 ,, 郝玉金,山东农业大学园艺科学与工程学院/作物生物学国家重点实验室,山东泰安 271018
,, 郝玉金,山东农业大学园艺科学与工程学院/作物生物学国家重点实验室,山东泰安 271018Genome-Wide Identification and Expression Pattern Analysis of NLP (Nin-Like Protein) Transcription Factor Gene Family in Apple
WANG Xun, CHEN XiXia, LI HongLiang, ZHANG FuJun, ZHAO XianYan, HAN YuePeng, WANG XiaoFei,, HAO YuJin,College of Horticulture Science and Engineering, Shandong Agricultural University/State Key Laboratory of Crop Biology, Tai’an 271018, Shandong通讯作者:
责任编辑: 赵伶俐
收稿日期:2019-04-28接受日期:2019-06-26网络出版日期:2019-12-01
| 基金资助: |
Received:2019-04-28Accepted:2019-06-26Online:2019-12-01
作者简介 About authors
王寻,E-mail:wx20145015@126.com

摘要
关键词:
Abstract
Keywords:
PDF (5665KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王寻, 陈西霞, 李宏亮, 张富军, 赵先炎, 韩月彭, 王小非, 郝玉金. 苹果NLP(Nin-Like Protein)转录因子基因家族全基因组鉴定及表达模式分析[J]. 中国农业科学, 2019, 52(23): 4333-4349 doi:10.3864/j.issn.0578-1752.2019.23.014
WANG Xun, CHEN XiXia, LI HongLiang, ZHANG FuJun, ZHAO XianYan, HAN YuePeng, WANG XiaoFei, HAO YuJin.
0 引言
【研究意义】植物氮素作为一种养分,对于植物生长发育具有十分重要的意义,近些年,氮素吸收转运相关基因的功能调控研究比较广泛,其中RWP-PK家族中的NLP转录因子亚家族响应氮饥饿并且在植物氮信号调节过程中处于中心地位[1]。因此,通过NLP的研究可以对氮素调节网络相关基因进一步认识,从而对植物氮素利用提供指导。尤其是在苹果等果树上,通过研究该基因,为提高果树氮肥利用效率与提质增效带来新的思考。【前人研究进展】关于NLP(Nin-like protein)最早的研究可以追溯到豆科模式植物百脉根Nin(nodule inception),起初Nin作为根瘤感受基因,被鉴定影响到根瘤的早期发育[2]。随后,在非豆科植物中对Nin同源基因Nin-like进行了大量鉴定,其中包括拟南芥[3]、水稻[3]、小麦[4]、玉米[5]、毛果杨[6]、枳[7]、甘蓝型油菜[8]等。随着对NLP研究的深入,包括该家族蛋白结构域、调控网络、表达模式等在一定程度上被揭示。前人通过比较不同物种NLP基因家族,发现NLP转录因子具有两个典型的特征结构域,高度保守的RWP-RK结构域以及一个位于羧基端的PB1结构域,除此之外,在氨基酸序列的氨基端还有一个类似于GAF的结构域;已知RWP-RK结构域可以与DNA结合发挥作用,PB1结构域被认为具有蛋白质与蛋白质相互作用的功能[3]。关于NLP的功能,模式植物拟南芥已有大量相关研究,MARCHIVE等[9]探究发现拟南芥中NLP7在早期响应硝酸盐中扮演了重要角色,YAN等[10]证明NLP8对于硝酸盐促进种子萌发至关重要,LIU等[11]发现在植株营养生长中重要的NO3--CPK-NLP信号通路,YU等[12]报道过表达拟南芥NLP7通过增强氮和碳的同化,在限制氮和充足氮的条件下均可以促进植物生长。上述研究证实,NLP转录因子在硝态氮响应与植株生长发育中发挥重要节点作用。【本研究切入点】许多豆科植物和非豆科植物中NLP转录因子家族基因已经进行了鉴定,苹果中NLP相关信息尚未见报道,本研究利用已发布的苹果参考基因组[13](Malus× domestica-genome.v1.0)对全基因组范围内的NLP转录因子成员进行搜索。【拟解决的关键问题】本研究采用生物信息学手段,首次对苹果NLP转录因子全基因组成员进行鉴定,并从基因和蛋白水平上系统地对其预测分析,此外运用荧光定量PCR技术检测MdNLPs的组织表达、氮响应过程及非生物胁迫变化情况,从而有助于苹果NLP转录因子特点与功能的进一步探究。1 材料与方法
试验于2018年9月至2019年4月在山东农业大学作物生物学国家重点实验室进行。1.1 植物材料与处理
试验材料为种植于山东农业大学园艺试验站(山东泰安)的十年生‘皇家嘎拉’苹果树和培养于实验室中的生长4个月左右的苹果砧木‘平邑甜茶’实生苗。在‘皇家嘎拉’苹果树上分别取根、幼嫩茎、叶、花(初花期)、果实(花后70 d)后,将各组织迅速于液氮中冷冻,然后放置到-80℃超低温冰箱贮藏,后续用于基因组织表达分析。从‘平邑甜茶’实生苗中选取生长状况等较为一致的幼苗分别用于3个试验处理:氮饥饿处理、干旱胁迫处理、ABA处理,其中对各个处理的每个取样时间节点设置3个生物学重复。氮饥饿处理:取培养于基质中的实生苗18株,转移至氮饥饿环境(蛭石+浇施缺氮营养液(成分:1.2 mol?L-1 KH2PO4、0.1 mol?L-1 CaCl2、1 mol?L-1 MgSO4、0.1 mmol?L-1 Fe盐、微量、1 mol?L-1 KCl))继续培养,0、3、6、12、24和36 h分别全株取样。干旱胁迫处理:取培养于基质中的实生苗24株,转移至干旱环境(干燥基质)继续培养,0、0.5、1、3、6、12、24和48 h分别全株取样。ABA处理:取培养于基质中的实生苗18株,转移至ABA处理环境(浇施0.1 mmol?L-1 ABA溶液)继续培养,0、1、3、6、12和24 h分别全株取样。3个试验处理所取样品迅速置于液氮中冷冻,而后储存于超低温冰箱,后续用于氮饥饿响应分析和非生物胁迫表达分析。1.2 苹果NLP转录因子家族成员的识别
从NCBI下载拟南芥9个NLPs成员蛋白序列,苹果蛋白数据库来自GDR[14](1.3 苹果NLP蛋白分析
1.3.1 苹果NLP蛋白理化特征 分子量大小、等电点等信息利用EXPASY中的在线工具ProtParam(1.3.2 苹果NLP多序列比对与结构域分析 蛋白质多序列比对采用在线软件Clustal Omega(
1.3.3 苹果NLP的系统进化分析 水稻6个NLP成员下载于Phytozome数据库(
1.3.4 苹果NLP保守基序分析 Linux系统中使用MEME[17](版本:MEME 5.0.2)工具进行保守模体分析,参数设置为“meme MdNLPs.fa-protein-oc motif.out- nostatus-mod zoops-nmotifs 15-minw 6-maxw 60”。
1.3.5 苹果NLP蛋白质结构分析 蛋白质二级结构预测利用SOPMA(
1.3.6 苹果NLP亚细胞定位预测 亚细胞定位预测使用在线程序WoLF PSORT:Protein Subcellular Localization Prediction[19](https://www.genscript.com/ wolf-psort.html),生物类型选择植物。
1.3.7 苹果NLP家族蛋白相互作用网络预测 通过功能蛋白关联网络在线网站STRING[20](
1.4 苹果NLP分析
1.4.1 苹果NLP染色体定位 在苹果基因组注释文件(Malus×domestica.v1.0.consensus.gff)中根据基因ID提取苹果NLP家族成员位置信息,提交至在线软件MG2C(1.4.2 苹果NLP结构分析 利用基因注释信息,在北京大学基因结构可视化服务器GSDS2.0[21](
1.4.3 苹果NLP基因GO功能注释 从注释文件中获取苹果NLP的GO条目信息,对功能注释进行分类统计。
1.4.4 苹果NLP的启动子分析 从苹果基因组中提取NLP翻译起始位点ATG上游约2 000 bp的片段作为启动子区域,将所有的MdNLP启动子序列提交至PlantCARE[22](
1.4.5 与苹果NLP相关miRNA的预测 利用植物小分子RNA靶标分析在线网站psRNATarget[23](
1.5 苹果NLP表达模式分析
‘皇家嘎拉’苹果树的根、茎、叶、花、果不同组织,‘平邑甜茶’苹果苗的氮饥饿处理、干旱胁迫处理、ABA处理各时间点样品,对它们的总RNA分别使用TRIzol Reagent试剂盒提取,RNA完整性和浓度分别利用琼脂糖凝胶电泳、Thermo Nano Drop 2000仪器检测。反转录合成cDNA过程采用PrimeScript RT reagent Kit with gDNA Eraser试剂盒,实时荧光定量PCR按照UltraSYBR Mixture(High ROX)试剂提供的方法进行,反应体系如下:2×UltraSYBR Mixture(High ROX)10 μL,ddH2O 7 μL,上、下游引物(10 mmol?L-1)各1 μL,cDNA模板为1 μL,总体系为20 μL。qRT-PCR反应过程为:95℃预变性10 min,95℃变性15 s,58℃退火15 s,65℃延伸10 s,进行40次循环,其中每次循环第3步采集荧光信号,每个反应进行3次重复。定量引物的设计利用在线网站Primer3Plus(Table 1
表1
表1苹果NLP相对表达实时荧光定量PCR引物
Table 1
| 基因Gene | 上游引物Forward primer (5′-3′) | 下游引物Reverse primer (5′-3′) |
|---|---|---|
| MdNLP2 | CTATGCATCGAGGAAACAGCTTG | CAATTCCCTCACCTTCCTCAAGA |
| MdNLP3 | GAAATGGAGAAAGAGGGCTCTGA | TTCAGAAGGGCTTGGATACTTCC |
| MdNLP4 | GTCAGTATGCTCTCGATCCTGATA | CAGTAAGATGTGTAGGATGTTGGC |
| MdNLP5 | GCTTGCTTCTGTGGAGACATTAC | TCCAGTATGAGTGCTCTGTAAGC |
| MdActin | GGACAGCGAGGACATTCAGC | CTGACCCATTCCAACCATAACA |
新窗口打开|下载CSV
2 结果
2.1 苹果NLP转录因子家族成员鉴定及基本信息
通过两种方法并进行严格筛选、确认,最终共获得6个MdNLP,且基因组注释文件(Malus×domestica. v1.0.consensus.gff)均注释到存在RWP-RK结构域、PB1结构域(MDP0000788505的蛋白氨基酸C末端缺失,只注释到RWP-RK结构域)。根据其在染色体上的位置信息,依次命名为MdNLP1(MDP0000788505)、MdNLP2(MDP0000265619)、MdNLP3(MDP0000246881)、MdNLP4(MDP0000239938)、MdNLP5(MDP0000132856)、MdNLP6(MDP0000584547)。经统计发现,6个MdNLP长度最小的为MdNLP1(3 856 bp),最长的为MdNLP6(10 212 bp),编码序列长度在2 460—5 121 bp,编码氨基酸数目在819—1 706,蛋白质的分子量在90 889.49—189 398.44 D,等电点5.45—7.9,具体信息见表2。2.2 MdNLP蛋白结构域分析与多序列比对
在鉴定MdNLP的过程中,通过NCBI的SMART蛋白结构域查询,可以发现苹果NLP蛋白与拟南芥NLP蛋白均含有两个典型的结构域(RWP-RK和PB1),使用Clustal Omega在线工具对9条拟南芥与6条苹果NLP蛋白序列进行多序列比对,并用Jalview对两个结构域进行编辑截取。由图1可知,RWP-PK结构域保守性非常高,由大约50多个氨基酸组成,其中包含“RWPXRK”特征核苷酸组成,该结构域中只有极少的氨基酸位点一致性较差。在NLP蛋白C端的PB1结构域保守性较高,由大约80个氨基酸组成,比较特殊的是MdNLP1的PB1结构域C端部分缺失,只有大约30个氨基酸。图1
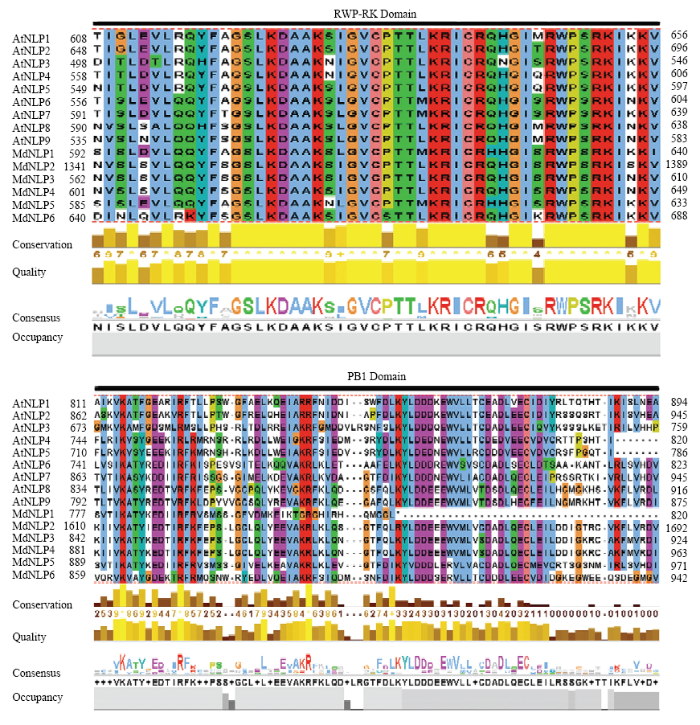 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1拟南芥与苹果NLP蛋白的多序列比对与典型保守结构域
Fig. 1Multi-sequence alignment and typical conserved domains of NLP proteins between Arabidopsis thaliana and apple
2.3 苹果NLP蛋白系统发生树及Motif分析
来自苹果、拟南芥、水稻的全部NLP蛋白序列(MdNLP1—6、AtNLP1—9、OsNLP1—6)用于构建系统发生树,以RWP-RK结构域部分缺失的OsNLP6作为外类群。由图2-a进化树分支结构可以看出NLP家族蛋白明显被分为3类,与SCHAUSER等[3]的研究结果一致,在苹果NLP家族中,MdNLP6为一类(I),MdNLP2、MdNLP3、MdNLP4为一类(II),MdNLP1、MdNLP5聚为一类(III),拟南芥中研究最多的NLP7位于分支的第III类。苹果与拟南芥NLP蛋白经MEME软件分析(图2-b),识别的15种保守基序在这些序列中基本均有分布。其中RWP-RK保守结构域由Motif1和Motif12组成,PB1结构域由Motif5、Motif9、Motif15构成。对于MdNLP1而言,由于其C端PB1结构域不完整,只检测到Motif9,而Motif15和Motif5两个基序缺失。除此之外,参照CHARDIN等[25]的研究,对NLP蛋白另外的一个N端GAF结构域进行标注,苹果NLP蛋白中GAF结构域包括Motif3、Motif4、Motif6、Motif8、Motif11、Motif13,与拟南芥的保守基序组成一致。图2
