 ,, 杨承槐,中国兽医药品监察所,北京 100081
,, 杨承槐,中国兽医药品监察所,北京 100081The Effects of Downstream 3513bp of UL56 on Characterization of Duck Enteritis Virus
MAO YaQing, ZHANG Bing, WANG TuanJie, HOU LiDan, HUANG XiaoJie, LIU Dan, ZHAO JunJie, LI QiHong, WANG LeYuan, LI JunPing,, YANG ChengHuai,China Institute of Veterinary Drug Control, Beijing 100081通讯作者:
责任编辑: 林鉴非
收稿日期:2019-04-22接受日期:2019-07-22网络出版日期:2019-12-01
| 基金资助: |
Received:2019-04-22Accepted:2019-07-22Online:2019-12-01
作者简介 About authors
毛娅卿,Tel:010-62103641;E-mail:wcpmyq @163.com。

摘要
关键词:
Abstract
Keywords:
PDF (5464KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
毛娅卿, 张兵, 王团结, 侯力丹, 黄小洁, 刘丹, 赵俊杰, 李启红, 王乐元, 李俊平, 杨承槐. UL56基因下游3513bp对鸭肠炎病毒生物特性的影响[J]. 中国农业科学, 2019, 52(23): 4390-4397 doi:10.3864/j.issn.0578-1752.2019.23.019
MAO YaQing, ZHANG Bing, WANG TuanJie, HOU LiDan, HUANG XiaoJie, LIU Dan, ZHAO JunJie, LI QiHong, WANG LeYuan, LI JunPing, YANG ChengHuai.
0 引言
【研究意义】鸭肠炎病毒(duck enteritis virus, DEV),又称鸭瘟病毒,属于疱疹病毒科,能引起鸭、鹅等雁形目禽类发生急性、热性、败血性的传染病。该病也称为鸭瘟,发病率和死亡率很高,对水禽养殖业危害很大[1,2]。本研究根据DEV强弱株基因组差异,拟分析DEV毒力相关基因,解析DEV致病机制,探索外源基因插入位点、活载体疫苗开发的可行性。【前人研究进展】目前已完成多株DEV全基因组测序[3,4,5,6,7]。DEV疫苗检验用参考强毒株(CVCC AV1221)于1962年从南京一鸭场分离,是我国较早分离的一个强毒,毒力稳定,是我国官方评价鸭瘟疫苗免疫效果的参考强毒,其基因组全长为162 131 bp[8]。与参考强毒株基因组相比,DEV疫苗株UL区5′端连续缺失了3 513 bp(2 716—6 228位核苷酸)导致UL56基因移框突变;德国分离DEV 2085株基因组在2 561位核苷酸后连续缺失1 170 bp[3-4,8-9],该区域是否与DEV毒力相关,尚未见报道。【本研究切入点】本研究以红色荧光蛋白(RFP)为报告基因,构建缺失UL56基因下游3 513 bp的重组病毒,比较缺失前后病毒在细胞中的生长特性以及对易感鸭的致病性、免疫原性。【拟解决的关键问题】本研究旨在分析UL56基因下游3 513 bp对DEV生物特性的影响,为揭示DEV UL56基因下游3 513 bp功能、DEV活载体疫苗研究奠定基础。1 材料与方法
1.1 试验时间、地点
试验于2015年6月至2017年7月在中国兽医药品监察所完成。1.2 试验材料
1.2.1 毒株和细胞 DEV参考强毒株(CVCCAV1221)由中国兽医微生物菌种保藏管理中心保存;SPF鸭胚购自中国农业科学院哈尔滨兽医研究所。1.2.2 试验动物 6周龄易感麻鸭(中和抗体<1﹕4)购自北京昌平某鸭场。
1.2.3 质粒和菌株 含有红色荧光蛋白RFP基因的载体pT-RFP由中国兽医药品监察所病毒制品检测室构建保存;pMD18T-Simple载体购自大连TaKaRa公司,DH5α受体菌购自天根生化科技(北京)有限公司。
1.2.4 主要试剂 Ex Taq DNA 聚合酶、限制性内切酶、T4 DNA 连接酶、T4 DNA 聚合酶购自大连TaKaRa公司;胶回收试剂盒等均购自天根生化科技(北京)有限公司;胎牛血清、M199培养液购自Hyclone公司;OPTI-MEM培养液购自Gibco公司;lipofectamine2000购自Invitrogen公司;无内毒素高纯质粒提取试剂盒Endo-free Plasmid Mini Kit II购自Omega公司。
1.2.5 引物试验所用PCR引物见表1,由上海Invitrogen生物公司合成。
Table 1
表1
表1目的基因的扩增引物及鉴定引物
Table 1
| 引物名称 Primer name | 序列(5′-3′) Sequence (5′-3′) |
|---|---|
| UL56-uF | GTCTGCTCTTCCGCCATTC |
| UL56-uR | GCACGCGTAAGATCAGACCGCCGCCTA |
| UL56-dF | GCACGCGTTTCATTGTTTACCGTGTC |
| UL56-dR | CATCTTGCTTATCGCTTT |
| Overlap-R | GACACGGTAAACAATGAAAAGATCAGACCGCCGCCTA |
| Overlap-F | TAGGCGGCGGTCTGATCTTTTCATTGTTTACCGTGTC |
新窗口打开|下载CSV
1.3 试验方法
1.3.1 基因组DNA的提取 参照文献[10,11,12] 进行。1.3.2 重组质粒的构建 重组质粒pT-UL56-RFP构建策略见图1。以提取的DEV基因组DNA为模板,分别扩增UL56基因上下游两端同源臂UL56-u、UL56-d。将两同源臂片段克隆到pMD18T-Simple载体,获得重组质粒pT-UL56ud。将重组质粒pT-UL56ud用MluI酶切,电泳回收去磷酸化后,与用MluI酶切的pT-RFP连接,将RFP表达盒插入到pT-UL56ud中,获得重组质粒pT-UL56-RFP。按Endo-free plasmid mini kit II说明书提取高纯度质粒备用。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1重组病毒的构建示意图
a:DEV基因组示意图,包括UL, IRS, US和TRS区域;b:PCR扩增UL56基因的上下游片段UL56-u,UL56-d;c:DEVΔ3513-RFP的构建,将RFP基因表达盒插入到DEV UL56基因组;d: 应用融合PCR从DEV亲本毒中扩增UL56-ud。PCR产物与DEVΔ3513-RFP进行同源重组,获得重组病毒DEVΔ3513;e: 应用PCR从DEV亲本毒中扩增UL56u-3 513 bp-UL56d。PCR产物与DEVΔ3513-RFP进行同源重组,获得重组病毒DEVΔ3513(R)
Fig. 1Construction of recombinant DEV
a : Map of the DEV genome, which consists of long (UL) and short (US) unique regions with inverted repeat sequences (IRS,TRS) flanking the US region; b : Regions upstream (UL56-u) and downstream (UL56-d) of UL56 were amplified by PCR; c: Construction of DEVΔ3513-RFP. An expression cassette encoding RFP was inserted in DEV genome; d: UL56-ud were amplified from wt DEV and assembled together by SOE PCR. The product was used to generate DEVΔ3513 by recombination with DEVΔ3513-RFP; e: The region of the DEV genome encompassing UL56u-3 513 bp-UL56d was amplified by PCR. The product was used to generate DEVΔ3513(R) by recombination with DEVΔ3513-RFP
1.3.3 重组病毒DEVΔ3513-RFP的构建 DEV接种DEF(MOI=0.1),吸附1—2 h后,按Lipofectamine 2000说明书转染高纯质粒pT-UL56-RFP。按文献[10, 13]进行蚀斑纯化。
