的抗性基因或采用多基因联合手段克服AVR-Pik的变异仍需进一步的研究。
AvrPiz-t是一个片段长度为327 bp,无内含子的基因,编码一个含有108个氨基酸的小分泌蛋白。它存在于有毒菌株guy11中,但在其启动子区域-462 bp处有转座子Pot3的插入,使基因表达不稳定无毒功能丧失[11]。AvrPiz-t的变异类型主要有:一是插入突变,如在启动子区域和ORF区域插入片段大小不等的重复序列,如转座子Pot2、Pot3,逆转录转座子Inago2、MGR583等。二是点突变,如编码区域第41位氨基酸的改变(A41V)[32]。本试验中插入突变和点突变均有发现。其中AvrPiz-t-B曾在2014和2015年黑龙江省稻区内被发现[33],但当时是否丧失无毒功能未被验证。AvrPiz-t-C的变异类型为点突变和单个碱基的插入,导致翻译提前终止,AvrPiz-t-D为-462 bp处插入Pot3转座子,通过基因功能验证得知AvrPiz-t(B、C、D)已丧失无毒功能。
4 结论
无毒基因AVR-Pib、AVR-Pik和AvrPiz-t在黑龙江省均有分布,并以不同分布频率与不同变异类型出现,且3种无毒基因型在黑龙江省稻瘟病菌中稳定遗传。无毒基因AVR-Pib在黑龙江省检测出4种带型(无带、高带、中高带与低带),同时检测出5种基因型AVR-Pib(1-1、1-2、2、3、3-1),其中AVR-Pib(1-1、1-2、2、3-1)无毒功能均已丧失;AVR-Pik检测出7种等位基因型,AVR-Pik(D、A、B、C、E、F、F2),均有文献报道;AvrPiz-t本试验检测出4种基因型AvrPiz-t(A、B、C、D),AvrPiz-t-C为新增基因型,AvrPiz-t(B、C、D)失去无毒功能。黑龙江省各稻区稻瘟病菌复杂多样,但能够通过及时地监测无毒基因在稻瘟病病原菌群体中的组成与变化,来指导田间抗性基因的合理布局和抗病品种的定期轮换,以有效地控制病害发生,并为抗病种质资源筛选提供参考依据。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1080/15572536.2003.11833196URLPMID:21156541 [本文引用: 1]

Magnaporthe oryzae is described as a new species distinct from M. grisea. Gene trees were inferred for Magnaporthe species using portions of three genes: actin, beta-tubulin, and calmodulin. These gene trees were found to be concordant and distinguished two distinct clades within M. grisea. One clade is associated with the grass genus Digitaria and is therefore nomenclaturally tied to M. grisea. The other clade is associated with Oryza sativa and other cultivated grasses and is described as a new species, M. oryzae. While no morphological characters as yet distinguish them, M. oryzae is distinguished from M. grisea by several base substitutions in each of three loci as well as results from laboratory matings; M.oryzae and M. grisea are not interfertile. Given that M. oryzae is the scientifically correct name for isolates associated with rice blast and grey leaf spot, continued use of M. grisea for such isolates would require formal nomenclatural conservation.
[本文引用: 1]
DOI:10.1146/annurev-phyto-072910-095339URLPMID:21599495 [本文引用: 1]
The apple scab (Venturia inaequalis-Malus) pathosystem was one of the first systems for which Flor's concept of gene-for-gene (GfG) relationships between the host plant and the pathogen was demonstrated. There is a rich resource of host resistance genes present in Malus germplasm that could potentially be marshalled to confer durable resistance against this most important apple disease. A comprehensive understanding of the host-pathogen interactions occurring in this pathosystem is a prerequisite for effectively manipulating these host resistance factors. An accurate means of identification of specific resistance and consistent use of gene nomenclature is critical for this process. A set of universally available, differentially resistant hosts is described, which will be followed by a set of defined pathogen races at a later stage. We review pertinent aspects of the history of apple scab research, describe the current status and future directions of this research, and resolve some outstanding issues.
DOI:10.1105/tpc.110.078048URLPMID:20858844 [本文引用: 1]
Magnaporthe oryzae causes rice blast, the most serious foliar fungal disease of cultivated rice (Oryza sativa). During hemibiotrophic leaf infection, the pathogen simultaneously combines biotrophic and necrotrophic growth. Here, we provide cytological and molecular evidence that, in contrast to leaf tissue infection, the fungus adopts a uniquely biotrophic infection strategy in roots for a prolonged period and spreads without causing a loss of host cell viability. Consistent with a biotrophic lifestyle, intracellularly growing hyphae of M. oryzae are surrounded by a plant-derived membrane. Global, temporal gene expression analysis used to monitor rice responses to progressive root infection revealed a rapid but transient induction of basal defense-related gene transcripts, indicating perception of the pathogen by the rice root. Early defense gene induction was followed by suppression at the onset of intracellular fungal growth, consistent with the biotrophic nature of root invasion. By contrast, during foliar infection, the vast majority of these transcripts continued to accumulate or increased in abundance. Furthermore, induction of necrotrophy-associated genes during early tissue penetration, previously observed in infected leaves, was not seen in roots. Collectively, our results not only report a global characterization of transcriptional root responses to a biotrophic fungal pathogen but also provide initial evidence for tissue-adapted fungal infection strategies.