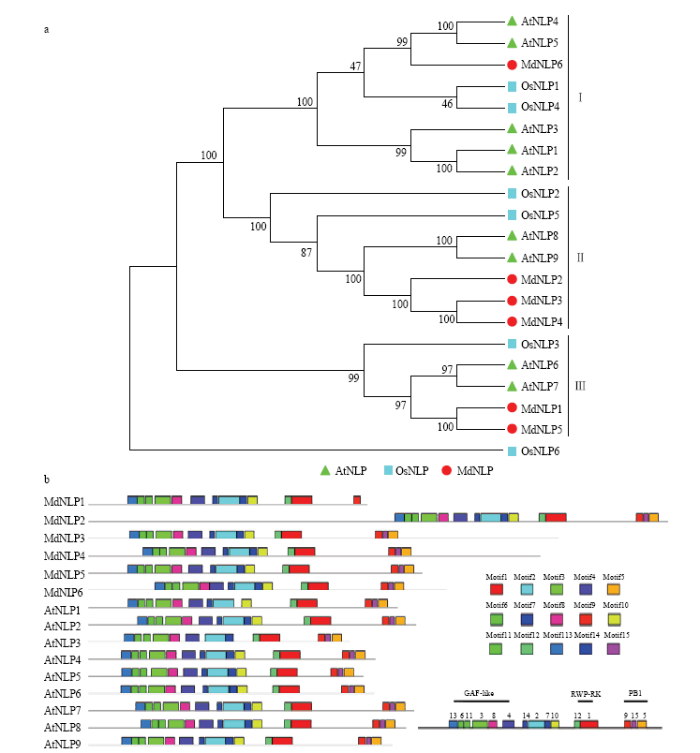 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2苹果、拟南芥、水稻NLP蛋白系统进化树(a)及苹果NLP蛋白保守基序(b)
a图中红色实心圆点代表苹果NLP蛋白,绿色实心三角代表拟南芥NLP蛋白,蓝色实心方形代表水稻NLP蛋白
Fig. 2Phylogenetic evolution of NLP proteins in apple, Arabidopsis thaliana and rice(a) and conserved motifs of NLP proteins in apple (b)
In figure 2-a, the red solid dot represents apple NLP protein, the green solid triangle represents Arabidopsis NLP protein, and the blue solid square represents rice NLP protein
2.4 苹果NLP蛋白结构预测
对MdNLP蛋白进行二级结构预测,从表3看出,6个蛋白质二级结构都以无规卷曲和α螺旋为主,其中无规卷曲占到最大比例,此外二级结构中还包含β转角,所占比例都是最小的,而且在这些蛋白质中各二级结构所占比例大致相似。为了比较苹果NLP蛋白中3种结构域(GAF-like、RWP-RK、PB1)的结构特征,本次研究采用同源建模的方法绘制出保守区段的三维蛋白模型(图3)。通过比较这些模型,很直观地观察到苹果NLP蛋白保守域结构的高度相似性,而往往特定结构与特殊功能相关联,对此猜测苹果中NLP家族的功能离不开这些区域。此外,注意到MdNLP1蛋白的PB1区段发生缺失,由于缺失序列较长,无法模拟出三级结构,这导致了其功能相较于其他家族成员可能会出现差异。Table 3
表3
表3苹果NLP蛋白质二级结构
Table 3
| 蛋白质Protein | α-螺旋α-helix | β-转角β-turn | 无规则卷曲Random coil | 延长链Extended strand |
|---|---|---|---|---|
| MdNLP1 | 28.69% | 3.79% | 53.11% | 14.41% |
| MdNLP2 | 33.94% | 5.57% | 43.14% | 17.35% |
| MdNLP3 | 23.40% | 6.40% | 55.89% | 14.32% |
| MdNLP4 | 27.46% | 5.90% | 50.23% | 16.41% |
| MdNLP5 | 26.52% | 4.01% | 55.50% | 13.98% |
| MdNLP6 | 28.33% | 6.27% | 49.90% | 15.49% |
新窗口打开|下载CSV
图3
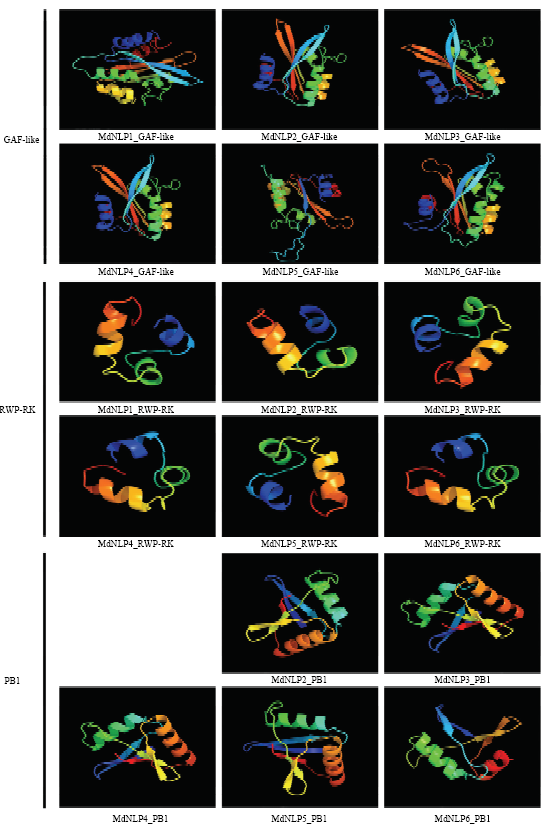 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3苹果NLP蛋白保守域三级结构的同源建模预测
Fig. 3Homologous modeling prediction of the three-level structure of the conservative domain of NLP protein in apple
2.5 苹果NLP蛋白的亚细胞定位预测
拟南芥中NLP7被证明,在没有硝酸盐信号时其存在于细胞质中,外部施加硝酸盐后,NLP7聚集到细胞核中[9],这是很明显的转录因子的特点。为了验证苹果中此类蛋白的定位情况,利用亚细胞定位预测在线网站WoLF PSORT对其进行鉴定,结果(表2)证实苹果NLP有极大可能性定位于细胞核,很低的比例预测到非细胞核区域,符合转录因子的特征。Table 2
表2
表2苹果NLP家族基因、蛋白特征及亚细胞定位预测
Table 2
| 基因 Gene | 登录号 Accession number | 基因长度 Gene length | 编码序列长度 CDS length | 氨基酸数目 Size of aa | 分子量 MW (D) | 等电点 pI | 亚细胞定位 Subcellular localization |
|---|---|---|---|---|---|---|---|
| MdNLP1 | MDP0000788505 | 3856 | 2460 | 819 | 90889.49 | 6.25 | 细胞核 Nucleus |
| MdNLP2 | MDP0000265619 | 9510 | 5121 | 1706 | 189398.44 | 7.9 | 细胞核 Nucleus |
| MdNLP3 | MDP0000246881 | 9366 | 4152 | 1383 | 153424.77 | 7.88 | 细胞核 Nucleus |
| MdNLP4 | MDP0000239938 | 8796 | 3990 | 1329 | 147220.18 | 5.5 | 细胞核 Nucleus |
| MdNLP5 | MDP0000132856 | 4454 | 2949 | 982 | 107556.64 | 5.45 | 细胞核 Nucleus |
| MdNLP6 | MDP0000584547 | 10212 | 3159 | 1052 | 117304.32 | 5.96 | 细胞核 Nucleus |
新窗口打开|下载CSV
2.6 苹果NLP蛋白相互作用蛋白网络构建
为了预测苹果中NLP转录因子潜在的功能,利用STRING网站根据模式物种拟南芥中的同源序列,构建了苹果NLP蛋白相互作用关系网络(图4),由图可知,MdNLP1对应AT1G64530(AtNLP6),MdNLP2对应AT3G59580(AtNLP9),MdNLP3与MdNLP4均对应AT2G43500(AtNLP8),MdNLP5对应NLP7(AT4G24020),MdNLP6对应AT1G76350(AtNLP5);网络图显示,与拟南芥NLP蛋白相互作用的有硝酸盐转运蛋白NRT1.1(AT1G12110)、NRT2.1(AT1G08090),胞质硝酸还原酶NIA1(AT1G77760)等,可以看出,NLP蛋白大多和氮素转运、同化等蛋白相关。此外,由拟南芥的相互作用蛋白,注意到AtNLP7和AtNLP6关系比较复杂,相关联蛋白较多,预测苹果中MDP0000132856和MDP0000788505相较于其他的苹果NLP家族成员可能具有更关键的作用。图4
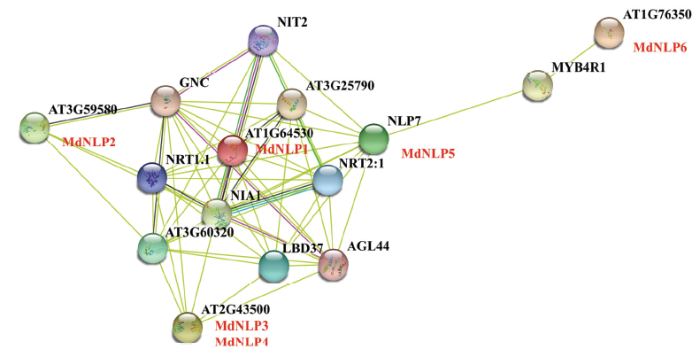 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4参考拟南芥NLP蛋白构建的MdNLPs蛋白相互作用网络
苹果同源蛋白编号以红色字体突出显示
Fig. 4Protein interaction network of MdNLPs constructed by referring to AtNLPs
The name of apple homologous proteins are highlighted in red
2.7 苹果NLP染色体定位、基因结构分析与GO功能注释
利用苹果基因组注释(Malus×domestica.v1.0. consensus.gff)对苹果NLP进行染色体定位及基因结构分析,通过比较发现苹果NLP分别定位于2号染色体(MdNLP1)、4号染色体(MdNLP2)、12号染色体(MdNLP3和MdNLP4)、15号染色体(MdNLP5),由于MdNLP6非染色体定位,没有在图中展示(图5-a)。由苹果NLP外显子-内含子分析可以得知外显子数目在5—14,另外,有意思的是MdNLP1和MdNLP5,MdNLP3和MdNLP4分别具有非常相似的基因组成(图5-b),加上前文对苹果NLP蛋白结构和系统进化关系的分析,推测这两组组内可能具有极其类似的功能。利用苹果基因组注释文件中GO(Gene Ontology)条目,对苹果NLP的GO分类功能注释进行了统计(表4),GFF文件共提取到3个MdNLP的9个GO编号,分别是MdNLP2(GO:0005524,GO:0017111,GO:0016887,GO:0016020,GO:0016021,GO:0006810,GO:0000166),MdNLP3(GO:0008270,GO:0000166,GO:0003676),
MdNLP4(GO:0000166,GO:0003676,GO:0008270),GO分类共分为分子功能、生物途径、细胞组分3大部分,MdNLP家族富集到的GO条目主要集中于分子功能中的核苷酸结合、锌离子结合、核酸结合等。此外,生物途径一类中注释到1个有关转运的功能,以上都间接证明苹果NLP家族是作为转录因子发挥作用的。
图5
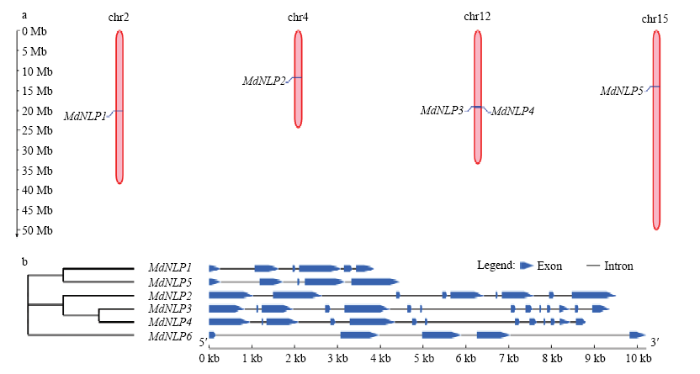 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5苹果NLP基因的染色体定位(a)与基因结构分析(b)
Fig. 5Chromosome localization (a) and gene structure analysis (b) of NLP gene in apple
Table 4
表4
表4苹果NLP GO分类统计列表
Table 4
| GO分类GO classification | GO条目GO term | 基因数目Gene number | 描述Description |
|---|---|---|---|
| 分子功能 Molecular Function | GO:0005524 GO:0017111 GO:0016887 GO:0000166 GO:0008270 GO:0003676 | 1 1 1 3 2 2 | ATP结合 ATP binding 核苷三磷酸酶活性 Nucleoside-triphosphatase activity ATP酶活性 ATPase activity 核苷酸结合 Nucleotide binding 锌离子结合 Zinc ion binding 核酸结合 Nucleic acid binding |
| 生物途径 Biological Process | GO:0006810 | 1 | 转运 Transport |
| 细胞组分 Cellular Component | GO:0016020 GO:0016021 | 1 1 | 膜 Membrane 膜的整体成分 Integral component of membrane |
新窗口打开|下载CSV
2.8 苹果NLP的启动子分析及其相关miRNA预测
通过对MdNLP启动子的分析(表5),鉴定到了大量和激素相关的顺式作用元件,其中包括响应生长素的TGA-element,响应赤霉素的GARE-motif,响应乙烯的ERE,响应脱落酸的ABRE,响应茉莉酸甲酯的CGTCA-motif,响应水杨酸的TCA-element等元件。另外,还在MdNLP5启动子中预测到了一个与氮素响应相关的GCN4元件[26],这与KUMAR等[4]在小麦中的研究结果相似。MicroRNA作为一种小分子调节物质,广泛参与到基因的表达调控过程中,本研究对苹果NLP基因家族成员相关联的miRNA进行预测。依据前人关于氮素响应相关miRNA的研究[27,28],在预测中发现了一些可能参与NLP介导的氮响应的miRNA,主要是mdm-miRNA169、mdm-miRNA171、mdm-miRNA395三个家族,结果列于表6,推测这些miRNA可能通过与MdNLP转录因子相互作用进而调控氮响应过程。Table 5
表5
表5苹果NLP启动子的顺式作用元件预测
Table 5
| 基因 Gene | 脱落酸 ABRE | 低氧 ARE | 茉莉酸甲酯 CGTCA | 乙烯 ERE | 赤霉素 GARE | 氮素 GCN4 | 干旱 MBS | 防御和胁迫 TC-rich repeats | 水杨酸 TCA | 生长素 TGA | 病原菌 W-box |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MdNLP1 | 0/1 | 2/0 | 1/1 | 1/1 | 0/1 | 1/0 | 1/0 | 0/1 | |||
| MdNLP2 | 2/3 | 1/0 | 3/0 | 0/1 | 0/1 | 0/1 | |||||
| MdNLP3 | 0/2 | 0/3 | 0/1 | 0/1 | 0/1 | 0/1 | 1/0 | 0/1 | |||
| MdNLP4 | 0/2 | 0/3 | 1/0 | 1/1 | 1/1 | 1/1 | 0/1 | ||||
| MdNLP5 | 1/1 | 2/0 | 1/2 | 0/1 | 0/1 | 1/0 | 0/1 | 0/1 | |||
| MdNLP6 | 4/3 | 1/0 | 3/3 | 1/0 | 0/1 |
新窗口打开|下载CSV
Table 6
表6
表6推测与苹果NLP相关的响应氮素的miRNA
Table 6
| 靶基因 Target gene | miRNA | 抑制类型 Inhibition | 靶基因 Target gene | miRNA | 抑制类型 Inhibition | 靶基因 Target gene | miRNA | 抑制类型 Inhibition | ||