1.3.4 重组病毒DEVΔ3513的构建 以DEV基因组DNA为模板,用引物UL56-uF、Overlap-R以及Overlap-F、UL56-dR进行融合PCR。纯化的PCR产物与重组病毒DEVΔ3513-RFP共转染。
1.3.5 重组病毒DEVΔ3513(R)的构建 以DEV基因组DNA为模板,用引物UL56-uF、UL56-dR进行PCR扩增。纯化的PCR产物与重组病毒DEVΔ3513-RFP共转染。
1.3.6 一步生长曲线 将重组病毒及其亲本病毒分别接种25cm2的细胞瓶中(moi=0.01),接种后每隔12h取出1瓶接毒细胞,测量其病毒含量,绘制一步生长曲线。
1.3.7 蚀斑大小分析 将重组病毒及其亲本病毒分别作适当稀释,接种已长成单层DEF,加入M199营养琼脂糖覆盖,培养5—7 d。随机选取20个蚀斑,应用CellSens v1.6软件(Olympus Company,Japan)对蚀斑面积进行测量[13]。
1.3.8 致病性试验 将20只6周龄易感麻鸭,随机分成4组,每组5只。第1组肌肉注射DEVΔ3513,每只105.0 TCID50;第2组肌肉注射DEVΔ3513(R),每只105.0TCID50;第3组肌肉注射DEV亲本毒,每只105.0TCID50;第4组注射生理盐水,作对照。每组单独隔离饲养,观察10 d,每天记录发病死亡情况。试验结束后剖检所有存活鸭。对全部动物取肝脏组织,固定于4%多聚甲醛溶液,按常规程序制备病理切片并进行H.E.染色。
1.3.9 免疫原性测定 将15只6周龄易感麻鸭,随机分成3组,每组5只。第1组肌肉注射DEVΔ3513,每只103.0 TCID50;第2组肌肉注射鸭瘟活疫苗种毒(CVCC AV1222),每只103.0 TCID50;第3组注射生理盐水,作对照。每组单独隔离饲养。在免疫后14天,腿部肌肉注射接种DEV强毒(CVCC AV1221),每只103.0MLD。观察10 d,每天记录发病死亡情况。
2 结果
2.1 重组病毒的构建及鉴定
DEV感染DEF细胞2 h后,用Lipofectamine 2000转染高纯质粒pT-UL56-RFP。转染后约10 h,即可见转染细胞中有带有红色荧光的梭形细胞。72 h左右80%细胞出现病变,收获病毒,冻融3次,用DEF细胞分离、纯化红色蚀斑,经过5代筛选,获得纯化的重组病毒DEVΔ3513-RFP。按照同样的方法,去除RFP获得了缺失UL56基因下游3 513 bp的重组病毒DEVΔ3513;用UL56基因下游3 513 bp替换病毒DEVΔ3513-RFP中的RFP,获得了回复突变株DEVΔ3513(R)。经测序鉴定,所构建的3个重组病毒均正确。
2.2 一步生长曲线
一步生长曲线结果表明,重组病毒DEVΔ3513、DEVΔ3513(R)及其亲本毒均在接种后72 h,病毒含量达到峰值(106.2-6.5TCID50/0.1mL),随后病毒含量下降(图5)。表明UL56下游3 513 bp的缺失对DEV的复制没有明显影响,缺失后病毒滴度无显著变化。图2
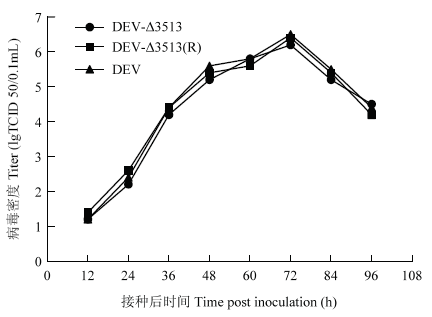 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2重组病毒的一步生长曲线
Fig. 2One step growth curve of recombinant DEV in DEF
2.3 蚀斑大小分析
重组病毒及其亲本毒均能在DEF细胞中形成清晰蚀斑;亲本毒、DEV-Δ3513、DEV-Δ3513(R)蚀斑面积分别为(1.91±0.28)mm2、(1.82±0.31)mm2、(1.98± 0.25)mm2,无显著差异(P>0.05)(图5-9),说明UL56基因下游3 513 bp缺失不影响DEV在细胞间的传播能力。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3重组毒及其亲本毒蚀斑面积
Fig. 3The plaque size of recombinant and its parental virus
2.4 致病性试验
DEV亲本毒、重组病毒DEV-Δ3513(R)在接种后3日麻鸭开始出现精神沉郁状;7日全部死亡,剖检肝脏表面有大量散在的白色坏死点。病理组织学观察可见:静脉和肝血窦淤血,充满大量红细胞;肝细胞内有大量脂滴呈空泡状,发生脂肪变性;部分区域出现局灶性肝细胞坏死,在坏死区域伴有炎性细胞,胞核溶解,与其他研究结果一致[14,15,16]。而DEV-Δ3513接种鸭和对照鸭在14 d观察期内,未出现任何临床症状和死亡。病理组织学诊断可见肝脏组织结构清晰,肝细胞形态正常,未见明显病理变化。说明UL56基因下游3 513 bp缺失后毒力明显减弱。图4
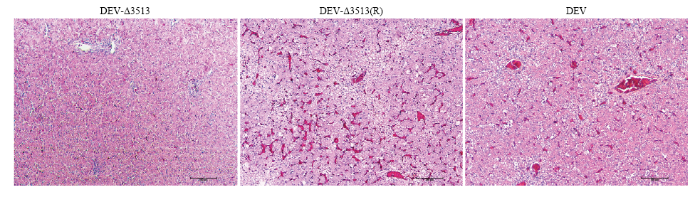 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4肝脏病理切片(H.E.染色)
Fig. 4Histopathologic lesion of liver
2.5 免疫原性测定
重组病毒DEVΔ3513以103.0 TCID50/只、104.0 TCID50/只剂量,免疫麻鸭后14 d,均能100%抵抗DEV强毒攻击(表2),表明UL56基因下游3 513 bp缺失后仍具有良好免疫原性。Table 2
表2
表2免疫后的攻毒保护效力
Table 2
| 组别 Groups | 毒株 Strains | 剂量 Doses | DEV攻毒后每天死亡鸭数 Numbers of dead ducks per day after DEV challenge | 累计死亡 Total death | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||||
| 1 | DEVΔ3513 | 105.0 TCID50 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0/5 |
| 2 | DEVΔ3513(R) | 105.0TCID50 | 0 | 0 | 0 | 1 | 3 | 0 | 1 | / | / | / | 5/5 |
| 3 | 亲本毒 Parent virus | 105.0TCID50 | 0 | 0 | 0 | 2 | 2 | 1 | / | / | / | / | 5/5 |
| 4 | 对照组 Control | / | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0/5 |
新窗口打开|下载CSV
3 讨论
本研究以红色荧光蛋白RFP为报告基因,以DEV参考强毒株为亲本毒,通过同源重组,构建了缺失UL56基因下游3 513 bp的重组病毒DEVΔ3513及其回复突变株DEVΔ3513(R)。并对其细胞生长特性、致病性和免疫原性进行研究。体外试验表明,重组病毒在细胞中的一步生长曲线、蚀斑大小与亲本毒相似,说明UL56基因下游3 513 bp缺失不影响DEV在细胞上的生长,是病毒复制非必需基因。动物试验结果表明,UL56基因下游3 513 bp缺失能显著降低DEV的毒力,是DEV的一个毒力决定因子;缺失后仍保留良好的免疫原性,能抵抗致死性强毒攻击,与现有商品化鸭瘟活疫苗免疫原性相当。Table 3
表3
表3免疫后的攻毒保护效力
Table 3
| 组别 Groups | 毒株 Strains | 剂量 Doses | DEV攻毒后每天死亡鸭数 Numbers of dead ducks pre day after DEV challenge | 累计 Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 死亡 Dead | 保护率 Protection ratio | |||
| 1 | DEVΔ3513 | 103.0TCID50 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0/5 | 100% |
| 2 | 鸭瘟活疫苗 DEV vaccines | 103.0TCID50 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0/5 | 100% |
| 3 | 对照组 Control | / | 0 | 0 | 0 | 2 | 2 | 1 | / | / | / | / | 5/5 | 0% |
新窗口打开|下载CSV