DOI:10.1371/journal.ppat.1001261URLPMID:21283781 [本文引用: 1]
Surface recognition and penetration are among the most critical plant infection processes in foliar pathogens. In Magnaporthe oryzae, the Pmk1 MAP kinase regulates appressorium formation and penetration. Its orthologs also are known to be required for various plant infection processes in other phytopathogenic fungi. Although a number of upstream components of this important pathway have been characterized, the upstream sensors for surface signals have not been well characterized. Pmk1 is orthologous to Kss1 in yeast that functions downstream from Msb2 and Sho1 for filamentous growth. Because of the conserved nature of the Pmk1 and Kss1 pathways and reduced expression of MoMSB2 in the pmk1 mutant, in this study we functionally characterized the MoMSB2 and MoSHO1 genes. Whereas the Momsb2 mutant was significantly reduced in appressorium formation and virulence, the Mosho1 mutant was only slightly reduced. The Mosho1 Momsb2 double mutant rarely formed appressoria on artificial hydrophobic surfaces, had a reduced Pmk1 phosphorylation level, and was nonresponsive to cutin monomers. However, it still formed appressoria and caused rare, restricted lesions on rice leaves. On artificial hydrophilic surfaces, leaf surface waxes and primary alcohols-but not paraffin waxes and alkanes- stimulated appressorium formation in the Mosho1 Momsb2 mutant, but more efficiently in the Momsb2 mutant. Furthermore, expression of a dominant active MST7 allele partially suppressed the defects of the Momsb2 mutant. These results indicate that, besides surface hydrophobicity and cutin monomers, primary alcohols, a major component of epicuticular leaf waxes in grasses, are recognized by M. oryzae as signals for appressorium formation. Our data also suggest that MoMsb2 and MoSho1 may have overlapping functions in recognizing various surface signals for Pmk1 activation and appressorium formation. While MoMsb2 is critical for sensing surface hydrophobicity and cutin monomers, MoSho1 may play a more important role in recognizing rice leaf waxes.
DOI:10.1007/s00122-006-0347-6URL [本文引用: 1]
Avirulence of Magnaporthe grisea isolate CHL346 on rice cultivar GA25 was studied with 242 ascospore progenies derived from the cross CHL346×CHL42. Segregation analysis of the avirulence in the progeny population was in agreement with the existence of a single avirulence (Avr) gene, designated as AvrPi15. For mapping the Avr gene, we developed a total of 121 microsatellite DNA markers [simple sequence repeat (SSR)], which evenly distributed in the whole-genome of M. grisea through bioinformatics analysis (BIA) using the publicly available sequence. Linkage analysis of the AvrPi15 gene with these SSR markers showed that six markers on chromosome 6, MS6-1, MS6-2, MS6-3, MS6-7, MS6-8 and MS6-10, were linked to the AvrPi15 locus. To further define the chromosomal location of the AvrPi15 locus, two additional markers, MS6-17 and STS6-6, which were developed based on the sequences of telomeric region 11 (TEL11), were subjected to linkage analysis. The results showed that MS6-17 and STS6-6 were associated with the locus by 3.3 and 0.8cM, respectively. To finely map the Avr gene, two additional candidate avirulence gene (CAG) markers, CAG6-1 and CAG6-2, were developed based on the gene annotation of the sequence of TEL 11. Linkage analysis of the Avr gene with these two markers revealed that both of them completely cosegregated with the AvrPi15 locus. Finally, this locus was physically mapped into ∼7.2-kb interval of the TEL11 by BIA using these sequence-ready markers. This is the key step toward positional cloning of the AvrPi15 gene.
DOI:10.1105/tpc.7.8.1221URLPMID:7549480 [本文引用: 1]
Genetic analysis of host specificity in the rice blast fungus (Magnaporthe grisea) identified a single gene, PWL2 (for Pathogenicity toward Weeping Lovegrass), that exerts a major effect on the ability of this fungus to infect weeping lovegrass (Eragrostis curvula). The allele of the PWL2 gene conferring nonpathogenicity was genetically unstable, with the frequent appearance of spontaneous pathogenic mutants. PWL2 was cloned based on its map position. Large deletions detected in pathogenic mutants guided the gene cloning efforts. Transformants harboring the cloned PWL2 gene lost pathogenicity toward weeping lovegrass but remained fully pathogenic toward other host plants. Thus, the PWL2 host species specificity gene has properties analogous to classical avirulence genes, which function to prevent infection of certain cultivars of a particular host species. The PWL2 gene encodes a glycine-rich, hydrophilic protein (16 kD) with a putative secretion signal sequence. The pathogenic allele segregating in the mapping population, pwl2-2, differed from PWL2 by a single base pair substitution that resulted in a loss of function. The PWL2 locus is highly polymorphic among rice pathogens from diverse geographic locations.
DOI:10.1094/mpmi-8-0939URLPMID:8664503 [本文引用: 1]
The PWL2 gene, isolated from a Magnaporthe grisea rice pathogen, prevents this fungus from infecting a second host grass, weeping lovegrass. We have investigated the distribution of sequences homologous to PWL2 in M. grisea strains isolated from diverse grass species. Multiple PWL2 homologs with varying degrees of sequence homology were identified. The presence of PWL2 homologs does not correlate with an avirulent phenotype on weeping lovegrass in many cases: some strains were fully pathogenic on weeping lovegrass although they carry multiple PWL2 homologs. Three weakly hybridizing PWL2 homologs were cloned and characterized. One of these, the PWL1 gene previously identified by genetic analysis, functioned to prevent infection of weeping lovegrass. Cloned PWL3 and PWL4 genes were nonfunctional, although PWL4 became functional if its expression was driven by either the PWL1 or the PWL2 promoter. The PWL1, PWL2, and PWL3/PWL4 genes map to different genomic locations. The amino acid sequences of the predicted PWL1, PWL3, and PWL4 proteins have 75, 51, and 57% identity, respectively, to the PWL2 protein. Our studies indicate that PWL genes are members of a dynamic, rapidly evolving gene family.