|---|---|---|---|---|---|---|---|---|---|---|
| MdNLP2 | mdm-miR171a | 直接降解Cleavage | MdNLP5 | mdm-miR169e | 直接降解Cleavage | MdNLP5 | mdm-miR395e | 直接降解Cleavage | ||
| mdm-miR171b | 直接降解Cleavage | mdm-miR169f | 直接降解Cleavage | mdm-miR395f | 直接降解Cleavage | |||||
| mdm-miR395a | 直接降解Cleavage | mdm-miR171c | 抑制翻译Translation | mdm-miR395g | 直接降解Cleavage | |||||
| mdm-miR395b | 直接降解Cleavage | mdm-miR171d | 抑制翻译Translation | mdm-miR395h | 直接降解Cleavage | |||||
| mdm-miR395c | 直接降解Cleavage | mdm-miR171e | 抑制翻译Translation | mdm-miR395i | 直接降解Cleavage | |||||
| mdm-miR395d | 直接降解Cleavage | mdm-miR171g | 抑制翻译Translation | |||||||
| mdm-miR395e | 直接降解Cleavage | mdm-miR171h | 抑制翻译Translation | |||||||
| mdm-miR395f | 直接降解Cleavage | mdm-miR395a | 直接降解Cleavage | |||||||
| mdm-miR395g | 直接降解Cleavage | mdm-miR395b | 直接降解Cleavage | |||||||
| mdm-miR395h | 直接降解Cleavage | mdm-miR395c | 直接降解Cleavage | |||||||
| mdm-miR395i | 直接降解Cleavage | mdm-miR395d | 直接降解Cleavage |
新窗口打开|下载CSV
2.9 苹果NLP转录因子家族基因表达模式
为了研究苹果NLP家族基因的时空表达规律,对苹果NLP的组织表达模式进行了定量分析。根据图6结果可知,苹果NLP基因家族成员不同发育时期的表达特征稍有差别,但大致相似。所验证的4个NLP(由于MdNLP1结构域缺失、MdNLP6非染色体定位,此处没有检测,后文定量研究均采用上述考虑)在根、茎、叶、花、果中均有不同程度的表达,明显可见苹果中NLP家族基因更偏向在叶片、茎等营养器官中表达,MdNLP2、MdNLP3、MdNLP4、MdNLP5基因表达量最高的部位分别是茎、叶片、叶片、叶片。图6
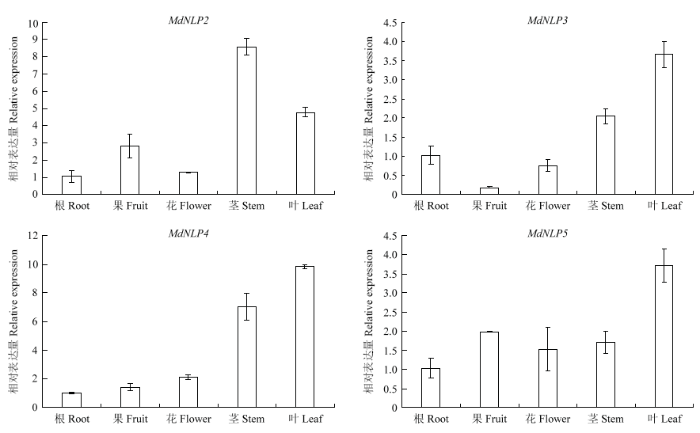 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6苹果NLP组织表达分析
Fig. 6Tissue expression analysis of NLP gene in apple
为了研究氮素匮乏条件下MdNLP的响应过程,对‘平邑甜茶’实生苗进行氮饥饿处理,结果发现(图7),在所取的氮饥饿处理的几个时间节点,所检测的4个基因,相较于最初的0 h,基因表达量均有不同程度的提高,表达变化趋势大致相似,先升高后降低。
图7
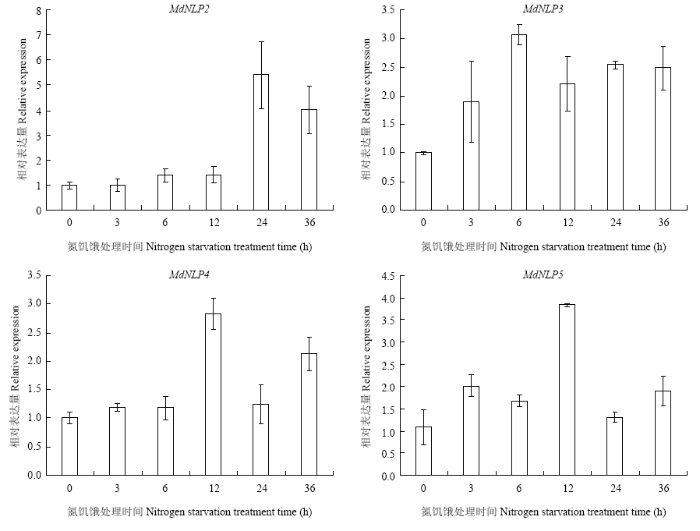 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7苹果NLP氮饥饿响应表达分析
Fig. 7Expression analysis of nitrogen starvation response of MdNLP gene
由于NLP转录因子可能与植物的抗旱性有关[7],对此,本研究进行了苹果NLP干旱胁迫表达分析。由图8可以发现,除了MdNLP4,其余3个基因(MdNLP2、MdNLP3、MdNLP5)在研究的时间范围内,随着干旱胁迫时间的增加,基因的表达量均呈现出先升高,到6 h表达量达最高,之后降低的一种表达情况。
图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8苹果NLP非生物胁迫(干旱处理和ABA处理)表达分析
Fig. 8Expression analysis of abiotic stress (drought treatment and ABA treatment) of MdNLP gene
除此之外,在对苹果NLP启动子进行分析时,找到大量与激素相关的调控元件,本研究还分析了ABA处理下苹果NLP的表达模式,结果如图8所示。MdNLP2、MdNLP3在ABA诱导下表达量总体上调,而MdNLP4、MdNLP5表达量则呈现总体下调的趋势。
3 讨论
一直以来,转录因子家族的研究都是一个热点话题,这是因为功能基因调控机制的阐释离不开转录因子。例如对成员数目较多的一些转录因子家族的分析:MYB[29]、NAC[30]、bHLH[31]、bZIP[32]、WRKY[33]、LBD[34]。本研究对一个响应氮饥饿的Nin-like转录因子家族在苹果全基因组中进行了鉴定,并且利用生物信息学的手段,系统分析了此类转录因子的基因与蛋白特征以及不同处理下的表达水平。NLP转录因子的功能已经进行了大量研究,除了已知的响应硝酸盐信号,还具有调节干旱胁迫抗性[35]的特点,另外有研究发现Nin通过与NLP相互作用介导硝酸盐对结瘤的抑制作用[36],GUAN等[37]证明NLP6/7与TCP20的相互作用可以促进N饥饿条件下根分生组织的生长。
本研究基于已公开发表的苹果基因组测序数据[13],一共识别到了6个苹果NLP基因家族成员,已知拟南芥中有9个NLP[3],水稻中有6个[3],GE等[5]鉴定了9个玉米NLP,曹雄军等[7]在甜橙蛋白质组数据中筛选到4个甜橙NLP,由此可见在不同物种中NLP数量存在差异。通过将拟南芥和苹果NLP蛋白的氨基酸序列进行多序列比对,发现在保守结构域RWP-RK附近除个别氨基酸位点外,其余位置保守程度很高,这一结构域对DNA结合很重要,另外位于C末端的PB1结构域相对较为保守,比较特殊的是,MdNLP1(MDP0000788505)C端缺失,关于N端附近的GAF-like结构域,朱新宇等[38]通过比较NLP与Nin的GAF区段二、三级结构,发现该区域存在变异,导致它们可能执行不同功能。
拟南芥中已知,NLP6和NLP7通过感知硝酸盐信号后活性被诱导,进而结合到具有硝酸盐响应元件(nitrate-responsive elements,NREs)的一系列和硝酸盐转运、同化相关的基因上,包括高亲和力硝酸盐转运基因NRT2.1、亚硝酸还原酶NIR1等基因的启动子上,硝酸还原酶NIA1的3′侧翼区等[39]。KONISHI等[40]报道两类转录因子基因LBD37-39和NIGT1也可能是NLPs的靶基因,它们可能在响应硝酸盐过程中触发二级转录事件。本次将苹果NLP蛋白映射到模式植物拟南芥中,同样地发现了这些与NLP相互联系的基因,其中涉及硝酸盐转运、同化以及受氮诱导调控、影响氮素代谢的部分基因。类比拟南芥NLP6和NLP7,苹果中MDP0000788505、MDP0000132856在关联网络中处于关键节点位置,推测也具有非常重要的功能。以MdNLP作靶基因,对其潜在microRNA进行预测,找到了大量的可能与其相互作用的miRNA,参考其他物种中的miRNA的作用规律,有意思的是在本研究发现了3个可能靶向于MdNLP并且响应硝酸盐的miRNA家族,分别是mdm-miR169、mdm-miR171和mdm-miR395,其中2个miR171成员和9个miR395靶向作用于MdNLP2,2个miRNA169、5个miRNA171和9个miRNA395成员靶向作用于MdNLP5,苹果中这些可能的调控作用有待进一步验证。
关于NLP的组织表达模式,CHARDIN等[25]通过分析GENEVESTIGATOR数据库中有关拟南芥和水稻不同组织中的NLP表达水平,发现NLP分布广泛,几乎分布于所有的已检测组织中(包括拟南芥的幼苗、根、茎、花,水稻的叶、节、花序等),其中在拟南芥中,AtNLP8和AtNLP9在衰老的叶片和种子中偏向表达,在水稻中,OsNLP1和OsNLP3偏向在源组织中表达;吴翔宇等[6]对毛果杨NLP表达芯片数据进行提取,结果显示毛果杨NLP偏向在嫩叶、根中表达,除此之外,部分NLP家族成员还在木质部这一输导组织中有表达。与以上研究结果类似,本研究检测的4个MdNLP几乎分布于苹果的根、茎、叶、花、果各组织中,不同部位表达存在较明显的差异,但更偏向于在茎、叶等器官中表达,这一特征可能与其参与转运硝酸盐的功能紧密联系。同时,苹果NLP相较于表达量较高的茎组织而言,在根中的表达要低得多,说明苹果NLP发挥作用涉及到硝酸盐的转运阶段,而不是硝酸盐的吸收过程,与KUMAR等[4]在小麦NLP中的研究观点相吻合。
在氮饥饿处理条件下,苹果NLP表达呈现先升高后降低的趋势(0—36 h),而LIU等[8]对甘蓝型油菜中NLP进行氮饥饿响应分析,发现在0—72 h阶段内随着时间延长,甘蓝型油菜幼苗叶片和根的BnaNLP表达主要存在两种趋势:逐渐上升和逐渐下降。由此可见,苹果NLP与甘蓝型油菜NLP的氮饥饿响应过程存在差异,可能是由于采样部位不同造成(本研究整株取样,甘蓝型油菜一文中为叶片和根分别取样),亦或是不同物种相同基因的表达存在差异。
对苹果苗干旱胁迫处理,发现3个苹果NLP(MdNLP2、MdNLP3、MdNLP5)随着干旱胁迫时间的延长,基因相对表达量逐渐增加,增大到最大值(6 h)后逐渐减少,这与曹雄军等[7]研究干旱条件下,在枳叶片中的NLP表达变化模式相似。氮饥饿处理和非生物胁迫处理结果表明,苹果NLP转录因子响应氮饥饿过程、参与干旱胁迫进程等,由此可见,MdNLP在氮代谢与逆境响应环节均发挥了重要的作用。考虑到MDP0000132856与拟南芥NLP7的高同源性,互作网络暗示其可能具有类似的调控关系,下一步可以围绕该基因重点展开研究。
4 结论
从苹果全部蛋白质序列鉴定MdNLP转录因子家族成员6个,启动子和miRNA预测分析表明它们响应氮素,并可能被其他基因或小分子RNA所调控。组织表达模式分析观察到苹果NLP偏向表达于茎、叶等地上部的营养器官,这也说明NLP蛋白在硝酸盐转运过程中的重要功能;氮饥饿响应表达分析,也再次证实苹果NLP转录因子参与到氮素的调节过程。综合比较发现苹果NLP不同的成员蛋白结构和表达模式大致相似,但存在部分差异,推测在响应硝酸盐的过程中扮演着不同但又有关联的角色。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/ncomms2621URLPMID:23511481 [本文引用: 1]

In plants, nitrate is not only a major nitrogen source but also a signalling molecule that modulates the expression of a wide range of genes and that regulates growth and development. The critical role of nitrate as a signalling molecule has been established for several decades. However, the molecular mechanisms underlying the nitrate response have remained elusive, as the transcription factor that primarily responds to nitrate signals has not yet been identified. Here we show that Arabidopsis NIN-LIKE PROTEIN (NLP) family proteins bind the nitrate-responsive cis-element and activate nitrate-responsive cis-element-dependent and nitrate-responsive transcription. Our results also suggest that the activity of NLPs is post-translationally modulated by nitrate signalling. Furthermore, the suppression of NLP function impairs the nitrate-inducible expression of a number of genes and causes severe growth inhibition. These results indicate that NLPs are the transcription factors mediating the nitrate signal and thereby function as master regulators of the nitrate response.
DOI:10.1038/46058URLPMID:10647012 [本文引用: 1]