我国鸭瘟活疫苗毒株,其亲本毒于1957年从我国广东分离,然后经鸡胚连续传代60多代致弱[17]。全基因组序列比较发现,与DEV参考强毒株相比,疫苗株基因组UL区5′端连续缺失了3 513 bp(2 716— 6 228位核苷酸)导致UL56蛋白C端第215位氨基酸后移框变和UL55缺失[1]。在α疱疹病毒中,除了牛疱疹病毒1型和5型外,均含有UL56同源物[18,19,20,21,22,23,24,25,26]。UL56蛋白存在病毒粒子中[27,28]。疱疹病毒UL56蛋白C端有一个疏水区,含跨膜区和多个PPxY 基序等,为典型的跨膜锚定功能域[29,30]。该功能域对维持单纯疱疹病毒1型(herpes simplex virus 1,HSV-1)毒力表型具有重要作用。致病性HSV-1的UL56被LacZ替代后,腹膜内注射不能引起树熊发病[29, 31-33];弱毒HSV-1 HFEM株UL56基因被强毒HSV-1 17株的UL56替换后,能恢复其毒力表型,进一步研究表明,缺失UL56蛋白C末端疏水区(217—234位氨基酸)导致HSV-1毒力丧失[31]。
本研究从DEV参考强毒与疫苗株基因组差异入手,分析了UL56基因下游3 513 bp对DEV参考强毒生物特性的影响,揭示了该区域对维持DEV毒力发挥了重要作用。下一步将缩小缺失范围,重点研究UL56蛋白C末端疏水区对DEV毒力的影响。
4 结论
4.1 重组病毒生长特性与亲本毒相似,首次证明UL56基因下游3 513 bp对DEV在细胞中的复制是非必需的。4.2 缺失UL56基因下游3 513 bp能显著降低DEV的毒力,首次证明是UL56基因下游3 513 bp是DEV的一个毒力决定因子。
4.3 缺失UL56基因下游3 513 bp后仍保留良好的免疫原性。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 1]
DOI:10.1128/JVI.00529-12URL [本文引用: 2]

The Chinese virulent (CHv) strain of duck enteritis virus (DEV) has a genome of approximately 162,175 nucleotides with a GC content of 44.89%. Here we report the complete genomic sequence and annotation of DEV CHv, which offer an effective platform for providing authentic research experiences to novice scientists. In addition, knowledge of this virus will extend our general knowledge of DEV and will be useful for further studies of the mechanisms of virus replication and pathogenesis.
DOI:10.1128/JVI.01517-12URLPMID:23166249 [本文引用: 2]
The icosahedral virion of duck enteritis virus (DEV) is roughly spherical and approximately 150 nm in diameter. Here, we describe the genomic features of DEV CHv together with a draft genome sequence and its annotation, highlighting the homogeneity and heterogeneity of this genome in comparison with its reference genomes.
DOI:10.1007/s00705-017-3491-1URLPMID:28730521 [本文引用: 1]
Here, we present the complete genomic sequence of an attenuated duck enteritis virus (DEV). The Chinese standard challenge strain of DEV (DEV CSC) was serially passaged 20 times in chick embryo fibroblasts and then 85 times in chick embryos. The virus was attenuated and was avirulent to 2-month-old ducks. The attenuated DEV genome is 162,131 base pairs (bp) in length and as long as the parental genomic sequence. There are only 22 nucleotide substitutions, resulting in single amino acid changes in open reading frames LORF5, LORF4, UL41, UL39, UL32, UL13, UL10, UL3, US3, US4 and US7. The genome sequence has been deposited in the GenBank database under accession number KU216226. This study provides genetic information about DEV attenuation and further advances our understanding of the molecular basis of DEV pathogenesis.
DOI:10.1007/s00705-014-2275-0URLPMID:25392272 [本文引用: 1]
To gain a better understanding of the genetic changes required for attenuation of duck enteritis virus (DEV), the Chinese standard challenge strain of DEV (DEV CSC) was serially passaged 80 times in chick embryo fibroblasts. We plaque-purified the virus after the 25th passage (DEV p25) and the 80th passage (DEV p80) and investigated its in vitro and in vivo properties. Average plaque sizes for DEV p25 and p80 were significantly smaller than those for their parental DEV CSC. The results from an in vivo experiment revealed that DEV p25 and p80 were avirulent in ducks and protected them from virulent DEV challenge. The complete genome sequence of DEV p80 was determined and compared with that of the parent virus. An 1801-bp deletion was identified in the genome of DEV p80, which affected the genes encoding gI and gE. Moreover, there were 11 base substitutions, which led to seven amino acid changes in open reading frames LORF9, UL51, UL9, UL7, UL4, ICP4 and US3. Further DNA sequence analysis showed that the 1801-bp deletion was also present in DEV p25. Our findings suggest that DEV gE and/or gI are nonessential for virus growth and might, as with other herpesviruses, play an important role in cell-to-cell spread and virulence. Our experiments provide more genetic information about DEV attenuation and further advance our understanding of the molecular basis of DEV pathogenesis.