DOI:10.1105/tpc.12.11.2019URLPMID:11090206 [本文引用: 1]
Genetic mapping showed that the rice blast avirulence gene AVR-Pita is tightly linked to a telomere on chromosome 3 in the plant pathogenic fungus Magnaporthe grisea. AVR-Pita corresponds in gene-for-gene fashion to the disease resistance (R) gene Pi-ta. Analysis of spontaneous avr-pita(-) mutants indicated that the gene is located in a telomeric 6.5-kb BglII restriction fragment. Cloning and DNA sequencing led to the identification of a candidate gene with features typical of metalloproteases. This gene is located entirely within the most distal 1.5 kb of the chromosome. When introduced into virulent rice pathogens, the cloned gene specifically confers avirulence toward rice cultivars that contain Pi-ta. Frequent spontaneous loss of AVR-Pita appears to be the result of its telomeric location. Diverse mutations in AVR-Pita, including point mutations, insertions, and deletions, permit the fungus to avoid triggering resistance responses mediated by Pi-ta. A point mutation in the protease consensus sequence abolishes the AVR-Pita avirulence function.
DOI:10.1111/j.1469-8137.2008.02459.xURLPMID:18433432 [本文引用: 1]
The avirulence gene ACE1 from the rice blast fungus Magnaporthe grisea encodes a polyketide synthase (PKS) fused to a nonribosomal peptide synthetase (NRPS) probably involved in the biosynthesis of a secondary metabolite recognized by Pi33 resistant rice (Oryza sativa) cultivars. Analysis of the M. grisea genome revealed that ACE1 is located in a cluster of 15 genes, of which 14 are potentially involved in secondary metabolism as they encode enzymes such as a second PKS-NRPS (SYN2), two enoyl reductases (RAP1 and RAP2) and a putative Zn(II)(2)Cys(6) transcription factor (BC2). These 15 genes are specifically expressed during penetration into the host plant, defining an infection-specific gene cluster. A pORF3-GFP transcriptional fusion showed that the highly expressed ORF3 gene from the ACE1 cluster is only expressed in appressoria, as is ACE1. Phenotypic analysis of deletion or disruption mutants of SYN2 and RAP2 showed that they are not required for avirulence in Pi33 rice cultivars, unlike ACE1. Inactivation of other genes was unsuccessful because targeted gene replacement and disruption were inefficient at this locus. Overall, the ACE1 gene cluster displays an infection-specific expression pattern restricted to the penetration stage which is probably controlled at the transcriptional level and reflects regulatory networks specific to early stages of infection.
URLPMID:9799257 [本文引用: 2]
The avrCO39 gene conferring avirulence toward rice cultivar CO39 was previously mapped to chromosome 1 of Magnaporthe grisea between cosegregating markers CH5-120H and 1.2H and marker 5-10-F. In the present study, this region of the chromosome was physically mapped using RecA-mediated Achilles' cleavage. Cleavage of genomic DNA sequences within CH5-120H and 5-10-F liberated a 610-kb restriction fragment, representing the physical distance between these markers. Chromosome walking was initiated from both markers but was curtailed due to the presence of repetitive DNA sequences and the absence of overlapping clones in cosmid libraries representing several genome equivalents. These obstacles were overcome by directly subcloning the target region after release by Achilles' cleavage and a contig spanning avrCO39 was thus assembled. Transformation of two cosmids into a virulent recipient strain conferred a cultivar-specific avirulence phenotype thus confirming the cloning of avrCO39. Meiotic crossover points were unevenly distributed across this chromosomal region and were clustered around the avrCO39 locus. A 14-fold variation in the relationship between genetic and physical distance was measured over the avrCO39 chromosomal region. Thus the poor correlation of physical to genetic distance previously observed in M. grisea appears to be manifested over relatively short distances.
DOI:10.1094/MPMI-22-4-0411URLPMID:19271956 [本文引用: 2]
The Magnaporthe oryzae avirulence gene AvrPiz-t activates immunity in a gene-for-gene fashion to rice mediated by the blast resistance gene Piz-t. To dissect the molecular mechanism underlying their recognition, we initiated the cloning of AvrPiz-t using a map-based cloning strategy. The AvrPiz-t gene was delimited to an approximately 21-kb genomic fragment, in which six genes were predicted. Complementation tests of each of these six candidate genes led to the final identification of AvrPiz-t, which encodes a 108-amino-acid predicted secreted protein with unknown function and no homologues in M. oryzae or in other sequenced fungi. We found that AvrPiz-t is present in the virulent isolate GUY11 but contains a Pot3 insertion at a position 462 bp upstream from the start codon. Complementation tests of AvrPiz-t genes driven by promoters of varying length revealed that a promoter larger than 462 bp is essential to maintain the AvrPiz-t function. These results suggest that a Pot3 insertion in GUY11 might interfere with the proper function of AvrPiz-t. Additionally, we found that AvrPiz-t can suppress the programmed cell death triggered by mouse BAX protein in Nicotiana benthamiana, identifying a mechanism by which AvrPiz-t may contribute virulence of M. oryzae.