Symbiotic nitrogen-fixing root nodules on legumes are founded by root cortical cells that de-differentiate and restart cell division to establish nodule primordia. Bacterial microsymbionts invade these primordia through infection threads laid down by the plant and, after endocytosis, membrane-enclosed bacteroids occupy cells in the nitrogen-fixing tissue of functional nodules. The bacteria excrete lipochitin oligosaccharides, triggering a developmental process that is controlled by the plant and can be suppressed. Nodule inception initially relies on cell competence in a narrow infection zone located just behind the growing root tip. Older nodules then regulate the number of nodules on a root system by suppressing the development of nodule primordia. To identify the regulatory components that act early in nodule induction, we characterized a transposon-tagged Lotus japonicus mutant, nin (for nodule inception), arrested at the stage of bacterial recognition. We show that nin is required for the formation of infection threads and the initiation of primordia. NIN protein has regional similarity to transcription factors, and the predicted DNA-binding/dimerization domain identifies and typifies a consensus motif conserved in plant proteins with a function in nitrogen-controlled development.
DOI:10.1007/s00239-004-0144-2URL [本文引用: 6]
Genetic studies in Lotus japonicus and pea have identified Nin as a core symbiotic gene required for establishing symbiosis between legumes and nitrogen fixing bacteria collectively called Rhizobium. Sequencing of additional Lotus cDNAs combined with analysis of genome sequences from Arabidopsis and rice reveals that Nin homologues in all three species constitute small gene families. In total, the Arabidopsis and rice genomes encode nine and three NIN-like proteins (NLPs), respectively. We present here a bioinformatics analysis and prediction of NLP evolution. On a genome scale we show that in Arabidopsis, this family has evolved through segmental duplication rather than through tandem amplification. Alignment of all predicted NLP protein sequences shows a composition with six conserved modules. In addition, Lotus and pea NLPs contain segments that might characterize NIN proteins of legumes and be of importance for their function in symbiosis. The most conserved region in NLPs, the RWP-RK domain, has secondary structure predictions consistent with DNA binding properties. This motif is shared by several other small proteins in both Arabidopsis and rice. In rice, the RWP-RK domain sequences have diversified significantly more than in Arabidopsis. Database searches reveal that, apart from its presence in Arabidopsis and rice, the motif is also found in the algae Chlamydomonas and in the slime mold Dictyostelium discoideum. Thus, the origin of this putative DNA binding region seems to predate the fungus–plant divide.
DOI:10.1371/journal.pone.0208409URLPMID:30540790 [本文引用: 3]
RWP-RKs represent a small family of transcription factors (TFs) that are unique to plants and function particularly under conditions of nitrogen starvation. These RWP-RKs have been classified in two sub-families, NLPs (NIN-like proteins) and RKDs (RWP-RK domain proteins). NLPs regulate tissue-specific expression of genes involved in nitrogen use efficiency (NUE) and RKDs regulate expression of genes involved in gametogenesis/embryogenesis. During the present study, using in silico approach, 37 wheat RWP-RK genes were identified, which included 18 TaNLPs (2865 to 7340 bp with 4/5 exons), distributed on 15 chromosomes from 5 homoeologous groups (with two genes each on 4B,4D and 5A) and 19 TaRKDs (1064 to 5768 bp with 1 to 6 exons) distributed on 12 chromosomes from 4 homoeologous groups (except groups 1, 4 and 5); 2-3 splice variants were also available in 9 of the 37 genes. Sixteen (16) of these genes also carried 24 SSRs (simple sequence repeats), while 11 genes had targets for 13 different miRNAs. At the protein level, MD simulation analysis suggested their interaction with nitrate-ions. Significant differences were observed in the expression of only two (TaNLP1 and TaNLP2) of the nine representative genes that were used for in silico expression analysis under varying levels of N at post-anthesis stage (data for other genes was not available for in silico expression analysis). Differences in expression were also observed during qRT-PCR, when expression of four representative genes (TaNLP2, TaNLP7, TaRKD6 and TaRKD9) was examined in roots and shoots of seedlings (under different conditions of N supply) in two contrasting genotypes which differed in NUE (C306 with low NUE and HUW468 with high NUE). These four genes for qRT-PCR were selected on the basis of previous literature, level of homology and the level of expression (in silico study). In particular, the TaNLP7 gene showed significant up-regulation in the roots and shoots of HUW468 (with higher NUE) during N-starvation; this gene has already been characterized in Arabidopsis and tobacco, and is known to be involved in nitrate-signal transduction pathway.
DOI:10.1007/s10725-017-0324-xURL [本文引用: 2]
URL [本文引用: 2]
NLP gene family is a special transcription factor, the initiation of nodule development is dependent on the function of this gene family, in other species plant it has the function of regulating plant nitrate absorption and assimilation. Genome-wide analysis in Populus trichocarpa had identified 14 NLP gene family members. These members have the characteristics of low hydrophilic, the conservative gene structure and contain RWP-RK and PB1 two conservative domain. The localization of all members of NLP genes in P.trichocarpa was predicted in the nucleus. Evolution analysis showed that NLP gene family members experience strict filter. Chromosome localization analysis showed that the members of P.trichocarpa NLP genes were located on 9 chromosomes, the expanding of NLPs in P.trichocarpa was caused by Salicaceae duplication events. Microarray data analysis showed that NLPs had a high transcript accumulation in leaf, root and male catkins, some genes were expressed in xylem, seed and female catkins, but no gene was detected in the mature leaf.