DOI:10.1016/j.virol.2009.06.018URLPMID:19595405 [本文引用: 1]
The genomic sequence of a strain of duck enteritis virus (DEV) was determined and analyzed in this study. The size of its genome is 158,091 bp in length and the genome is predicted to encode 78 putative proteins and resembles the members of the Alphaherpesvirinae in genomic organization and gene composition. The genome of the virus is composed of a unique long (UL) region, a unique short (US) region, a unique short internal repeat (IRS) region and a unique short terminal repeat (TRS) region. Its genomic arrangement pattern (UL-IRS-US-TRS) corresponds to D-type herpesvirus and is consistent with the members of Varicellovirus and Iltovirus genera. Sequence analysis reveals that the genome of the virus contains 67 genes having homologs in most members of the Alphaherpesvirinae. Out of these genes, one gene has a homolog in cercopithecine herpesvirus 8 which is a virus of Betaherpesvirinae, and 5 genes have homologs in avian herpesviruses. Furthermore, the genome possesses three unique genes without homologs in any other herpesviruses. Like most members of the Alphaherpesvirinae, the genes in the UL region of its genome are well conserved, whereas the gene arrangement of IRS-US is similar to that of Marek's disease virus and equine herpesviruses 1. Therefore, our data based on the genomic analysis suggest that DEV represents an osculant taxonomic entity within the Alphaherpesvirinae.
DOI:10.1007/s11262-013-1009-9URLPMID:24287923 [本文引用: 2]
Here, we present the complete genomic sequence of the Chinese standard challenge strain (CSC) of duck enteritis virus (DEV), which was isolated in China in 1962. The DEV CSC genome is 162,131 bp long and contains 78 predicted open reading frames (ORFs). Comparison of the genomic sequences of DEV CSC and DEV live vaccine strain K at passage 63 (DEV K p63) revealed that the DEV CSC genome is 4,040 bp longer than the DEV K p63 genome, mainly because of 3,513-bp and 528-bp insertions at the 5' and 3' ends of the unique long segment, respectively. At the nucleotide level, 63 of the 76 ORFs in the DEV CSC genome were 100 % identical to the ORFs in the DEV K p63 genome. Two ORFs (UL56 and US10) had frameshift mutations in the C-terminal regions, while LORF5 was unique to the DEV K p63 genome. It is difficult to assign attenuated virulence to changes in specific genes. However, the complete DEV CSC genome will further advance our understanding of the genes involved in virulence and evolution. The DEV CSC genome sequence has been deposited in GenBank under accession number JQ673560.
DOI:10.1016/j.virusres.2011.07.004URLPMID:21802458 [本文引用: 1]
We here report the complete genome sequence of the duck enteritis virus (DEV) wild-type strain 2085, an avian herpesvirus (GenBank ID: JF999965). The nucleotide sequence was derived from the 2085 genome cloned as an infectious bacterial artificial chromosome (BAC) clone. The DEV 2085 genome is 160,649-bp in length and encodes 78 predicted open reading frames (ORFs), a number identical to that identified for the attenuated DEV VAC strain (GenBank ID: EU082088.2). Comparison of the genome sequences DEV 2085 and VAC with partial sequences of the virulent CHv strain and the attenuated strain Clone-03 was carried out to identify nucleotide or amino acid polymorphisms that potentially contribute to DEV virulence. No amino acid changes were identified in 24 of the 78 ORFs, a result indicating high conservation in DEV independently of strain origin or virulence. In addition, 39 ORFs contain non-synonymous nucleotide substitutions, while 15 ORFs had nucleotide insertions or deletions, frame-shift mutations and/or non-synonymous nucleotide substitutions with an effect on ORF initiation or termination. In 7 of the 15 ORFs with high and 27 of the 39 ORFs with low variability, polymorphisms were exclusively found in DEV 2085, a finding that likely is a result of a different origin of 2085 (Europe) or VAC, Clone-03 and CHv (Eastern Asia). Five ORFs (UL2, UL12, US10, UL47 and UL41) with polymorphisms were identical between the virulent DEV 2085 and CHv but different from VAC or Clone-03. They, individually or in combination, may therefore represent DEV virulence factors. Our comparative analysis of four DEV sequences provides a comprehensive overview of DEV genome structure and identifies ORFs that are changed during serial virus passage.
DOI:10.3864/j.issn.0578-1752.2016.14.014URL [本文引用: 1]
【目的】鸭肠炎病毒(duck enteritis virus, DEV)不同毒株间存在明显差异,DEV疫苗株的UL2基因在195bp后连续缺失528bp,导致第65位氨基酸后连续缺失176aa[1]。将绿色荧光蛋白(GFP)基因插入DEV UL2基因中,获得表达绿色荧光蛋白的重组病毒,以研究UL2基因对DEV生物特性的影响和探讨DEV作为载体表达外源基因的可行性。【方法】以实验室保存的DEV细胞适应株DNA为模板,利用PCR技术扩增出病毒UL2基因上下游序列并克隆入pMD-18T载体;以UL2基因作为外源基因插入靶点及同源重组臂,将CMV启动子控制的含有GFP-gpt基因表达盒克隆入DEV UL2基因中,构建含GFP基因的转移质粒载体pT-UL2-GFP-gpt;用脂质体将其与DEV细胞适应株共转染CEF细胞,待80%细胞出现病变后,冻融3次,接种到新鲜CEF细胞单层的6孔培养板中,用含5%血清、1%双抗、1%琼脂的M199培养液覆盖,在荧光显微镜下挑取单个有绿色荧光的蚀斑,再接到新的细胞上,重复蚀斑筛选、纯化表达绿色荧光蛋白的重组病毒;利用PCR、基因测序技术鉴定重组病毒;重组病毒接种CEF(moi=0.01),每12h取出1瓶接毒细胞,分别收集上清和细胞,测量其病毒含量,绘制一步生长曲线;重组病毒在CEF中连续传代20次,在荧光显微镜下观察绿色荧光蛋白表达情况,并用PCR检测GFP的传代稳定性;重组病毒免疫4周龄SPF鸭后14d,肌肉注射接种DEV强毒(CVCC AV1221),观察免疫保护情况。【结果】经双酶切鉴定,成功构建了含绿色荧光蛋白报告基因的转移质粒载体pT-UL2-GFP-gpt,将其与DEV共转染CEF细胞后8h,即可见转染细胞中有带有绿色荧光的梭形细胞,经过8轮蚀斑筛选,获得纯化的重组病毒rDEV-△UL2-GFP-gpt;PCR鉴定及基因测序结果显示,GFP标记基因成功地插入到DEV基因组中,替换了DEV UL2基因的196—723位核苷酸;一步生长曲线结果显示,重组病毒在细胞和上清中的病毒含量分别在36h和72h达到峰值,为106.2TCID50/0.1mL、105.5TCID50/0.1mL,与亲本毒无明显差异;重组病毒在CEF中连续传代,1—5代可以稳定表达GFP基因,第6代起,开始出现少量没有荧光的细胞病变,15—20代中绝大部分细胞病变无绿色荧光,GFP在细胞连续传代过程中容易出现突变;重组病毒以103.0TCID50/只免疫麻鸭,免疫后14d能完全抵抗DEV强毒株的攻击,与亲本毒免疫原性一致。【结论】成功构建了表达绿色荧光蛋白的DEV,首次证实UL2基因缺失不影响其在细胞中的复制,也不影响其免疫原性,为DEV UL2基因功能、活载体疫苗研究奠定了基础。
DOI:10.3864/j.issn.0578-1752.2016.14.014URL [本文引用: 1]