DOI:10.1105/tpc.109.066324URLPMID:19454732 [本文引用: 7]
To subvert rice (Oryza sativa) host defenses, the devastating ascomycete fungus pathogen Magnaporthe oryzae produces a battery of effector molecules, including some with avirulence (AVR) activity, which are recognized by host resistance (R) proteins resulting in rapid and effective activation of innate immunity. To isolate novel avirulence genes from M. oryzae, we examined DNA polymorphisms of secreted protein genes predicted from the genome sequence of isolate 70-15 and looked for an association with AVR activity. This large-scale study found significantly more presence/absence polymorphisms than nucleotide polymorphisms among 1032 putative secreted protein genes. Nucleotide diversity of M. oryzae among 46 isolates of a worldwide collection was extremely low (theta=8.2x10(-5)), suggestive of recent pathogen dispersal. However, no association between DNA polymorphism and AVR was identified. Therefore, we used genome resequencing of Ina168, an M. oryzae isolate that contains nine AVR genes. Remarkably, a total of 1.68 Mb regions, comprising 316 candidate effector genes, were present in Ina168 but absent in the assembled sequence of isolate 70-15. Association analyses of these 316 genes revealed three novel AVR genes, AVR-Pia, AVR-Pii, and AVR-Pik/km/kp, corresponding to five previously known AVR genes, whose products are recognized inside rice cells possessing the cognate R genes. AVR-Pia and AVR-Pii have evolved by gene gain/loss processes, whereas AVR-Pik/km/kp has evolved by nucleotide substitutions and gene gain/loss.
DOI:10.1111/nph.13310URLPMID:25659573 [本文引用: 1]
We identified the Magnaporthe oryzae avirulence effector AvrPi9 cognate to rice blast resistance gene Pi9 by comparative genomics of requisite strains derived from a sequential planting method. AvrPi9 encodes a small secreted protein that appears to localize in the biotrophic interfacial complex and is translocated to the host cell during rice infection. AvrPi9 forms a tandem gene array with its paralogue proximal to centromeric region of chromosome 7. AvrPi9 is expressed highly at early stages during initiation of blast disease. Virulent isolate strains contain Mg-SINE within the AvrPi9 coding sequence. Loss of AvrPi9 did not lead to any discernible defects during growth or pathogenesis in M. oryzae. This study reiterates the role of diverse transposable elements as off-switch agents in acquisition of gain-of-virulence in the rice blast fungus. The prevalence of AvrPi9 correlates well with the avirulence pathotype in diverse blast isolates from the Philippines and China, thus supporting the broad-spectrum resistance conferred by Pi9 in different rice growing areas. Our results revealed that Pi9 and Piz-t at the Pi2/9 locus activate race specific resistance by recognizing sequence-unrelated AvrPi9 and AvrPiz-t genes, respectively.
DOI:10.1016/j.pep.2010.04.020URL [本文引用: 1]
Abstract
The rice blast disease caused by the ascomycete Magnaporthe grisea continues to cause a tremendous impact in rice (Oryza sativa) cultures around the world. Elucidating the molecular basis of the fungus interactions with its host might help increase the general understanding of the pathogen–host relationship. At the moment of invasion, the fungus secretes effectors that modify host defenses and cellular processes as they successively invade living rice cells. PWL2, an effector protein, is a known AVR (avirulence) gene product. The PWL2 gene prevents the fungus from infecting weeping lovegrass (Eragrostis curvula). In this study, we identified a PWL2 allele gene (which we termed PWL2D) in a strain of M. grisea. The sequence of PWL2D has only two bases different from that of PWL2, producing alterations in residue 90 and residue 142. However, the alteration of residue 90 (from D90 to N90) is critical to gene function. Here, we cloned the gene PWL2D in a pET System vector, expressed the gene product in Escherichia coli and evaluated by spectroscopic techniques some aspects of the PWL2D structure. While TRX-tagged PWL2D is prone to aggregation, the solubility of PWL2D is improved when it is overexpressed without its original signal peptide. Expression and purification procedures for these constructs are described. Finally, we found out that the protein seems to be an intrinsically disordered protein. Results from these studies will facilitate structural analysis of PWL2D and might contribute to understanding the gene’s function and of fungal/plant interactions.DOI:10.1038/srep11642URLPMID:26109439 [本文引用: 1]
Magnaporthe oryzae (Mo) is the causative pathogen of the damaging disease rice blast. The effector gene AvrPib, which confers avirulence to host carrying resistance gene Pib, was isolated via map-based cloning. The gene encodes a 75-residue protein, which includes a signal peptide. Phenotyping and genotyping of 60 isolates from each of five geographically distinct Mo populations revealed that the frequency of virulent isolates, as well as the sequence diversity within the AvrPib gene increased from a low level in the far northeastern region of China to a much higher one in the southern region, indicating a process of host-driven selection. Resequencing of the AvrPiballele harbored by a set of 108 diverse isolates revealed that there were four pathoways, transposable element (TE) insertion (frequency 81.7%), segmental deletion (11.1%), complete absence (6.7%), and point mutation (0.6%), leading to loss of the avirulence function. The lack of any TE insertion in a sample of non-rice infecting Moisolates suggested that it occurred after the host specialization of Mo. Both the deletions and the functional point mutation were confined to the signal peptide. The reconstruction of 16 alleles confirmed seven functional nucleotide polymorphisms for the AvrPiballeles, which generated three distinct expression profiles.