URL [本文引用: 2]
NLP gene family is a special transcription factor, the initiation of nodule development is dependent on the function of this gene family, in other species plant it has the function of regulating plant nitrate absorption and assimilation. Genome-wide analysis in Populus trichocarpa had identified 14 NLP gene family members. These members have the characteristics of low hydrophilic, the conservative gene structure and contain RWP-RK and PB1 two conservative domain. The localization of all members of NLP genes in P.trichocarpa was predicted in the nucleus. Evolution analysis showed that NLP gene family members experience strict filter. Chromosome localization analysis showed that the members of P.trichocarpa NLP genes were located on 9 chromosomes, the expanding of NLPs in P.trichocarpa was caused by Salicaceae duplication events. Microarray data analysis showed that NLPs had a high transcript accumulation in leaf, root and male catkins, some genes were expressed in xylem, seed and female catkins, but no gene was detected in the mature leaf.
DOI:10.3864/j.issn.0578-1752.2016.02.018URL [本文引用: 4]
【Objective】The objective of this study was to analyze the expression of Poncirus trifoliata (L.) Raf. NIN-like transcription factors to discuss the regulation mechanism of nitrogen assimilation under different water conditions in citrus.【Method】 Primers were designed to amplify the sequences of the NLP genes CDS of Poncirus trifoliate (L.) Raf.based on orange genome databases and molecular biology. Alignment of the sequences was performed using ClustalX and phylogenetic analysis of this alignment was conducted using MEGA. Analysis of the Relative NLP genes expression data under different water conditions using Real-Time Quantitative PCR.【Result】Four Poncirus trifoliata (L.) Raf. NLP genes: PtNLP2, PtNLP4, PtNLP7 and PtNLP8 were obtained. The sequence alignment analysis showed that the identity of all NIN-like proteins was 45.13%, and possessed both the RWP-RK and PB1 domain, and all NIN-like proteins were high identity to sweet orange, respectively 97.57%, 96.47%, 99% and 97.33%. Phylogenic analysis has indicated that the 4 Poncirus trifoliata (L.) Raf. NLP genes can be classified into four groups with Arabidopsis (PtNLP2 and AtNLP1/2, PtNLP4 and AtNLP4/5, PtNLP7 and AtNLP6/7, PtNLP8 and AtNLP8/9). There were differences in the expression pattern of NLP genes in Poncirus trifoliata (L.) Raf. leaves and roots. The results indicated that with the decrease of soil water, the expression level of NLP genes in Poncirus trifoliata (L.) Raf. leaves were up-regulated. The PtNLP2, PtNLP4, PtNLP7 and PtNLP8 expression level were the highest when the relative water capacity was 15.4%, they were 2.9, 3.5, 5.9 and 2.8 folds higher respectively compared to the control (the relative water holding capacity was 61.0%). After that, the gene expression level began to down-regulate and had no statistically significant difference between the control and the relative water holding capacity of 9.4%. While the expression level of NLP genes in Poncirus trifoliata (L.) Raf. roots of control were the highest, and the gene expression pattern showed a down-regulated trend accompanied by the loss of soil water and the difference was statistically significant. The PtNLP2, PtNLP4, PtNLP7 and PtNLP8 expression level of roots were maximum down-regulated by 6.7, 2.8, 4.8 and 2.3 folds respectively compared with the control. The expression of NLP genes in Poncirus trifoliata (L.) Raf. leaves and roots remained at the low expression level after rewatering and had statistically significant difference compared with the control.【Conclusion】The expression of Poncirus trifoliata (L.) Raf. NLP genes is closely related to the water condition of soil. The expression level of NLP genes in Poncirus trifoliata (L.) Raf. leaves were up-regulated by drought stress, and then down-regulated when the water deficit was very serious. But the expression level of NLP genes in Poncirus trifoliata (L.) Raf. roots were down-regulated continuously accompanied by the loss of soil water, and expression of the PtNLP2 and PtNLP7 had a great change in roots.
DOI:10.3864/j.issn.0578-1752.2016.02.018URL [本文引用: 4]
【Objective】The objective of this study was to analyze the expression of Poncirus trifoliata (L.) Raf. NIN-like transcription factors to discuss the regulation mechanism of nitrogen assimilation under different water conditions in citrus.【Method】 Primers were designed to amplify the sequences of the NLP genes CDS of Poncirus trifoliate (L.) Raf.based on orange genome databases and molecular biology. Alignment of the sequences was performed using ClustalX and phylogenetic analysis of this alignment was conducted using MEGA. Analysis of the Relative NLP genes expression data under different water conditions using Real-Time Quantitative PCR.【Result】Four Poncirus trifoliata (L.) Raf. NLP genes: PtNLP2, PtNLP4, PtNLP7 and PtNLP8 were obtained. The sequence alignment analysis showed that the identity of all NIN-like proteins was 45.13%, and possessed both the RWP-RK and PB1 domain, and all NIN-like proteins were high identity to sweet orange, respectively 97.57%, 96.47%, 99% and 97.33%. Phylogenic analysis has indicated that the 4 Poncirus trifoliata (L.) Raf. NLP genes can be classified into four groups with Arabidopsis (PtNLP2 and AtNLP1/2, PtNLP4 and AtNLP4/5, PtNLP7 and AtNLP6/7, PtNLP8 and AtNLP8/9). There were differences in the expression pattern of NLP genes in Poncirus trifoliata (L.) Raf. leaves and roots. The results indicated that with the decrease of soil water, the expression level of NLP genes in Poncirus trifoliata (L.) Raf. leaves were up-regulated. The PtNLP2, PtNLP4, PtNLP7 and PtNLP8 expression level were the highest when the relative water capacity was 15.4%, they were 2.9, 3.5, 5.9 and 2.8 folds higher respectively compared to the control (the relative water holding capacity was 61.0%). After that, the gene expression level began to down-regulate and had no statistically significant difference between the control and the relative water holding capacity of 9.4%. While the expression level of NLP genes in Poncirus trifoliata (L.) Raf. roots of control were the highest, and the gene expression pattern showed a down-regulated trend accompanied by the loss of soil water and the difference was statistically significant. The PtNLP2, PtNLP4, PtNLP7 and PtNLP8 expression level of roots were maximum down-regulated by 6.7, 2.8, 4.8 and 2.3 folds respectively compared with the control. The expression of NLP genes in Poncirus trifoliata (L.) Raf. leaves and roots remained at the low expression level after rewatering and had statistically significant difference compared with the control.【Conclusion】The expression of Poncirus trifoliata (L.) Raf. NLP genes is closely related to the water condition of soil. The expression level of NLP genes in Poncirus trifoliata (L.) Raf. leaves were up-regulated by drought stress, and then down-regulated when the water deficit was very serious. But the expression level of NLP genes in Poncirus trifoliata (L.) Raf. roots were down-regulated continuously accompanied by the loss of soil water, and expression of the PtNLP2 and PtNLP7 had a great change in roots.
DOI:10.1016/j.gene.2019.144275URLPMID:31809843 [本文引用: 2]
Major latex protein/ripening-related proteins (MLP/RRP) subfamily are a class of proteins that play crucial roles in response to defense and stress response. However, their biological function is still not clear, the identification and characterization will provide essential information for understanding their roles. Here, we carried out a genome-wide evolutionary characteristics and gene expression analysis of the MLP family in apple (Malus domestica, Borkh.). A total of 36 MdMLP genes were screened in apple genome. They were uneven located on 5 chromosomes, where were mainly arranged in tandem clusters, and the phylogenetic analysis put forward further views on the evolutionary relationship and putative functions among the genes. The conserved motifs showed that the MLP proteins which contained motif 1 had the potential function, and tissue-specific expression analysis showed that apple MLP members had diverse biological roles. Furthermore, the results showed seven of the MdMLPs that harbored cis-acting regulatory elements in response to defense and stress, and our expression data proved that they were involved in biotic stresses. The present study provides new views to the evolution and regulation of MdMLP genes, which represent objectives of future research and incorporate in resistance-related molecular breeding projects.
DOI:10.1038/ncomms2650URLPMID:23591880 [本文引用: 2]
Nitrate is both an important nutrient and a signalling molecule for plants. Although several components of the nitrate signalling pathway have been identified, their hierarchical organization remains unclear. Here we show that the localization of NLP7, a member of the RWP-RK transcription factor family, is regulated by nitrate via a nuclear retention mechanism. Genome-wide analyses revealed that NLP7 binds and modulates a majority of known nitrate signalling and assimilation genes. Our findings indicate that plants, like fungi and mammals, rely on similar nuclear retention mechanisms to instantaneously respond to the availability of key nutrients.
DOI:10.1038/ncomms13179URLPMID:27731416 [本文引用: 1]
Seeds respond to multiple different environmental stimuli that regulate germination. Nitrate stimulates germination in many plants but how it does so remains unclear. Here we show that the Arabidopsis NIN-like protein 8 (NLP8) is essential for nitrate-promoted seed germination. Seed germination in nlp8 loss-of-function mutants does not respond to nitrate. NLP8 functions even in a nitrate reductase-deficient mutant background, and the requirement for NLP8 is conserved among Arabidopsis accessions. NLP8 reduces abscisic acid levels in a nitrate-dependent manner and directly binds to the promoter of CYP707A2, encoding an abscisic acid catabolic enzyme. Genetic analysis shows that NLP8-mediated promotion of seed germination by nitrate requires CYP707A2. Finally, we show that NLP8 localizes to nuclei and unlike NLP7, does not appear to be activated by nitrate-dependent nuclear retention of NLP7, suggesting that seeds have a unique mechanism for nitrate signalling.
DOI:10.1038/nature22077URLPMID:28489820 [本文引用: 1]
Nutrient signalling integrates and coordinates gene expression, metabolism and growth. However, its primary molecular mechanisms remain incompletely understood in plants and animals. Here we report unique Ca2+ signalling triggered by nitrate with live imaging of an ultrasensitive biosensor in Arabidopsis leaves and roots. A nitrate-sensitized and targeted functional genomic screen identifies subgroup III Ca2+-sensor protein kinases (CPKs) as master regulators that orchestrate primary nitrate responses. A chemical switch with the engineered mutant CPK10(M141G) circumvents embryo lethality and enables conditional analyses of cpk10 cpk30 cpk32 triple mutants to define comprehensive nitrate-associated regulatory and developmental programs. Nitrate-coupled CPK signalling phosphorylates conserved NIN-LIKE PROTEIN (NLP) transcription factors to specify the reprogramming of gene sets for downstream transcription factors, transporters, nitrogen assimilation, carbon/nitrogen metabolism, redox, signalling, hormones and proliferation. Conditional cpk10 cpk30 cpk32 and nlp7 mutants similarly impair nitrate-stimulated system-wide shoot growth and root establishment. The nutrient-coupled Ca2+ signalling network integrates transcriptome and cellular metabolism with shoot-root coordination and developmental plasticity in shaping organ biomass and architecture.
DOI:10.1038/srep27795URLPMID:27293103 [本文引用: 1]
Nitrogen is essential for plant survival and growth. Excessive application of nitrogenous fertilizer has generated serious environment pollution and increased production cost in agriculture. To deal with this problem, tremendous efforts have been invested worldwide to increase the nitrogen use ability of crops. However, only limited success has been achieved to date. Here we report that NLP7 (NIN-LIKE PROTEIN 7) is a potential candidate to improve plant nitrogen use ability. When overexpressed in Arabidopsis, NLP7 increases plant biomass under both nitrogen-poor and -rich conditions with better-developed root system and reduced shoot/root ratio. NLP7-overexpressing plants show a significant increase in key nitrogen metabolites, nitrogen uptake, total nitrogen content, and expression levels of genes involved in nitrogen assimilation and signalling. More importantly, overexpression of NLP7 also enhances photosynthesis rate and carbon assimilation, whereas knockout of NLP7 impaired both nitrogen and carbon assimilation. In addition, NLP7 improves plant growth and nitrogen use in transgenic tobacco (Nicotiana tabacum). Our results demonstrate that NLP7 significantly improves plant growth under both nitrogen-poor and -rich conditions by coordinately enhancing nitrogen and carbon assimilation and sheds light on crop improvement.