【目的】鸭肠炎病毒(duck enteritis virus, DEV)不同毒株间存在明显差异,DEV疫苗株的UL2基因在195bp后连续缺失528bp,导致第65位氨基酸后连续缺失176aa[1]。将绿色荧光蛋白(GFP)基因插入DEV UL2基因中,获得表达绿色荧光蛋白的重组病毒,以研究UL2基因对DEV生物特性的影响和探讨DEV作为载体表达外源基因的可行性。【方法】以实验室保存的DEV细胞适应株DNA为模板,利用PCR技术扩增出病毒UL2基因上下游序列并克隆入pMD-18T载体;以UL2基因作为外源基因插入靶点及同源重组臂,将CMV启动子控制的含有GFP-gpt基因表达盒克隆入DEV UL2基因中,构建含GFP基因的转移质粒载体pT-UL2-GFP-gpt;用脂质体将其与DEV细胞适应株共转染CEF细胞,待80%细胞出现病变后,冻融3次,接种到新鲜CEF细胞单层的6孔培养板中,用含5%血清、1%双抗、1%琼脂的M199培养液覆盖,在荧光显微镜下挑取单个有绿色荧光的蚀斑,再接到新的细胞上,重复蚀斑筛选、纯化表达绿色荧光蛋白的重组病毒;利用PCR、基因测序技术鉴定重组病毒;重组病毒接种CEF(moi=0.01),每12h取出1瓶接毒细胞,分别收集上清和细胞,测量其病毒含量,绘制一步生长曲线;重组病毒在CEF中连续传代20次,在荧光显微镜下观察绿色荧光蛋白表达情况,并用PCR检测GFP的传代稳定性;重组病毒免疫4周龄SPF鸭后14d,肌肉注射接种DEV强毒(CVCC AV1221),观察免疫保护情况。【结果】经双酶切鉴定,成功构建了含绿色荧光蛋白报告基因的转移质粒载体pT-UL2-GFP-gpt,将其与DEV共转染CEF细胞后8h,即可见转染细胞中有带有绿色荧光的梭形细胞,经过8轮蚀斑筛选,获得纯化的重组病毒rDEV-△UL2-GFP-gpt;PCR鉴定及基因测序结果显示,GFP标记基因成功地插入到DEV基因组中,替换了DEV UL2基因的196—723位核苷酸;一步生长曲线结果显示,重组病毒在细胞和上清中的病毒含量分别在36h和72h达到峰值,为106.2TCID50/0.1mL、105.5TCID50/0.1mL,与亲本毒无明显差异;重组病毒在CEF中连续传代,1—5代可以稳定表达GFP基因,第6代起,开始出现少量没有荧光的细胞病变,15—20代中绝大部分细胞病变无绿色荧光,GFP在细胞连续传代过程中容易出现突变;重组病毒以103.0TCID50/只免疫麻鸭,免疫后14d能完全抵抗DEV强毒株的攻击,与亲本毒免疫原性一致。【结论】成功构建了表达绿色荧光蛋白的DEV,首次证实UL2基因缺失不影响其在细胞中的复制,也不影响其免疫原性,为DEV UL2基因功能、活载体疫苗研究奠定了基础。
DOI:10.1016/s0166-0934(99)00058-0URLPMID:10488769 [本文引用: 1]
In the field of human cytomegalovirus pathogenesis there is growing interest in analyzing recent clinical isolates rather than cell culture adapted laboratory strains. However, true low passage isolates are strictly cell associated prior to cell culture adaptation and only a minor fraction of cells are infected at low passage number. Both conditions hinder the preparation of pure viral DNA. To date, genetic analyses had been carried out mostly with supernatant associated cytomegalovirus. A rapid and simple method is described for preparation of viral DNA from low passage cell associated isolates with little cytopathogenic effect. The protocol is based on a combination of Triton X-100 lysis, nuclease treatment, and subsequent phenol chloroform extraction. Cellular background was reduced significantly to enable clear detection of all viral DNA fragments in restriction fragment length analysis. The method yielded DNA which was suitable for downstream applications like cloning of viral DNA fragments or transfection of genomic viral DNA. This method may facilitate genomic analyses of pathogenic cell associated recent cytomegalovirus isolates.
DOI:10.1007/s00705-016-3077-3URLPMID:27709401 [本文引用: 1]
H9 subtype avian influenza viruses (AIVs) remain a significant burden in the poultry industry and are considered to be one of the most likely causes of any new influenza pandemic in humans. As ducks play an important role in the maintenance of H9 viruses in nature, successful control of the spread of H9 AIVs in ducks will have significant beneficial effects on public health. Duck enteritis virus (DEV) may be a promising candidate viral vector for aquatic poultry vaccination. In this study, we constructed a recombinant DEV, rDEV-?UL2-HA, inserting the hemagglutinin (HA) gene from duck-origin H9N2 AIV into the UL2 gene by homologous recombination. One-step growth analyses showed that the HA gene insertion had no effect on viral replication and suggested that the UL2 gene was nonessential for virus growth in vitro. In vivo tests further showed that the insertion of the HA gene in place of the UL2 gene did not affect the immunogenicity of the virus. Moreover, a single dose of 103 TCID50 of rDEV-?UL2-HA induced solid protection against lethal DEV challenge and completely prevented H9N2 AIV viral shedding. To our knowledge, this is the first report of a DEV-vectored vaccine providing robust protection against both DEV and H9N2 AIV virus infections in ducks.
DOI:10.1007/s00705-014-2275-0URLPMID:25392272 [本文引用: 1]
To gain a better understanding of the genetic changes required for attenuation of duck enteritis virus (DEV), the Chinese standard challenge strain of DEV (DEV CSC) was serially passaged 80 times in chick embryo fibroblasts. We plaque-purified the virus after the 25th passage (DEV p25) and the 80th passage (DEV p80) and investigated its in vitro and in vivo properties. Average plaque sizes for DEV p25 and p80 were significantly smaller than those for their parental DEV CSC. The results from an in vivo experiment revealed that DEV p25 and p80 were avirulent in ducks and protected them from virulent DEV challenge. The complete genome sequence of DEV p80 was determined and compared with that of the parent virus. An 1801-bp deletion was identified in the genome of DEV p80, which affected the genes encoding gI and gE. Moreover, there were 11 base substitutions, which led to seven amino acid changes in open reading frames LORF9, UL51, UL9, UL7, UL4, ICP4 and US3. Further DNA sequence analysis showed that the 1801-bp deletion was also present in DEV p25. Our findings suggest that DEV gE and/or gI are nonessential for virus growth and might, as with other herpesviruses, play an important role in cell-to-cell spread and virulence. Our experiments provide more genetic information about DEV attenuation and further advance our understanding of the molecular basis of DEV pathogenesis.
DOI:10.1080/03079450020016869URLPMID:19184858 [本文引用: 1]
Duck enteritis virus (DEV), a herpesvirus, has been shown to cause lymphoid organ atrophy and immunosuppression in white Pekin ducklings. The cells that support virus replication and could be important for immunosuppression were identified in this study. Lymphoid organs of white Pekin ducks infected at 2 weeks of age were collected at 3, 6, 8 and 10 days post-inoculation (d.p.i.). Frozen sections were double-stained for DEV-infected (DEV+) and epithelial cells, DEV+ and CD3+ cells or DEV+ and B cells. DEV antigen was detected in the spleen, thymus and bursa for 3, 6 and 8 d.p.i., respectively. DEV antigen was demonstrated in epithelial cells of all examined lymphoid organs. B cells were irreversibly depleted from bursa; however, the depletion in the spleen was only for 8 d.p.i. Depletion of CD3+ cells was only observed in the thymus. These data show that the target cells for DEV are epithelial and B cells.