DOI:10.3724/SP.J.1006.2012.02192URL [本文引用: 1]
Theavirulence genes from 177 single spore strains of Magnaporthe grisea isolated from different rice varieties in Heilongjiang province were detected by specific primers. The results indicated that Avr-pita, ACE1, Avr-pia, Avr-pit, Avr1-co39, Avr-pik, and Avr-pizt could be detected in the strains from different rice-growing regions, and the amplification frequencies of avirurence genes were associated with the geographical orgin and rice variety, ACE1 and Avr1-co39 showed the highest and lowest frequency, that was 61.6%and 31.6%, respectively. Moreover, 20 monogenic lines with known blast resistance genes from International Rice Research Institute (IRRI) were used to analyze the genes which were valuable in breeding program in Heilongjiang Province by artificial inoculation. The resistance gene Pi-9 had the broadest resistance spectrum to theisolates from six regions.
DOI:10.3724/SP.J.1006.2012.02192URL [本文引用: 1]
Theavirulence genes from 177 single spore strains of Magnaporthe grisea isolated from different rice varieties in Heilongjiang province were detected by specific primers. The results indicated that Avr-pita, ACE1, Avr-pia, Avr-pit, Avr1-co39, Avr-pik, and Avr-pizt could be detected in the strains from different rice-growing regions, and the amplification frequencies of avirurence genes were associated with the geographical orgin and rice variety, ACE1 and Avr1-co39 showed the highest and lowest frequency, that was 61.6%and 31.6%, respectively. Moreover, 20 monogenic lines with known blast resistance genes from International Rice Research Institute (IRRI) were used to analyze the genes which were valuable in breeding program in Heilongjiang Province by artificial inoculation. The resistance gene Pi-9 had the broadest resistance spectrum to theisolates from six regions.
DOI:10.3864/j.issn.0578-1752.2014.03.006URL [本文引用: 3]
【Objective】The objective of this study is to identify avirulence genes (Avr-genes), which existed in the prevalent fungus of Magnaporthe oryzae, understand the distribution of Avr-genes in the epidemic strains of different regions, and to provide references for rice cultivars distribution.【Method】According to the sequences of 6 Avr-genes which had been cloned and were associated with the rice blast pathogenicity, the primers were designed to amplify the target sequences of the epidemic rice blast fungus in Liaoning Province. The DNA, extracted from the 26 strains of rice blast fungi, was used as a template for PCR amplification. By analyzing the PCR products by AGE (agarose gel electrophoresis) and sequencing, the sequences of base and amino acid for each Avr-gene were examined and compared. Some candidate primers were designed to check out the Avr-gene which had no PCR product.【Result】There was no PCR product when amplified Avr1-CO39, Avr-pia and Avr-pii by the primers, and there was also no PCR product by using the candidate primers, which showed that Avr1-CO39, Avr-pia and Avr-pii didn’t exist in most of the M. oryzae isolated from the main rice growing regions in Liaoning Province. On the contrary, the PCR products of AvrPiz-t, Avr-pik and Avr-pita were detected by AGE, and different genome types showed the 3 genes below were carried by the fungus in different mutated types and rates. AvrPiz-t related to the resistant genes including Pi2, Pi9 and Piz-t was detected in 22 M. oryzae strains, and the sequences of 21 strains were same to which exited in AvrPiz-t. The results of this experiment illustrated that the rice varieties carrying Pi2, Pi9 or Piz-t could have broad-spectrum disease resistance to rice blast fungi. The sequence of No. 16 strain was not the same as others, because it lost a single base C in the position of 192 bp, which caused the frame-shift mutation. The mutation led the AA (amino acid) sequence to terminate in position of 72nd AA, and made the secretory protein lose the function of avirulence. Avr-pik associated to the resistant genes like Pik, Pik-p, Pik-m and Pik-s were detected by AGE. The results showed that all the strains had the target bands. Their sequence showed that there were 4 genotypes of Avr-pik allele in the strains (B, D, F and G), and genotype D which carried by 12 strains could be recognized by rice blast disease resistance genes of Pik and its alleles as Pik-m and Pik-p. The 9 strains which carried genotype B of Avr-pik had been reported before, but its function of virulence had not been tested. The genotype F which exited in two strains was firstly found to be the new allele of Avr-pik, and they appeared in Dandong and Panjin, respectively. The specific point mutation, the 143 (A/G) of the base sequence, was the difference between genotype F and D. Genotype G which was also firstly detected was carried by the remaining 3 strains which only appeared in the area of Xinbin County in Fushun. The difference between genotype G and D was the point mutation of 168 (G/A) in the base sequence, which caused the protein translation early termination, therefore, genotype G lost its avirulent function. According to the AGE results, the same bands of Avr-pita were detected in all the 26 strains, and the sequences were tested and analyzed. The results showed that the genes in 26 strains had 5 genotypes of alleles, and there were several point mutations between the 5 alleles and the Avr-pita sequence, and the changes of the sequences caused the missense mutation. The difference of AA among the 5 genotypes were the 3 point mutation of 83 (D/N), 192 (Y/C) and 207 (K/R), which had been found in a structural domain of Avr-pita. The genotypes of allele all had been reported before. 【Conclusion】 The M. oryzae who carried Avr-pik, Avr-pita and AvrPiz-t distributed widely in rice growing regions of Liaoning Province. Breeding and cultivating the rice varieties carrying the corresponding resistance genes could alleviate the damage of the rice blast.