DOI:10.1038/ng.654URLPMID:20802477 [本文引用: 2]
We report a high-quality draft genome sequence of the domesticated apple (Malus × domestica). We show that a relatively recent (>50 million years ago) genome-wide duplication (GWD) has resulted in the transition from nine ancestral chromosomes to 17 chromosomes in the Pyreae. Traces of older GWDs partly support the monophyly of the ancestral paleohexaploidy of eudicots. Phylogenetic reconstruction of Pyreae and the genus Malus, relative to major Rosaceae taxa, identified the progenitor of the cultivated apple as M. sieversii. Expansion of gene families reported to be involved in fruit development may explain formation of the pome, a Pyreae-specific false fruit that develops by proliferation of the basal part of the sepals, the receptacle. In apple, a subclade of MADS-box genes, normally involved in flower and fruit development, is expanded to include 15 members, as are other gene families involved in Rosaceae-specific metabolism, such as transport and assimilation of sorbitol.
DOI:10.1093/nar/gky1000URLPMID:30357347 [本文引用: 1]
The Genome Database for Rosaceae (GDR, https://www.rosaceae.org) is an integrated web-based community database resource providing access to publicly available genomics, genetics and breeding data and data-mining tools to facilitate basic, translational and applied research in Rosaceae. The volume of data in GDR has increased greatly over the last 5?years. The GDR now houses multiple versions of whole genome assembly and annotation data from 14 species, made available by recent advances in sequencing technology. Annotated and searchable reference transcriptomes, RefTrans, combining peer-reviewed published RNA-Seq as well as EST datasets, are newly available for major crop species. Significantly more quantitative trait loci, genetic maps and markers are available in MapViewer, a new visualization tool that better integrates with other pages in GDR. Pathways can be accessed through the new GDR Cyc Pathways databases, and synteny among the newest genome assemblies from eight species can be viewed through the new synteny browser, SynView. Collated single-nucleotide polymorphism diversity data and phenotypic data from publicly available breeding datasets are integrated with other relevant data. Also, the new Breeding Information Management System allows breeders to upload, manage and analyze their private breeding data within the secure GDR server with an option to release data publicly.
DOI:10.1093/nar/gkt1223URLPMID:24288371 [本文引用: 1]
Pfam, available via servers in the UK (http://pfam.sanger.ac.uk/) and the USA (http://pfam.janelia.org/), is a widely used database of protein families, containing 14 831 manually curated entries in the current release, version 27.0. Since the last update article 2 years ago, we have generated 1182 new families and maintained sequence coverage of the UniProt Knowledgebase (UniProtKB) at nearly 80%, despite a 50% increase in the size of the underlying sequence database. Since our 2012 article describing Pfam, we have also undertaken a comprehensive review of the features that are provided by Pfam over and above the basic family data. For each feature, we determined the relevance, computational burden, usage statistics and the functionality of the feature in a website context. As a consequence of this review, we have removed some features, enhanced others and developed new ones to meet the changing demands of computational biology. Here, we describe the changes to Pfam content. Notably, we now provide family alignments based on four different representative proteome sequence data sets and a new interactive DNA search interface. We also discuss the mapping between Pfam and known 3D structures.
DOI:10.1093/molbev/msw054URLPMID:27004904 [本文引用: 1]
We present the latest version of the Molecular Evolutionary Genetics Analysis (Mega) software, which contains many sophisticated methods and tools for phylogenomics and phylomedicine. In this major upgrade, Mega has been optimized for use on 64-bit computing systems for analyzing larger datasets. Researchers can now explore and analyze tens of thousands of sequences in Mega The new version also provides an advanced wizard for building timetrees and includes a new functionality to automatically predict gene duplication events in gene family trees. The 64-bit Mega is made available in two interfaces: graphical and command line. The graphical user interface (GUI) is a native Microsoft Windows application that can also be used on Mac OS X. The command line Mega is available as native applications for Windows, Linux, and Mac OS X. They are intended for use in high-throughput and scripted analysis. Both versions are available from www.megasoftware.net free of charge.
DOI:10.1093/nar/gkl198URLPMID:16845028 [本文引用: 1]
MEME (Multiple EM for Motif Elicitation) is one of the most widely used tools for searching for novel 'signals' in sets of biological sequences. Applications include the discovery of new transcription factor binding sites and protein domains. MEME works by searching for repeated, ungapped sequence patterns that occur in the DNA or protein sequences provided by the user. Users can perform MEME searches via the web server hosted by the National Biomedical Computation Resource (http://meme.nbcr.net) and several mirror sites. Through the same web server, users can also access the Motif Alignment and Search Tool to search sequence databases for matches to motifs encoded in several popular formats. By clicking on buttons in the MEME output, users can compare the motifs discovered in their input sequences with databases of known motifs, search sequence databases for matches to the motifs and display the motifs in various formats. This article describes the freely accessible web server and its architecture, and discusses ways to use MEME effectively to find new sequence patterns in biological sequences and analyze their significance.
DOI:10.1038/nprot.2015.053URLPMID:25950237 [本文引用: 1]
Phyre2 is a suite of tools available on the web to predict and analyze protein structure, function and mutations. The focus of Phyre2 is to provide biologists with a simple and intuitive interface to state-of-the-art protein bioinformatics tools. Phyre2 replaces Phyre, the original version of the server for which we previously published a paper in Nature Protocols. In this updated protocol, we describe Phyre2, which uses advanced remote homology detection methods to build 3D models, predict ligand binding sites and analyze the effect of amino acid variants (e.g., nonsynonymous SNPs (nsSNPs)) for a user's protein sequence. Users are guided through results by a simple interface at a level of detail they determine. This protocol will guide users from submitting a protein sequence to interpreting the secondary and tertiary structure of their models, their domain composition and model quality. A range of additional available tools is described to find a protein structure in a genome, to submit large number of sequences at once and to automatically run weekly searches for proteins that are difficult to model. The server is available at http://www.sbg.bio.ic.ac.uk/phyre2. A typical structure prediction will be returned between 30 min and 2 h after submission.
DOI:10.1104/pp.110.156851URLPMID:20647376 [本文引用: 1]
A complete map of the Arabidopsis (Arabidopsis thaliana) proteome is clearly a major goal for the plant research community in terms of determining the function and regulation of each encoded protein. Developing genome-wide prediction tools such as for localizing gene products at the subcellular level will substantially advance Arabidopsis gene annotation. To this end, we performed a comprehensive study in Arabidopsis and created an integrative support vector machine-based localization predictor called AtSubP (for Arabidopsis subcellular localization predictor) that is based on the combinatorial presence of diverse protein features, such as its amino acid composition, sequence-order effects, terminal information, Position-Specific Scoring Matrix, and similarity search-based Position-Specific Iterated-Basic Local Alignment Search Tool information. When used to predict seven subcellular compartments through a 5-fold cross-validation test, our hybrid-based best classifier achieved an overall sensitivity of 91% with high-confidence precision and Matthews correlation coefficient values of 90.9% and 0.89, respectively. Benchmarking AtSubP on two independent data sets, one from Swiss-Prot and another containing green fluorescent protein- and mass spectrometry-determined proteins, showed a significant improvement in the prediction accuracy of species-specific AtSubP over some widely used "general" tools such as TargetP, LOCtree, PA-SUB, MultiLoc, WoLF PSORT, Plant-PLoc, and our newly created All-Plant method. Cross-comparison of AtSubP on six nontrained eukaryotic organisms (rice [Oryza sativa], soybean [Glycine max], human [Homo sapiens], yeast [Saccharomyces cerevisiae], fruit fly [Drosophila melanogaster], and worm [Caenorhabditis elegans]) revealed inferior predictions. AtSubP significantly outperformed all the prediction tools being currently used for Arabidopsis proteome annotation and, therefore, may serve as a better complement for the plant research community. A supplemental Web site that hosts all the training/testing data sets and whole proteome predictions is available at http://bioinfo3.noble.org/AtSubP/.
DOI:10.1093/nar/gky1131URLPMID:30476243 [本文引用: 1]
Proteins and their functional interactions form the backbone of the cellular machinery. Their connectivity network needs to be considered for the full understanding of biological phenomena, but the available information on protein-protein associations is incomplete and exhibits varying levels of annotation granularity and reliability. The STRING database aims to collect, score and integrate all publicly available sources of protein-protein interaction information, and to complement these with computational predictions. Its goal is to achieve a comprehensive and objective global network, including direct (physical) as well as indirect (functional) interactions. The latest version of STRING (11.0) more than doubles the number of organisms it covers, to 5090. The most important new feature is an option to upload entire, genome-wide datasets as input, allowing users to visualize subsets as interaction networks and to perform gene-set enrichment analysis on the entire input. For the enrichment analysis, STRING implements well-known classification systems such as Gene Ontology and KEGG, but also offers additional, new classification systems based on high-throughput text-mining as well as on a hierarchical clustering of the association network itself. The STRING resource is available online at https://string-db.org/.
DOI:10.1093/bioinformatics/btu817URLPMID:25504850 [本文引用: 1]
: Visualizing genes' structure and annotated features helps biologists to investigate their function and evolution intuitively. The Gene Structure Display Server (GSDS) has been widely used by more than 60?000 users since its first publication in 2007. Here, we reported the upgraded GSDS 2.0 with a newly designed interface, supports for more types of annotation features and formats, as well as an integrated visual editor for editing the generated figure. Moreover, a user-specified phylogenetic tree can be added to facilitate further evolutionary analysis. The full source code is also available for downloading.
DOI:10.1093/nar/30.1.325URLPMID:11752327 [本文引用: 1]
PlantCARE is a database of plant cis-acting regulatory elements, enhancers and repressors. Regulatory elements are represented by positional matrices, consensus sequences and individual sites on particular promoter sequences. Links to the EMBL, TRANSFAC and MEDLINE databases are provided when available. Data about the transcription sites are extracted mainly from the literature, supplemented with an increasing number of in silico predicted data. Apart from a general description for specific transcription factor sites, levels of confidence for the experimental evidence, functional information and the position on the promoter are given as well. New features have been implemented to search for plant cis-acting regulatory elements in a query sequence. Furthermore, links are now provided to a new clustering and motif search method to investigate clusters of co-expressed genes. New regulatory elements can be sent automatically and will be added to the database after curation. The PlantCARE relational database is available via the World Wide Web at http://sphinx.rug.ac.be:8080/PlantCARE/.
DOI:10.1093/nar/gky316URLPMID:29718424 [本文引用: 1]
Plant regulatory small RNAs (sRNAs), which include most microRNAs (miRNAs) and a subset of small interfering RNAs (siRNAs), such as the phased siRNAs (phasiRNAs), play important roles in regulating gene expression. Although generated from genetically distinct biogenesis pathways, these regulatory sRNAs share the same mechanisms for post-translational gene silencing and translational inhibition. psRNATarget was developed to identify plant sRNA targets by (i) analyzing complementary matching between the sRNA sequence and target mRNA sequence using a predefined scoring schema and (ii) by evaluating target site accessibility. This update enhances its analytical performance by developing a new scoring schema that is capable of discovering miRNA-mRNA interactions at higher 'recall rates' without significantly increasing total prediction output. The scoring procedure is customizable for the users to search both canonical and non-canonical targets. This update also enables transmitting and analyzing 'big' data empowered by (a) the implementation of multi-threading chunked file uploading, which can be paused and resumed, using HTML5 APIs and (b) the allocation of significantly more computing nodes to its back-end Linux cluster. The updated psRNATarget server has clear, compelling and user-friendly interfaces that enhance user experiences and present data clearly and concisely. The psRNATarget is freely available at http://plantgrn.noble.org/psRNATarget/.
DOI:10.1006/meth.2001.1262URLPMID:11846609 [本文引用: 1]
The two most commonly used methods to analyze data from real-time, quantitative PCR experiments are absolute quantification and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control. The 2(-Delta Delta C(T)) method is a convenient way to analyze the relative changes in gene expression from real-time quantitative PCR experiments. The purpose of this report is to present the derivation, assumptions, and applications of the 2(-Delta Delta C(T)) method. In addition, we present the derivation and applications of two variations of the 2(-Delta Delta C(T)) method that may be useful in the analysis of real-time, quantitative PCR data.