URLPMID:11007006 [本文引用: 1]
Duck enteritis virus (DEV) was isolated from commercial 2-to-6-wk-old white Pekin ducks experiencing 25%-30% mortality and high morbidity. Secondary infections with Pasteurella multocida, Riemerella anatipestifer, and Escherichia coli were frequently seen in affected ducks. The isolated virus was identical to the prototype DEV by virus neutralization test but differed from the classic DEV by causing lymphoid organ atrophy and inconsistent hemorrhagic lesions in the intestinal annular bands. Attempts to reproduce the disease in white Pekin ducks were unsuccessful until the virulence of the virus was increased by three passages in Muscovy ducklings. Significant thymic atrophy (P &lt; or = 0.001) was detected during the first 10 days postinfection (DPI), but thymus size returned to normal by 17-24 DPI. However, bursal atrophy increased significantly (P &lt; or = 0.001) from 4 DPI until the end of the experiment (39 DPI). Reduction in body weight was significant (P &lt; or = 0.05) between 4 and 6 DPI. There was massive depletion of thymic and bursal lymphocytes with lymphoid necrosis in the thymus, bursa, spleen, and Harderian gland. Eosinophilic intranuclear inclusions were observed in thymus, bursa, spleen, esophagus, cloaca, liver, conjunctiva, and Harderian gland. Occasional intracytoplasmic inclusions were also found scattered in the epithelial cells of conjunctiva, esophagus, bursa of Fabricius, and cloaca. Virus was recovered from experimentally infected ducks from thymus, bursa, spleen, liver, kidneys, trigeminal ganglion, and cloaca during the first 10 days of infection. These findings suggest that a low-virulent DEV can cause a massive lymphoid atrophy and can sustain immunosuppression as noted by the secondary bacterial infection.
DOI:10.1637/8120-100207-ResNote.1URLPMID:18646467 [本文引用: 1]
To better understand the pathogenesis of duck virus enteritis (DVE), the levels of viral DNA in various tissues of ducklings during acute stage of virulent duck enteritis virus (DEV) infection were investigated by using quantitative real-time polymerase chain reaction. The results show that the viral levels of DEV in systemic organs have a close correlation with the progression of disease. The rapid dissemination and active replication of virulent DEV in multiple systemic organs at the early phase of acute infection accelerate the progression of disease. The levels of viral DNA increase sharply soon after developed clinical signs of disease, and the extent of increase and the magnitude of DEV DNA load in various tissues of ducklings after the exhibition of clinical signs may be a critical determinant of the outcome of DEV infection. The relatively high levels of DEV in bursa and small intestine tissues of dead ducklings most likely reflect the abundance of target epithelial and lymphoid cells in these tissues, which therefore play a key role in the pathogenesis of acute DVE and manifest as severe tissue lesions on the bursa and small intestine.
[本文引用: 1]
URLPMID:7637001 [本文引用: 1]
We have determined the nucleotide sequence and transcriptional pattern of a group of open reading frames in the pseudorabies virus (PrV) genome located near the left end of the unique long region within BamHI 5' fragment at map positions 0.01 to 0.06. The 7,412-bp BamHI 5' fragment was found to contain five complete open reading frames and part of a sixth whose deduced amino acid sequences showed homology to the UL50 (partial), UL51, UL52, UL53, and UL54 gene products of herpes simplex virus type 1 (HSV-1) and corresponding genes identified in other alphaherpesviruses. Homologs to the UL55 and UL56 genes of HSV-1 were not detected. However, we identified a gene with homology only to the first open reading frame (ORF-1) of the equine herpesvirus 1 strain Ab4 (E. A. Telford, M. S. Watson, K. McBride, and A. J. Davison, Virology 189:304-316, 1992). Northern blot analyses revealed unique mRNAs for the UL51, UL54, and ORF-1 genes and a set of 3'-coterminal mRNAs for the UL52 to UL54 genes. A PrV mutant lacking ORF-1 was isolated after deletion of ORF-1 coding sequences and insertion of a lacZ expression cassette. The ORF-1- PrV mutant was able to productively replicate in noncomplementing cells to levels similar to those of wild-type PrV, proving that ORF-1 is not essential for replication of PrV in cell culture. The conservation of this gene between PrV and equine herpesvirus 1 documents the close evolutionary relationship between these animal herpesviruses and points to a possible function of the respective proteins in infection of the natural host.
DOI:10.1099/0022-1317-67-9-1759URLPMID:3018124 [本文引用: 1]
The entire DNA sequence of varicella-zoster virus (VZV) was determined using the M13-dideoxynucleotide technology. The genome is variable in size, but the sequence which was obtained comprises 124884 bp. Analysis of the sequence indicated that the genome contains 70 genes distributed about equally between the two DNA strands. The genes are organized compactly, but regions of overlap between protein-coding regions are not extensive. Many of the genes are arranged in 3'-coterminal families, and at least one is spliced. The discerned organization of VZV genes and that deduced for herpes simplex virus type 1 (HSV-1) from published transcript mapping data indicate that these two members of the Alphaherpesvirinae are very similar in gene layout. Comparisons of the predicted amino acid sequences of VZV proteins with those available for HSV-1 proteins generally suggest evolution from an ancestral genome, and allow the functions of several VZV genes to be deduced, although limited regions where the genomes differ in functional organization were also identified.
DOI:10.1016/0042-6822(92)90706-uURLPMID:1318606 [本文引用: 1]
The complete DNA sequence was determined of a pathogenic British isolate of equine herpesvirus-1, a respiratory virus which can cause abortion and neurological disease. The genome is 150,223 bp in size, has a base composition of 56.7% G + C, and contains 80 open reading frames likely to encode protein. Since four open reading frames are duplicated in the major inverted repeat, two are probably expressed as a spliced mRNA, and one may contain an internal transcriptional promoter, the genome is considered to contain 76 distinct genes. The genes are arranged collinearly with those in the genomes of the two previously sequenced alphaherpesviruses, varicella-zoster virus, and herpes simplex virus type-1, and comparisons of predicted amino acid sequences allowed the functions of many equine herpesvirus 1 proteins to be assigned.
DOI:10.1099/0022-1317-69-7-1575URLPMID:2839595 [本文引用: 1]
The genome structure of equine herpesvirus 1 (EHV-1) subtype 2 was shown by electron microscopic studies and restriction endonuclease site mapping to comprise two covalently linked segments (L, 109 kbp; S, 35 kbp). The S segment contains a unique sequence (US) flanked by a substantial inverted repeat (TRS/IRS). Thus, the genome structure of EHV-1 subtype 2 is similar to that published previously for EHV-1 subtype 1, but the two subtypes differ in the occurrences of EcoRI and BamHI restriction sites. Hybridization studies using cloned EHV-1 DNA showed that the genome of EHV-1 subtype 2 is colinear with the genomes of EHV-1 subtype 1 and herpes simplex virus type 1. DNA sequence data for four EHV-1 subtype 2 genes, including one potentially encoding a glycoprotein, were obtained by sequencing a 4574 bp BamHI fragment containing the junction between US and TRS. The genome structure, hybridization and sequence data confirm that EHV-1 subtype 2 is of the alphaherpesvirus lineage.