DOI:10.3864/j.issn.0578-1752.2014.03.006URL [本文引用: 3]
【Objective】The objective of this study is to identify avirulence genes (Avr-genes), which existed in the prevalent fungus of Magnaporthe oryzae, understand the distribution of Avr-genes in the epidemic strains of different regions, and to provide references for rice cultivars distribution.【Method】According to the sequences of 6 Avr-genes which had been cloned and were associated with the rice blast pathogenicity, the primers were designed to amplify the target sequences of the epidemic rice blast fungus in Liaoning Province. The DNA, extracted from the 26 strains of rice blast fungi, was used as a template for PCR amplification. By analyzing the PCR products by AGE (agarose gel electrophoresis) and sequencing, the sequences of base and amino acid for each Avr-gene were examined and compared. Some candidate primers were designed to check out the Avr-gene which had no PCR product.【Result】There was no PCR product when amplified Avr1-CO39, Avr-pia and Avr-pii by the primers, and there was also no PCR product by using the candidate primers, which showed that Avr1-CO39, Avr-pia and Avr-pii didn’t exist in most of the M. oryzae isolated from the main rice growing regions in Liaoning Province. On the contrary, the PCR products of AvrPiz-t, Avr-pik and Avr-pita were detected by AGE, and different genome types showed the 3 genes below were carried by the fungus in different mutated types and rates. AvrPiz-t related to the resistant genes including Pi2, Pi9 and Piz-t was detected in 22 M. oryzae strains, and the sequences of 21 strains were same to which exited in AvrPiz-t. The results of this experiment illustrated that the rice varieties carrying Pi2, Pi9 or Piz-t could have broad-spectrum disease resistance to rice blast fungi. The sequence of No. 16 strain was not the same as others, because it lost a single base C in the position of 192 bp, which caused the frame-shift mutation. The mutation led the AA (amino acid) sequence to terminate in position of 72nd AA, and made the secretory protein lose the function of avirulence. Avr-pik associated to the resistant genes like Pik, Pik-p, Pik-m and Pik-s were detected by AGE. The results showed that all the strains had the target bands. Their sequence showed that there were 4 genotypes of Avr-pik allele in the strains (B, D, F and G), and genotype D which carried by 12 strains could be recognized by rice blast disease resistance genes of Pik and its alleles as Pik-m and Pik-p. The 9 strains which carried genotype B of Avr-pik had been reported before, but its function of virulence had not been tested. The genotype F which exited in two strains was firstly found to be the new allele of Avr-pik, and they appeared in Dandong and Panjin, respectively. The specific point mutation, the 143 (A/G) of the base sequence, was the difference between genotype F and D. Genotype G which was also firstly detected was carried by the remaining 3 strains which only appeared in the area of Xinbin County in Fushun. The difference between genotype G and D was the point mutation of 168 (G/A) in the base sequence, which caused the protein translation early termination, therefore, genotype G lost its avirulent function. According to the AGE results, the same bands of Avr-pita were detected in all the 26 strains, and the sequences were tested and analyzed. The results showed that the genes in 26 strains had 5 genotypes of alleles, and there were several point mutations between the 5 alleles and the Avr-pita sequence, and the changes of the sequences caused the missense mutation. The difference of AA among the 5 genotypes were the 3 point mutation of 83 (D/N), 192 (Y/C) and 207 (K/R), which had been found in a structural domain of Avr-pita. The genotypes of allele all had been reported before. 【Conclusion】 The M. oryzae who carried Avr-pik, Avr-pita and AvrPiz-t distributed widely in rice growing regions of Liaoning Province. Breeding and cultivating the rice varieties carrying the corresponding resistance genes could alleviate the damage of the rice blast.
DOI:10.3864/j.issn.0578-1752.2018.24.005URL [本文引用: 1]
【Objective】 The objective of this study is to analyze the avirulent genotypes of Magnaporthe oryzae derived from the hybrid rice combinations of Guang 8 A, and to provide a reference for rational distribution of cultivars with different blast resistant genes in South China. 【Method】 A set of single-spore strain was subjected to pathotype analysis using blast monogenic differential lines. According to the functional markers of 8 Avr genes, which had been cloned and were associated with the rice blast pathogenicity, the haplotypes of Avr genes were analyzed. The DNA, which extracted from the 27 strains of rice blast fungi, was used as a template for PCR amplification. The PCR products of avirulent full gene or CDS region were analyzed by agarose gel electrophoresis and sequencing, and their sequences were compared to corresponding avirulent genes.【Result】 The tested strains showed high frequency (>85%) of avirulence to the 5 monogenic differential lines, such as IRBLkh-K3 (Pikh), IRBLb-B (Pib), IRBLz5-CA (Pi2), NIL-e1 (Pi50) and IRBL9-W (Pi9), all strains were avirulent to IRBL9-W (Pi9) and NIL-e1 (Pi50). These strains showed relatively low frequency of avirulence (<20%) to the 7 monogenic differential lines, such as IRBLks-F5 (Piks), IRBLa-A (Pia), IRBL19-A (Pi19) and so on, indicating that the strains from the combination of Guang 8 A showed different pathotypes to different rice blast-resistant lines. AvrPi9 and AvrPik fragments were almost present in all strains, and two haplotypes (AvrPik-D and AvrPik-E) of AvrPik were identified. But none of Avr1-CO39, AvrPii or AvrPia was amplified in any strain. The expected size products of AvrPiz-t, AvrPib and AvrPita could be detected in some strains, but they all appeared in lower frequency and different mutation types in the tested strain. Amplicon sequencing of 7 strains (GD13-621, GD14-349, GD15-291, et al.) revealed that the sequences of AvrPi9, AvrPita and AvrPiz-t were identical to those of the respective avirulent strains. This result indicated that these 3 Avr genes had a stable gene structure. Compared to AvrPib in an avirulent strain, AvrPib in the tested strains contained 2 nucleotide changes or 3 consecutive nucleotide insertions in the gene upstream region. The results indicated that AvrPib had a rich haplotype. The sequences of AvrPik in 6 strains were highly consistent, only contained one nucleotide changes in the coding region (136C/A), resulting in one amino acid substitutions (46H/N), which showed two haplotypes of AvrPik (AvrPik-D or AvrPik-E).【Conclusion】 In the strains from the major rice cultivars of Guang 8 A in South China, the distribution of AvrPi9 and AvrPik was relatively wide. In the susceptible areas of the above cultivars, the resistant cultivars carrying Pi9 and Pik could be used as rotation planting cultivars.