DOI:10.1093/jxb/eru261URL [本文引用: 2]
The plant specific RWP-RK family of transcription factors, initially identified in legumes and Chlamydomonas, are found in all vascular plants, green algae, and slime molds. These proteins possess a characteristic RWP-RK motif, which mediates DNA binding. Based on phylogenetic and domain analyses, we classified the RWP-RK proteins of six different species in two subfamilies: the NIN-like proteins (NLPs), which carry an additional PB1 domain at their C-terminus, and the RWP-RK domain proteins (RKDs), which are divided into three subgroups. Although, the functional analysis of this family is still in its infancy, several RWP-RK proteins have a key role in regulating responses to nitrogen availability. The nodulation-specific NIN proteins are involved in nodule organogenesis and rhizobial infection under nitrogen starvation conditions. Arabidopsis NLP7 in particular is a major player in the primary nitrate response. Several RKDs act as transcription factors involved in egg cell specification and differentiation or gametogenesis in algae, the latter modulated by nitrogen availability. Further studies are required to extend the general picture of the functional role of these exciting transcription factors.
DOI:10.1046/j.1365-313x.1993.04020343.xURLPMID:8220485 [本文引用: 1]
The 431 bp C-hordein promoter of lambda-1-17 exhibits a specific response to amino acids and NH4NO3 in developing barley (Hordeum vulgare L.) endosperms. With the aid of particle bombardment it is shown that the GCN4 motif ATGA(C/G)TCAT is the dominating cis-acting element in this response. But synergistic interaction with the neighbouring endosperm motif TGTAAAGT within the bifactorial prolamin element and cooperation with upstream sequences including a second prolamin-like element is an absolute requirement for a strong, positive regulation by an optimal nitrogen regime. Low nitrogen levels convert the GCN4 box into a negative motif. In contrast the endosperm box on its own exerted a silencing activity, independent of nitrogen nutrition. Sequence comparisons revealed that GCN4- and endosperm-like motifs are widely distributed among plant promoters. Their putative role in nitrogen regulation is discussed.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
DOI:10.1093/aob/mct133URLPMID:23788746 [本文引用: 1]
MicroRNAs (miRNAs) play an important role in the responses and adaptation of plants to many stresses including low nitrogen (LN). Characterizing relevant miRNAs will improve our understanding of nitrogen (N) use efficiency and LN tolerance and thus contribute to sustainable maize production. The objective of this study was to identify novel and known miRNAs and their targets involved in the response and adaptation of maize (Zea mays) to LN stress.
[D].
DOI:10.1093/aob/mct133URLPMID:23788746 [本文引用: 1]
MicroRNAs (miRNAs) play an important role in the responses and adaptation of plants to many stresses including low nitrogen (LN). Characterizing relevant miRNAs will improve our understanding of nitrogen (N) use efficiency and LN tolerance and thus contribute to sustainable maize production. The objective of this study was to identify novel and known miRNAs and their targets involved in the response and adaptation of maize (Zea mays) to LN stress.
DOI:10.3864/j.issn.0578-1752.2017.20.013URL [本文引用: 1]
【Objective】 MYB is one of the most common transcription factor families in plants. It is widely involved in plant growth, and metabolic regulations. So far, there is no systematic analysis of the MYB transcription factor family of tree crops. Analysis of MYB family based on the transcriptome data in Lycium ruthenicum Murr. was conducted in this study, which laid a foundation for the research on biological function, and mechanism of metabolic regulations of MYB genes. 【Method】 Based on the transcriptome sequencing (RNA-Seq) data, the NR, NT, Swiss-Prot, PFAM and NCBI sites were used at the same time to screen and classify the MYB genes of L. ruthenicum Murr. The Web Logo3, Prot Comp 9.0, and MEGA5.0 were also applied to conservative structure prediction, subcellular localization, and phylogenetic analysis. The expression pattern of MYB genes related to fruit development was obtained and Real-time fluorescence quantitative PCR was used to detect the specific expression of those genes. 【Result】 Based on the transcriptome sequencing (RNA-Seq) data, 83 transcription factors of MYB family were annotated, selected, and divided into four categories (R2R3-MYB, 1R-MYB, 3R-MYB and 4R-MYB) according to their structural characteristics. The R2 MYB motif of the R2R3-MYB transcription factor contains three highly conserved tryptophan residues, and the first tryptophan residue in the R3 MYB motif is replaced by some hydrophobic amino acids. The phylogenetic trees of MYB family of L. ruthenicum Murr. and Arabidopsis thaliana were constructed, which showed that the MYB family of L. ruthenicum Murr. contained three major branches, and six evolutionary branches. The result of the subcellular localization demonstrated that 44 MYB transcription factors were located in the cytoplasm, and 37 MYB transcription factors were located in the nucleus. The analysis of differential expression of MYB genes of L. ruthenicum Murr. based on transcriptome sequencing (RNA-Seq) showed that MYB genes might be involved in the regulation of anthocyanin in three fruit development periods. Additionally, differential expression data based on fluorescence quantitative PCR confirmed that some MYB transcription factors might play a role in the regulation of anthocyanin synthesis in different fruit development periods ofL. ruthenicum Murr.. 【Conclusion】 83 transcription factors of MYB family were annotated of L. ruthenicum Murr. The findings have laid a foundation for further studies of the structures and biological functions of MYB family.
DOI:10.3864/j.issn.0578-1752.2017.20.013URL [本文引用: 1]
【Objective】 MYB is one of the most common transcription factor families in plants. It is widely involved in plant growth, and metabolic regulations. So far, there is no systematic analysis of the MYB transcription factor family of tree crops. Analysis of MYB family based on the transcriptome data in Lycium ruthenicum Murr. was conducted in this study, which laid a foundation for the research on biological function, and mechanism of metabolic regulations of MYB genes. 【Method】 Based on the transcriptome sequencing (RNA-Seq) data, the NR, NT, Swiss-Prot, PFAM and NCBI sites were used at the same time to screen and classify the MYB genes of L. ruthenicum Murr. The Web Logo3, Prot Comp 9.0, and MEGA5.0 were also applied to conservative structure prediction, subcellular localization, and phylogenetic analysis. The expression pattern of MYB genes related to fruit development was obtained and Real-time fluorescence quantitative PCR was used to detect the specific expression of those genes. 【Result】 Based on the transcriptome sequencing (RNA-Seq) data, 83 transcription factors of MYB family were annotated, selected, and divided into four categories (R2R3-MYB, 1R-MYB, 3R-MYB and 4R-MYB) according to their structural characteristics. The R2 MYB motif of the R2R3-MYB transcription factor contains three highly conserved tryptophan residues, and the first tryptophan residue in the R3 MYB motif is replaced by some hydrophobic amino acids. The phylogenetic trees of MYB family of L. ruthenicum Murr. and Arabidopsis thaliana were constructed, which showed that the MYB family of L. ruthenicum Murr. contained three major branches, and six evolutionary branches. The result of the subcellular localization demonstrated that 44 MYB transcription factors were located in the cytoplasm, and 37 MYB transcription factors were located in the nucleus. The analysis of differential expression of MYB genes of L. ruthenicum Murr. based on transcriptome sequencing (RNA-Seq) showed that MYB genes might be involved in the regulation of anthocyanin in three fruit development periods. Additionally, differential expression data based on fluorescence quantitative PCR confirmed that some MYB transcription factors might play a role in the regulation of anthocyanin synthesis in different fruit development periods ofL. ruthenicum Murr.. 【Conclusion】 83 transcription factors of MYB family were annotated of L. ruthenicum Murr. The findings have laid a foundation for further studies of the structures and biological functions of MYB family.
[本文引用: 1]
[本文引用: 1]
DOI:10.3389/fpls.2017.00480URLPMID:28443104 [本文引用: 1]
The bHLH (basic helix-loop-helix) transcription factor family is the second largest in plants. It occurs in all three eukaryotic kingdoms, and plays important roles in regulating growth and development. However, family members have not previously been studied in apple. Here, we identified 188 MdbHLH proteins in apple "Golden Delicious" (Malus × domestica Borkh.), which could be classified into 18 groups. We also investigated the gene structures and 12 conserved motifs in these MdbHLHs. Coupled with expression analysis and protein interaction network prediction, we identified several genes that might be responsible for abiotic stress responses. This study provides insight and rich resources for subsequent investigations of such proteins in apple.
DOI:10.3864/j.issn.0578-1752.2016.07.010URL [本文引用: 1]
【Objective】In this study, putative bZIP transcription factor-encoding genes in the apple (Malus×domestica Borkh.) genome were identified, so as to provide a basis for studying the theoretical roles of bZIP genes in the bud dormancy, and to provide valuable information for bZIP genes in apple. 【Method】 The Hidden Markov Model profiles of the bZIP domains (PF00170 and PF07716) downloaded from Pfam were used to search the database using HMMER, ver. 3.0 with the default E-value. The obtained amino acid sequences were analyzed with the bioinformatics softwares, including Clustal Omega, MEGA6.0, MapInspect, DNAMAN 6.0 and MEME4.10.2. Furthermore, Microarray analysis and qRT-PCR results indicated that many MdbZIP genes may be involved in regulating bud dormancy in different cultivar of apples. 【Result】 In Total 120 MdbZIP genes were found in the apple genome. Phylogenetic analyses of the genes based on A. thaliana counterparts indicated that the transcription factors can be categorized into 10 different groups (Groups A–I and S), Chromosome mapping analysis showed that 109 MdbZIP genes were distributed unevenly on 17 chromosomes. Except for 11 genes that were not located on chromosomes, the largest number of MdbZIP genes were found on chromosomes 8 (thirteen genes), only 1 genes located on chromosome 1, some chromosomal regions had a relatively high density of MdbZIP genes. The results of gene structure analysis revealed that MdbZIP gene contained 0-23 exons, 23 bZIP genes were intronless and were found in Groups S (nine genes) and F (one gene), bZIP gene structure were highly conserved in apples. Conserved motif analysis showed that the conserved motifs 1, which specify the bZIP domain were observed in all apple bZIP proteins, motif 10 and 14 as the known domain were observed in Group D and G, respectively. Furthermore, microarray data indicated that many MdbZIP genes may be involved in regulating bud dormancy release. And qRT-PCR results indicated that ABA induce eight members of group A expression in high-chill and low-chill apples, but the largest number of differentially expressed genes was found in group D with cold temperatures. 【Conclusion】 These results suggested that MdbZIP gene family was highly and structurally conserved, and involved in abscisic acid (ABA) signaling and cold stresses thereby possibly involved into the regulation of bud dormancy in apples.
DOI:10.3864/j.issn.0578-1752.2016.07.010URL [本文引用: 1]
【Objective】In this study, putative bZIP transcription factor-encoding genes in the apple (Malus×domestica Borkh.) genome were identified, so as to provide a basis for studying the theoretical roles of bZIP genes in the bud dormancy, and to provide valuable information for bZIP genes in apple. 【Method】 The Hidden Markov Model profiles of the bZIP domains (PF00170 and PF07716) downloaded from Pfam were used to search the database using HMMER, ver. 3.0 with the default E-value. The obtained amino acid sequences were analyzed with the bioinformatics softwares, including Clustal Omega, MEGA6.0, MapInspect, DNAMAN 6.0 and MEME4.10.2. Furthermore, Microarray analysis and qRT-PCR results indicated that many MdbZIP genes may be involved in regulating bud dormancy in different cultivar of apples. 【Result】 In Total 120 MdbZIP genes were found in the apple genome. Phylogenetic analyses of the genes based on A. thaliana counterparts indicated that the transcription factors can be categorized into 10 different groups (Groups A–I and S), Chromosome mapping analysis showed that 109 MdbZIP genes were distributed unevenly on 17 chromosomes. Except for 11 genes that were not located on chromosomes, the largest number of MdbZIP genes were found on chromosomes 8 (thirteen genes), only 1 genes located on chromosome 1, some chromosomal regions had a relatively high density of MdbZIP genes. The results of gene structure analysis revealed that MdbZIP gene contained 0-23 exons, 23 bZIP genes were intronless and were found in Groups S (nine genes) and F (one gene), bZIP gene structure were highly conserved in apples. Conserved motif analysis showed that the conserved motifs 1, which specify the bZIP domain were observed in all apple bZIP proteins, motif 10 and 14 as the known domain were observed in Group D and G, respectively. Furthermore, microarray data indicated that many MdbZIP genes may be involved in regulating bud dormancy release. And qRT-PCR results indicated that ABA induce eight members of group A expression in high-chill and low-chill apples, but the largest number of differentially expressed genes was found in group D with cold temperatures. 【Conclusion】 These results suggested that MdbZIP gene family was highly and structurally conserved, and involved in abscisic acid (ABA) signaling and cold stresses thereby possibly involved into the regulation of bud dormancy in apples.