DOI:10.3389/fvets.2017.00211URLPMID:29312962 [本文引用: 1]
Equine herpesvirus 1 (EHV-1) is a major pathogen affecting equines worldwide. The virus causes respiratory disease, abortion, and, in some cases, neurological disease. EHV-1 Kentucky A (KyA) is attenuated in the mouse and equine, whereas wild-type pathogenic strain RacL11 induces severe inflammatory infiltration of the lung, causing infected mice to succumb. The complete DNA sequencing of the KyA genome revealed that genes UL17 (ORF17), US6 (ORF73; gI), US7 (ORF74; gE), and US8 (ORF75; 10?K) are deleted as compared to the RacL11 and Ab4 genomes. In-frame deletions in the US1 (ORF68), US4 (ORF71; gp2), and UL63 (ORF63; EICP0) genes and point mutations in 14 different open reading frames (ORFs) were detected in the KyA genome. Interestingly, UL1 (ORF1) and UL2 (ORF2) were deleted in both KyA and RacL11. Our previous studies showed that EHV-1 glycoproteins gI, gE, and full-length gp2 contribute to the pathogenesis of the RacL11 strain. The confirmation of these gene deletions in KyA suggests their contribution to the attenuation of this virus. The growth kinetics results revealed that KyA replicates to high titers in cell culture as compared to RacL11 and Ab4, indicating that the above genomic deletions and mutations in KyA do not have an inhibitory effect on KyA replication in cells of mouse, rabbit, equine, or human origin. Studies of EHV-1 pathogenesis in CBA mice showed that KyA is attenuated whereas mice infected with RacL11 succumbed by 3-6?days post-infection, which is consistent with our previous results.
DOI:10.1016/j.vaccine.2013.01.013URLPMID:23333211 [本文引用: 1]
Bovine herpesvirus-1 (BoHV-1) causes significant disease in cattle including respiratory, fetal diseases, and reproductive tract infections. Control programs usually include vaccination with a modified live viral (MLV) vaccine. On occasion BoHV-1 strains are isolated from diseased animals or fetuses postvaccination. Currently there are no markers for differentiating MLV strains from field strains of BoHV-1. In this study several BoHV-1 strains were sequenced using whole-genome sequencing technologies and the data analyzed to identify single nucleotide polymorphisms (SNPs). Strains sequenced included the reference BoHV-1 Cooper strain (GenBank Accession JX898220), eight commercial MLV vaccine strains, and 14 field strains from cases presented for diagnosis. Based on SNP analyses, the viruses could be classified into groups having similar SNP patterns. The eight MLV strains could be differentiated from one another although some were closely related to each other. A number of field strains isolated from animals with a history of prior vaccination had SNP patterns similar to specific MLV viruses, while other field isolates were very distinct from all vaccine strains. The results indicate that some BoHV-1 isolates from clinically ill cattle/fetuses can be associated with a prior MLV vaccination history, but more information is needed on the rate of BoHV-1 genome sequence change before irrefutable associations can be drawn.
DOI:10.1128/JVI.00646-12URL [本文引用: 1]
DOI:10.1128/JVI.00445-12URL [本文引用: 1]
A new strain of equine herpesvirus type 8 (EHV-8), Wh, has been isolated from horses in China, and its complete genome has been sequenced and analyzed. The result indicates that the new strain has the same constitution and arrangement of open read frames as EHV-1 and EHV-9. This work is the first announced complete genome sequence of EHV-8.
DOI:10.1099/jgv.0.000521URLPMID:27283114 [本文引用: 1]
Infectious laryngotracheitis (ILT) is a highly contagious respiratory disease of chickens caused by infectious laryngotracheitis virus (ILTV). The disease is controlled by the use of live-attenuated vaccines. Previously we reported the complete nucleotide sequence of the ILTV vaccine strain (TCO) and identified a nonsense mutation in the gene encoding the ORF C protein. This suggested that the ORF C protein might be associated with viral virulence. To investigate this, an ILTV recombinant with a deletion in the gene encoding ORF C was constructed using the genome of the virulent United States Department of Agriculture (USDA) challenge strain (USDAch). Compared to the parental virus, the ΔORF C recombinant replicated in chicken kidney (CK) cells with similar kinetics and generated similar titres. This demonstrated that the ORF C deletion had no deleterious effects on replication efficacy in vitro. In chickens, the recombinant induced only minor microscopic tracheal lesions when inoculated via the intra-tracheal/ocular route, while the parental strain induced moderate to severe microscopic tracheal lesions, even though virus load in the tracheas were comparable. Groups of chickens vaccinated via eye-drop with the ?ORFC-ILTV were protected to levels comparable to those elicited by TCO vaccination. To our knowledge, this is the first report that demonstrates the suitability of ?ORFC as a live-attenuated vaccine to prevent the losses caused by ILTV.
DOI:10.1016/0168-1702(94)90017-5URLPMID:7941700 [本文引用: 1]
In order to investigate the functional properties of the UL56 gene of herpes simplex virus type 1 (HSV-1), it was necessary to express the UL56 protein in vitro. The DNA sequences corresponding to the open reading frame of the UL56 gene of HSV-1 strain F were amplified from genomic viral DNA by PCR using primers corresponding to the translational start and termination regions of the UL56 ORF. The PCR product (705 bp) was inserted into the EcoRI/XbaI recognition sites of the bacterial expression vector pMal-c2. This procedure allowed the expression of the viral UL56 gene fused to the maltose-binding protein (MBP) of Escherichia coli, and subsequent cleavage of the fusion protein with the specific protease factor Xa. The induced fusion protein was purified by affinity chromatography using amylose columns. The apparent molecular weight of the fusion protein was about 70 kDa. Factor Xa cleaves the fusion protein into two subfragments of 42 kDa (MBP) and 30 kDa (UL56). Rabbit antisera induced against recombinant UL56 protein were used for detection of the UL56 gene product during the infection cycles of HSV-1. The presence of the UL56 protein was detected in infected cells and in HSV-1 virions by Western blot experiments and by immunofluorescence assays. A strong and increasing cytoplasmic fluorescence was observed in RC-37 cells infected with HSV-1 strain F between 6 and 16 h post-infection. In addition it was found that human HSV-1 IgM/IgG positive convalescent sera recognized the recombinant UL56 protein.
DOI:10.1023/a:1008053017716URLPMID:9778788 [本文引用: 1]
Recently the UL56 protein of herpes simplex virus type 1 (HSV-1) was shown to be associated with the virion of HSV-1 as determined by Western blot analysis. The detection of the UL56 protein in infected cells and its association with virions of HSV-1 is of particular importance, pointing to a possible involvement of UL56 protein in virus-host interactions. In order to investigate the properties of the UL56 protein further immuno-localization was performed using rabbit hyperimmune serum against fusion recombinant UL56 protein and purified virions of HSV-1 strain F. The UL56 protein was detected in the HSV-1 virions by immuno gold negative staining.