DOI:10.3864/j.issn.0578-1752.2018.24.005URL [本文引用: 1]
【Objective】 The objective of this study is to analyze the avirulent genotypes of Magnaporthe oryzae derived from the hybrid rice combinations of Guang 8 A, and to provide a reference for rational distribution of cultivars with different blast resistant genes in South China. 【Method】 A set of single-spore strain was subjected to pathotype analysis using blast monogenic differential lines. According to the functional markers of 8 Avr genes, which had been cloned and were associated with the rice blast pathogenicity, the haplotypes of Avr genes were analyzed. The DNA, which extracted from the 27 strains of rice blast fungi, was used as a template for PCR amplification. The PCR products of avirulent full gene or CDS region were analyzed by agarose gel electrophoresis and sequencing, and their sequences were compared to corresponding avirulent genes.【Result】 The tested strains showed high frequency (>85%) of avirulence to the 5 monogenic differential lines, such as IRBLkh-K3 (Pikh), IRBLb-B (Pib), IRBLz5-CA (Pi2), NIL-e1 (Pi50) and IRBL9-W (Pi9), all strains were avirulent to IRBL9-W (Pi9) and NIL-e1 (Pi50). These strains showed relatively low frequency of avirulence (<20%) to the 7 monogenic differential lines, such as IRBLks-F5 (Piks), IRBLa-A (Pia), IRBL19-A (Pi19) and so on, indicating that the strains from the combination of Guang 8 A showed different pathotypes to different rice blast-resistant lines. AvrPi9 and AvrPik fragments were almost present in all strains, and two haplotypes (AvrPik-D and AvrPik-E) of AvrPik were identified. But none of Avr1-CO39, AvrPii or AvrPia was amplified in any strain. The expected size products of AvrPiz-t, AvrPib and AvrPita could be detected in some strains, but they all appeared in lower frequency and different mutation types in the tested strain. Amplicon sequencing of 7 strains (GD13-621, GD14-349, GD15-291, et al.) revealed that the sequences of AvrPi9, AvrPita and AvrPiz-t were identical to those of the respective avirulent strains. This result indicated that these 3 Avr genes had a stable gene structure. Compared to AvrPib in an avirulent strain, AvrPib in the tested strains contained 2 nucleotide changes or 3 consecutive nucleotide insertions in the gene upstream region. The results indicated that AvrPib had a rich haplotype. The sequences of AvrPik in 6 strains were highly consistent, only contained one nucleotide changes in the coding region (136C/A), resulting in one amino acid substitutions (46H/N), which showed two haplotypes of AvrPik (AvrPik-D or AvrPik-E).【Conclusion】 In the strains from the major rice cultivars of Guang 8 A in South China, the distribution of AvrPi9 and AvrPik was relatively wide. In the susceptible areas of the above cultivars, the resistant cultivars carrying Pi9 and Pik could be used as rotation planting cultivars.
[本文引用: 2]
[本文引用: 2]
[D].
[本文引用: 2]
[D].
[本文引用: 2]
DOI:10.11847/zgggws2006-22-03-30URLPMID:19443872 [本文引用: 1]
The recent report of the Intergovernmental Panel on Climate Change confirmed that there is overwhelming evidence that the global climate will severely affect human health. Climate change might have severe consequences on public health in Bangladesh, especially in light of the poor state of the country's public health infrastructure. A number of possible direct and indirect impacts of climate change on public health in Bangladesh have been identified in this article. Adaptive measures that should be taken to reduce the negative consequences of climate change on public health have also been discussed.