[本文引用: 1]
[本文引用: 1]
DOI:10.3864/j.issn.0578-1752.2013.12.011URL [本文引用: 1]
【Objective】Identification of LBD genes from tomato genome, and analysis of phylogeny,gene structure, chromosome location, phylogenetic and tissue expression pattern analysis of LBD family genes in tomato will be useful to the functions identification of plant LBD genes.【Method】Based on tomato genome database and bioinformatic method, tomato LBD family genes were identified and the genes were sequenced. A phylogenetic tree was created using the MEGA5 program. Gene structure and chromosomes location were done by Perl-based program, MapDraw and GSDS. Expression pattern of LBD genes at different development stages was analyzed based on the existing microarray database.【Result】A total of 46 LBD genes were systematically identified from tomato and classified into 2 classes (class I and class II), then was classified into 5 subfamilies (Ia, Ib, Ic, Id and II) according to the gene structure and conserved domain phylogeny relationship. They were distributed on 10 of tomato chromosomes, suggesting that they have an extensive distribution on the tomato chromosomes. Most of the LBD genes had differential expression pattern and response to external stimulation. 【Conclusion】Forty-six LBD family genes in tomato were identified by genome-wide screening. They were classified into 2 classes, 5 subfamilies and distributed on 10 chromosomes with different expression patterns in different tissues and developmental stages. They had special response to external stimulation. These results are helpful for the functional analysis of LBD genes in plants.
DOI:10.3864/j.issn.0578-1752.2013.12.011URL [本文引用: 1]
【Objective】Identification of LBD genes from tomato genome, and analysis of phylogeny,gene structure, chromosome location, phylogenetic and tissue expression pattern analysis of LBD family genes in tomato will be useful to the functions identification of plant LBD genes.【Method】Based on tomato genome database and bioinformatic method, tomato LBD family genes were identified and the genes were sequenced. A phylogenetic tree was created using the MEGA5 program. Gene structure and chromosomes location were done by Perl-based program, MapDraw and GSDS. Expression pattern of LBD genes at different development stages was analyzed based on the existing microarray database.【Result】A total of 46 LBD genes were systematically identified from tomato and classified into 2 classes (class I and class II), then was classified into 5 subfamilies (Ia, Ib, Ic, Id and II) according to the gene structure and conserved domain phylogeny relationship. They were distributed on 10 of tomato chromosomes, suggesting that they have an extensive distribution on the tomato chromosomes. Most of the LBD genes had differential expression pattern and response to external stimulation. 【Conclusion】Forty-six LBD family genes in tomato were identified by genome-wide screening. They were classified into 2 classes, 5 subfamilies and distributed on 10 chromosomes with different expression patterns in different tissues and developmental stages. They had special response to external stimulation. These results are helpful for the functional analysis of LBD genes in plants.
DOI:10.1111/j.1365-313X.2008.03695.xURLPMID:18826430 [本文引用: 1]
Nitrate is an essential nutrient, and is involved in many adaptive responses of plants, such as localized proliferation of roots, flowering or stomatal movements. How such nitrate-specific mechanisms are regulated at the molecular level is poorly understood. Although the Arabidopsis ANR1 transcription factor appears to control stimulation of lateral root elongation in response to nitrate, no regulators of nitrate assimilation have so far been identified in higher plants. Legume-specific symbiotic nitrogen fixation is under the control of the putative transcription factor, NIN, in Lotus japonicus. Recently, the algal homologue NIT2 was found to regulate nitrate assimilation. Here we report that Arabidopsis thaliana NIN-like protein 7 (NLP7) knockout mutants constitutively show several features of nitrogen-starved plants, and that they are tolerant to drought stress. We show that nlp7 mutants are impaired in transduction of the nitrate signal, and that the NLP7 expression pattern is consistent with a function of NLP7 in the sensing of nitrogen. Translational fusions with GFP showed a nuclear localization for the NLP7 putative transcription factor. We propose NLP7 as an important element of the nitrate signal transduction pathway and as a new regulatory protein specific for nitrogen assimilation in non-nodulating plants.
DOI:10.1038/s41477-018-0261-3URLPMID:30297831 [本文引用: 1]
Legume plants can assimilate inorganic nitrogen and have access to fixed nitrogen through symbiotic interaction with diazotrophic bacteria called rhizobia. Symbiotic nitrogen fixation is an energy-consuming process and is strongly inhibited when sufficient levels of fixed nitrogen are available, but the molecular mechanisms governing this regulation are largely unknown. The transcription factor nodule inception (NIN) is strictly required for nodulation and belongs to a family of NIN-like proteins (NLPs), which have been implicated in the regulation of nitrogen homeostasis in Arabidopsis. Here, we show that mutation or downregulation of NLP genes prevents nitrate inhibition of infection, nodule formation and nitrogen fixation. We find that NIN and NLPs physically interact through their carboxy-terminal PB1 domains. Furthermore, we find that NLP1 is required for the expression of nitrate-responsive genes and that nitrate triggers NLP1 re-localization from the cytosol to the nucleus. Finally, we show that NLP1 can suppress NIN activation of CRE1 expression in Nicotiana benthamiana and Medicago truncatula. Our findings highlight a central role for NLPs in the suppression of nodulation by nitrate.
DOI:10.1073/pnas.1615676114URLPMID:28202720 [本文引用: 1]
Plants have evolved adaptive strategies that involve transcriptional networks to cope with and survive environmental challenges. Key transcriptional regulators that mediate responses to environmental fluctuations in nitrate have been identified; however, little is known about how these regulators interact to orchestrate nitrogen (N) responses and cell-cycle regulation. Here we report that teosinte branched1/cycloidea/proliferating cell factor1-20 (TCP20) and NIN-like protein (NLP) transcription factors NLP6 and NLP7, which act as activators of nitrate assimilatory genes, bind to adjacent sites in the upstream promoter region of the nitrate reductase gene, NIA1, and physically interact under continuous nitrate and N-starvation conditions. Regions of these proteins necessary for these interactions were found to include the type I/II Phox and Bem1p (PB1) domains of NLP6&7, a protein-interaction module conserved in animals for nutrient signaling, and the histidine- and glutamine-rich domain of TCP20, which is conserved across plant species. Under N starvation, TCP20-NLP6&7 heterodimers accumulate in the nucleus, and this coincides with TCP20 and NLP6&7-dependent up-regulation of nitrate assimilation and signaling genes and down-regulation of the G2/M cell-cycle marker gene, CYCB1;1 TCP20 and NLP6&7 also support root meristem growth under N starvation. These findings provide insights into how plants coordinate responses to nitrate availability, linking nitrate assimilation and signaling with cell-cycle progression.
DOI:10.3724/SP.J.1259.2013.00519URL [本文引用: 1]
豆科植物百脉根(Lotus japonicus)的根瘤感受基因Nin与根瘤的早期发育有关。Nin的同源基因(Nin-like基因)功能上涉及氮代谢过程。从完成测序的豆科和非豆科植物基因组中获取Nin-like基因并进行系统发育分析。在此基础上, 追踪基因和蛋白质结构的歧异式样, 尝试建立结构歧异和功能分化的联系。通过比较, 新的Nin-like基因被鉴别。系统发育分析不仅重现了以前分辨的直系同源群(分支I、II和III), 且识别了它们之间的姐妹群关系。Nin-like基因的结构呈现多样性, 支持系统发育分析的结果。水稻OsNLP5基因缺乏内含子, 可能起源于基因返座事件。NIN-like蛋白结构域组织和功能位点在不同分支中存在差异, 提示它们的功能发生了分化。根瘤固氮植物NIN-like蛋白的GAF结构域中存在一个显著变异区, 三级建模分析显示这个变异区对应于百脉根非固氮NIN-like蛋白的一段保守构象, 这一变异可能使豆科植物具有根瘤固氮能力。研究结果为阐明Nin-like基因的功能提供了新的研究思路。
DOI:10.3724/SP.J.1259.2013.00519URL [本文引用: 1]
豆科植物百脉根(Lotus japonicus)的根瘤感受基因Nin与根瘤的早期发育有关。Nin的同源基因(Nin-like基因)功能上涉及氮代谢过程。从完成测序的豆科和非豆科植物基因组中获取Nin-like基因并进行系统发育分析。在此基础上, 追踪基因和蛋白质结构的歧异式样, 尝试建立结构歧异和功能分化的联系。通过比较, 新的Nin-like基因被鉴别。系统发育分析不仅重现了以前分辨的直系同源群(分支I、II和III), 且识别了它们之间的姐妹群关系。Nin-like基因的结构呈现多样性, 支持系统发育分析的结果。水稻OsNLP5基因缺乏内含子, 可能起源于基因返座事件。NIN-like蛋白结构域组织和功能位点在不同分支中存在差异, 提示它们的功能发生了分化。根瘤固氮植物NIN-like蛋白的GAF结构域中存在一个显著变异区, 三级建模分析显示这个变异区对应于百脉根非固氮NIN-like蛋白的一段保守构象, 这一变异可能使豆科植物具有根瘤固氮能力。研究结果为阐明Nin-like基因的功能提供了新的研究思路。
DOI:10.1016/j.plantsci.2014.09.006URL [本文引用: 1]
Nitrate reductase is a key enzyme in nitrogen assimilation, and it catalyzes the nitrate-to-nitrite reduction process in plants. A variety of factors, including nitrate, light, metabolites, phytohormones, low temperature, and drought, modulate the expression levels of nitrate reductase genes as well as nitrate reductase activity, which is consistent with its physiological role. Recently, several transcription factors involved in controlling the expression of nitrate reductase genes have been identified in Arabidopsis. NODULE-INCEPTION-like proteins (NLPs) are transcription factors responsible for nitrate-inducible expression of nitrate reductase genes. Since NLPs also control nitrate-inducible expression of genes encoding nitrate transporter, nitrite transporter, and nitrite reductase, the expression levels of nitrate reduction pathway-associated genes are coordinately modulated by NLPs in response to nitrate. LATERAL ORGAN BOUNDARIES DOMAIN (LBD) transcription factors (LBD37-LBD39) are strong candidates for transcription factors mediating negative feedback regulation in response to increases in the contents of nitrogen-containing metabolites, whereas LONG HYPOCOTYL 5 (HY5) that promotes photomorphogenesis in light may be a transcription factor involved in light-induced expression of a nitrate reductase gene. Furthermore, unidentified transcription factors likely mediate other signals and regulate the expression of nitrate reductase genes. This review presents a summary of our current knowledge of such transcription factors. (C) 2014 Elsevier Ireland Ltd.
DOI:10.1093/jxb/eru267URL [本文引用: 1]
Nitrogen is one of the primary macronutrients of plants, and nitrate is the most abundant inorganic form of nitrogen in soils. Plants take up nitrate in soils and utilize it both for nitrogen assimilation and as a signalling molecule. Thus, an essential role for nitrate in plants is triggering changes in gene expression patterns, including immediate induction of the expression of genes involved in nitrate transport and assimilation, as well as several transcription factor genes and genes related to carbon metabolism and cytokinin biosynthesis and response. Significant progress has been made in recent years towards understanding the molecular mechanisms underlying nitrate-regulated gene expression in higher plants; a new stage in our understanding of this process is emerging. A key finding is the identification of NIN-like proteins (NLPs) as transcription factors governing nitrate-inducible gene expression. NLPs bind to nitrate-responsive DNA elements (NREs) located at nitrate-inducible gene loci and activate their NRE-dependent expression. Importantly, post-translational regulation of NLP activity by nitrate signalling was strongly suggested to be a vital process in NLP-mediated transcriptional activation and subsequent nitrate responses. We present an overview of the current knowledge of the molecular mechanisms underlying nitrate-regulated gene expression in higher plants.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}