DOI:10.1016/j.vetmic.2010.02.014URLPMID:20346601 [本文引用: 2]
This paper is about the taxonomy and genomics of herpesviruses. Each theme is presented as a digest of current information flanked by commentaries on past activities and future directions. The International Committee on Taxonomy of Viruses recently instituted a major update of herpesvirus classification. The former family Herpesviridae was elevated to a new order, the Herpesvirales, which now accommodates 3 families, 3 subfamilies, 17 genera and 90 species. Future developments will include revisiting the herpesvirus species definition and the criteria used for taxonomic assignment, particularly in regard to the possibilities of classifying the large number of herpesviruses detected only as DNA sequences by polymerase chain reaction. Nucleotide sequence accessions in primary databases, such as GenBank, consist of the sequences plus annotations of the genetic features. The quality of these accessions is important because they provide a knowledge base that is used widely by the research community. However, updating the accessions to take account of improved knowledge is essentially reserved to the original depositors, and this activity is rarely undertaken. Thus, the primary databases are likely to become antiquated. In contrast, secondary databases are open to curation by experts other than the original depositors, thus increasing the likelihood that they will remain up to date. One of the most promising secondary databases is RefSeq, which aims to furnish the best available annotations for complete genome sequences. Progress in regard to improving the RefSeq herpesvirus accessions is discussed, and insights into particular aspects of herpesvirus genomics arising from this work are reported.
DOI:10.1128/jvi.76.13.6718-6728.2002URLPMID:12050385 [本文引用: 1]
The UL56 gene product of herpes simplex virus (HSV) has been shown to play an important role in viral pathogenicity. However, the properties and functions of the UL56 protein are little understood. We raised rabbit polyclonal antisera specific for the UL56 protein of HSV type 2 (HSV-2) and examined its expression and properties. The gene product was identified as three polypeptides with apparent molecular masses ranging from 32 to 35 kDa in HSV-2-infected cells, and at least one species was phosphorylated. Studies of their origins showed that the UL56 protein of HSV-2 is also translated from the upstream in-frame methionine codon that is not present in the HSV-1 genome. Synthesis was first detected at 6 h postinfection and was not abolished by the viral DNA synthesis inhibitor phosphonoacetic acid. Indirect immunofluorescence studies revealed that the UL56 protein localized to both the Golgi apparatus and cytoplasmic vesicles in HSV-2-infected and single UL56-expressing cells. Deletion mutant analysis showed that the C-terminal hydrophobic region of the protein was required for association with the cytoplasmic membrane and that the N-terminal proline-rich region was important for its translocation to the Golgi apparatus and cytoplasmic vesicles. Moreover, the results of protease digestion assays and sucrose gradient fractionation strongly suggested that UL56 is a tail-anchored type II membrane protein associated with lipid rafts. We thus hypothesized that the UL56 protein, as a tail-anchored type II membrane protein, may be involved in vesicular trafficking in HSV-2-infected cells.
DOI:10.1016/0168-1702(96)80248-6URLPMID:8725118 [本文引用: 2]
Recently it was shown that the avirulent phenotype of HSV-1 strain HFEM is correlated to the lack of DNA sequences of the promoter region of the UL56 gene. In order to investigate the role of the UL56 gene of HSV-1 in the process of viral pathogenicity in more detail, a complete copy of the UL56 gene of the virulent HSV-1 strain 17 was inserted within the DNA sequences of the incomplete UL56 gene of the genome of HSV-1 strain HFEM. The UL56 gene of HSV-1 strain 17 comprises 1428 bp corresponding to the nucleotide positions (NP) 11,5967-117,395 of the genome of HSV-1 strain 17 (SacII-DNA fragment) containing the promoter region and the entire UL56 gene with identical transcription termination signals. This particular DNA fragment was inserted into the corresponding region of the genome of HSV-1 strain HFEM by co-transfection experiments in which the beta-galactosidase gene served as reporter gene. Those recombinant viruses with the ability to express the UL56 gene were tested for their pathogenicity in vivo. The results of these experiments indicate that the restoration of the viral UL56 gene expression led to the restitution of the virulent phenotype of HSV-1 strain HFEM. The UL56 protein which has been shown to be a component of the virion possesses several characteristic signatures e.g. a hydrophobic domain at the carboxy-terminus between amino acid residues 217 and 234 (VFGVVAIVVVIILVFLWR). In order to investigate the role of this particular signature of the UL56 protein in the process of viral pathogenicity, site-specific mutagenesis was performed for removing the carboxy-terminus of the UL56 protein. The deleted region of the DNA sequences of the UL56 gene between NP 1122-1175 corresponds to NP 116 220-116 373 of the viral genome. The DNA sequences of the UL56 gene of virulent HSV-1 strain 17 and F were replaced by DNA sequences of the truncated UL56 gene by co-transfection experiments in which the beta-galactosidase gene served as a reporter gene. Those recombinant viruses with the ability to express the truncated UL56 gene were examined for their pathogenicity in vivo. The analysis revealed that the expression of the truncated UL56 protein (without hydrophobic domain 217-234 aa) was not sufficient for the maintenance of the virulent phenotype of HSV-1 strains.
DOI:10.1016/j.micinf.2005.05.007URLPMID:16054416
Herpes simplex virus (HSV), a neurotropic virus, establishes life-long and, although rare, life-threatening infection in humans, and it may precipitate substantial medical and psychosocial morbidity. Here we show that HSV-1 strain HF clone 10 (HF10) exhibits impaired neuroinvasiveness in peripheral olfactory, vomeronasal and trigeminal conduits following intranasal as well as corneal inoculation. HF10 attenuation likely arises from multiple defects of HSV genes, so that HF10 will not revert to a virulent phenotype. Intranasal vaccination of mice with HF10 conferred significant protection against lethal challenge with HSV-1 and HSV-2 via the intranasal and intravaginal routes. Thus, we propose that HF10 explicitly meets the prerequisites for a candidate live attenuated HSV vaccine.
DOI:10.1016/0168-1702(91)90076-8URLPMID:1662844 [本文引用: 1]
In order to investigate whether or not the UL56 gene is involved in those processes determining the viral pathogenicity and latency, a recombinant virus HSV-1-M-LacZ was constructed in which the DNA sequences between nucleotide position (np) 116030 and 121753 were replaced by the E. coli beta-galactosidase (LacZ) gene. This deletion spans from the carboxyterminus of UL55 (np 116030) to the second exon of IE110 (np 121753) eliminating UL56 and the variable region of the BamHI DNA fragment B which were implicated in intraperitoneal pathogenicity and latency. The host range and growth kinetics of the recombinant virus HSV-1 M-LacZ were comparable to the parental strain HSV-1 F. As expected it was found that HSV-1-M-LacZ lost its virulent phenotype and was not able to develop acute infection in animals. The state of the UL56 gene was investigated by determining the cDNA sequence of the UL56 gene transcript of HSV-1 F using PCR products obtained after amplification of the cDNA with oligonucleotide primers corresponding to the translational start and stop codons of this gene. This analysis revealed that the DNA sequence of the UL56 gene of HSV-1 F differed from those DNA sequences determined for the genomic DNA of HSV-1 strain 17. Between nucleotide position 116343 and 116344 two nucleotides -AG- are inserted which prolong the ORF of the UL56 gene to 233 amino acids with a predicted molecular weight of 30 kDa.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}