DOI:10.11847/zgggws2006-22-03-30URLPMID:19443872 [本文引用: 1]
The recent report of the Intergovernmental Panel on Climate Change confirmed that there is overwhelming evidence that the global climate will severely affect human health. Climate change might have severe consequences on public health in Bangladesh, especially in light of the poor state of the country's public health infrastructure. A number of possible direct and indirect impacts of climate change on public health in Bangladesh have been identified in this article. Adaptive measures that should be taken to reduce the negative consequences of climate change on public health have also been discussed.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1094/MPMI-09-18-0245-RURLPMID:30601714 [本文引用: 3]
Higher yield potential and greater yield stability are common targets for crop breeding programs, including those in rice. Despite these efforts, biotic and abiotic stresses continue to impact rice production. Rice blast disease, caused by Magnaporthe oryzae, is the most devastating disease affecting rice worldwide. In the field, resistant varieties are unstable and can become susceptible to disease within a few years of release due to the adaptive potential of the blast fungus, specifically in the effector (avirulence [AVR]) gene pool. Here, we analyzed genetic variation of the effector gene AVR-Pik in 58 rice blast isolates from Thailand and examined the interaction between AVR-Pik and the cognate rice resistance gene Pik. Our results reveal that Thai rice blast isolates are very diverse. We observe four AVR-Pik variants in the population, including three previously identified variants, AVR-PikA, AVR-PikD, and AVR-PikE, and one novel variant, which we named AVR-PikF. Interestingly, 28 of the isolates contained two copies of AVR-Pik, always in the combination of AVR-PikD and AVR-PikF. Blast isolates expressing only AVR-PikF show high virulence to rice cultivars encoding allelic Pik resistance genes, and the AVR-PikF protein does not interact with the integrated heavy metal-associated domain of the Pik resistance protein in vitro, suggesting a mechanism for immune evasion.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/j.1365-313X.2012.05110.xURLPMID:22805093 [本文引用: 2]
Attack and counter-attack impose strong reciprocal selection on pathogens and hosts, leading to development of arms race evolutionary dynamics. Here we show that Magnaporthe oryzae avirulence gene AVR-Pik and the cognate rice resistance (R) gene Pik are highly variable, with multiple alleles in which DNA replacements cause amino acid changes. There is tight recognition specificity of the AVR-Pik alleles by the various Pik alleles. We found that AVR-Pik physically binds the N-terminal coiled-coil domain of Pik in a yeast two-hybrid assay as well as in an in planta co-immunoprecipitation assay. This binding specificity correlates with the recognition specificity between AVR and R genes. We propose that AVR-Pik and Pik are locked into arms race co-evolution driven by their direct physical interactions.
URL [本文引用: 1]
Magnaporthe grisea is one of the most devastating pathogens of rice. The specific interaction between rice and M. grisea follows the gene\|for\|gene hypothesis, and the cognate genes in M. grisea were called AVR genes. When the corresponding host gene, known as resistance (R) genes recognize the products of the cognate AVR genes, the immune responses of rice which are always associated with hypersensitive response are trigged. During the interaction between M. grisea and its host, AVR genes always exhibit the characterization of instability. The mutation of AVR genes usually result in the loss of resistance of rice. The mutations of AVR genes involve several mechanisms, including point mutations, deletions, insertion, duplication and frame\|shift mutation. The mutation mechanisms of AVR1\|CO39, PWL, AVR\|Pita are well studied, the mutation pattern of other AVR genes including ACE1, AvrPiz\|t, AVR\|Pia, AVR\|Pii and AVR\|Pik/km/kp are less known and need further research.
URL [本文引用: 1]
Magnaporthe grisea is one of the most devastating pathogens of rice. The specific interaction between rice and M. grisea follows the gene\|for\|gene hypothesis, and the cognate genes in M. grisea were called AVR genes. When the corresponding host gene, known as resistance (R) genes recognize the products of the cognate AVR genes, the immune responses of rice which are always associated with hypersensitive response are trigged. During the interaction between M. grisea and its host, AVR genes always exhibit the characterization of instability. The mutation of AVR genes usually result in the loss of resistance of rice. The mutations of AVR genes involve several mechanisms, including point mutations, deletions, insertion, duplication and frame\|shift mutation. The mutation mechanisms of AVR1\|CO39, PWL, AVR\|Pita are well studied, the mutation pattern of other AVR genes including ACE1, AvrPiz\|t, AVR\|Pia, AVR\|Pii and AVR\|Pik/km/kp are less known and need further research.
DOI:10.1038/srep11642URLPMID:26109439 [本文引用: 1]
Magnaporthe oryzae (Mo) is the causative pathogen of the damaging disease rice blast. The effector gene AvrPib, which confers avirulence to host carrying resistance gene Pib, was isolated via map-based cloning. The gene encodes a 75-residue protein, which includes a signal peptide. Phenotyping and genotyping of 60 isolates from each of five geographically distinct Mo populations revealed that the frequency of virulent isolates, as well as the sequence diversity within the AvrPib gene increased from a low level in the far northeastern region of China to a much higher one in the southern region, indicating a process of host-driven selection. Resequencing of the AvrPiballele harbored by a set of 108 diverse isolates revealed that there were four pathoways, transposable element (TE) insertion (frequency 81.7%), segmental deletion (11.1%), complete absence (6.7%), and point mutation (0.6%), leading to loss of the avirulence function. The lack of any TE insertion in a sample of non-rice infecting Moisolates suggested that it occurred after the host specialization of Mo. Both the deletions and the functional point mutation were confined to the signal peptide. The reconstruction of 16 alleles confirmed seven functional nucleotide polymorphisms for the AvrPiballeles, which generated three distinct expression profiles.
DOI:10.4238/2014.November.24.1URL [本文引用: 1]
The in vitro sensitivity of AvrPik allele isolates of Magnaporthe oryzae to isoprothiolane was examined and the virulence fitness costs of AvrPik allele isolates to isoprothiolane were assessed. Isoprothiolane was found to suppress the radial growth of AvrPik allele isolates at all concentrations (1, 5, 10, 15, and 20 mu g/mL). Generally, a higher isoprothiolane concentration has a stronger inhibitory effect on mycelial growth in AvrPik allele isolates at 6 and 10 days after inoculation. The inhibitory effect of isoprothiolane also increased with treatment time. To determine whether a correlation existed between the in vitro sensitivity of AvrPik allele isolates and virulence, the half-maximal inhibitor concentration and 75% of the maximum inhibitor concentration were calculated for each mutation isolate and wild-type isolate. Based on these values and virulence, no significant correlation between the susceptibility of AvrPik allele isolates and virulence was detected. In summary, no fitness costs were associated with sensitivity of blast isolates carrying specific AvrPik alleles to different virulence.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}