 ,, 吴贤锋, 高承芳, 黄勤楼,福建省农业科学院畜牧兽医研究所,福州 350013
,, 吴贤锋, 高承芳, 黄勤楼,福建省农业科学院畜牧兽医研究所,福州 350013Transcriptome Analysis of Differentially Gene Expression Associated with Ovary Tissue During the Follicular Stage in Fuqing Goat and Nubian Black Goat
LI WenYang, LIU Yuan,, WU XianFeng, GAO ChengFang, HUANG QinLou,Institute of animal husbandry and veterinary medicine, Fujian Academy of Agricultural Sciences, Fuzhou 350013通讯作者:
责任编辑: 林鉴非
收稿日期:2018-07-15接受日期:2019-04-15网络出版日期:2019-06-16
| 基金资助: |
Received:2018-07-15Accepted:2019-04-15Online:2019-06-16
作者简介 About authors
李文杨,Tel:0591-87902045;E-mail: 516316606@qq.com。

摘要
关键词:
Abstract
Keywords:
PDF (2572KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李文杨, 刘远, 吴贤锋, 高承芳, 黄勤楼. 福清山羊与努比亚黑山羊发情期卵巢组织RNA-Seq分析[J]. 中国农业科学, 2019, 52(12): 2171-2182 doi:10.3864/j.issn.0578-1752.2019.12.014
LI WenYang, LIU Yuan, WU XianFeng, GAO ChengFang, HUANG QinLou.
0 引言
【研究意义】繁殖性能是养羊业的重要经济性状,能繁母羊产仔数的多少直接影响养羊业的经济效益。由于羊繁殖性状受到遗传力低、限性性状等因素制约,常规选育存在周期长、遗传进展慢的缺点,最经济有效的方法是通过选择控制多胎性能的主效基因(分子标记)提高母羊的产羔率[1,2]。因此研究羊繁殖性状的分子调控机理,挖掘、鉴定和充分利用控制羊繁殖性状的主效基因,并应用于羊繁殖性状的遗传改良具有重要意义[3]。【前人研究进展】转录组是特定组织或细胞在某一环境或生理条件下所转录表达的所有RNA总和,是连接基因组遗传信息与蛋白质组生物功能的纽带[4]。高通量转录组测序技术(RNA sequencing,RNA-Seq)的发展和成熟为系统研究基因表达及调控提供了重要的手段和方法,在畜禽重要经济性状分子机制研究中得到了广泛使用[5,6,7]。基于高通量测序技术,研究者采用不同的策略开展了羊繁殖性状候选基因的筛选与鉴定。罗庆苗等[8]利用数字基因表达谱技术(digital gene expression tag profiling,DGE)分别检测了处于自然发情期的FecBBFecBB型与FecB+FecB+型小尾寒羊卵巢组织的基因表达情况,认为类固醇快速调节蛋白(steroidogenic acute regulatory protein,STAR)、卵巢滤泡激素(Folliculin,FLCN)等5个基因与Notch信号通路可能是参与调控小尾寒羊高低繁殖性状差异的重要基因和调控通路。DI等[9]利用RNA-Seq技术获得了绵羊不同繁殖状态下的基因表达模式。LING等[3]比较了发情期多羔和单羔安徽白山羊卵巢组织的差异表达情况,筛选出了12个可能与山羊高繁殖力有关的基因。FENG等[10]研究了高产和低产湖羊卵泡期卵巢组织RNA表达差异,筛选出了76个差异表达基因和5个差异表达长链非编码RNA(Long non-coding RNA, lncRNA)。付绍印等[11]对巴美肉羊性腺轴进行了转录组分析,功能富集分析表明,神经活性的配体-受体相互作用信号通路在下丘脑-垂体-卵巢性腺轴调控排卵及卵泡发育方面发挥着重要调控作用。【本研究切入点】目前对部分绵羊、山羊品种的繁殖力相关基因研究较多,但对福清山羊的相关研究较少。而影响不同品种繁殖力的基因也不尽相同,李文杨等[12]证实控制部分绵羊、山羊多胎性状主效基因骨形态发生蛋白15(bone morphogenetic protein 15,BMP15)、骨形态发生蛋白受体1B(bone morphogenetic protein receptor 1B,BMPR-1B)和生长分化因子9(growth differentiation factor 9,GDF9)并非控制福清山羊繁殖性状的主效基因。努比亚黑山羊母羊繁殖指数(dam reproduction index,DRI)高达0.99,而福清山羊母羊DRI仅为0.45[13]。同时有研究表明利用努比亚黑山羊改良贵州黑山羊可提高后者产羔率37.2%[14]。因此,导入努比亚黑山羊基因改良福清山羊品种,对其繁殖、生产性能的提高具有重要的生产实践意义。【拟解决的关键问题】本研究拟以福建地方山羊品种——福清山羊和福建省引进的努比亚黑山羊为材料,采用RNA-Seq技术比较发情期卵巢组织的差异表达基因(differentially expressed genes, DEGs)及新转录本分析,并对筛选的DEGs进行GO(gene ontology)、KEGG(kyoto encyclopediaof genes and genomes)功能富集和COG(Cluster of Orthologous Groups of proteins)分类,挖掘控制福清山羊繁殖性能的相关候选基因,一方面为地方品种种质资源的遗传机制研究提供理论依据,另一方面通过测序数据与参考基因组的比对,筛选潜在可能影响福清山羊繁殖性能的单核苷酸多态性(single nucleotide polymorphisms,SNP)和插入缺失标记(insertion-deletion,InDel)分子遗传标记,为福清山羊繁殖性能分子遗传改良提供新的育种素材。1 材料与方法
1.1 试验材料
本研究选用的福清山羊和努比亚黑山羊由福建省农业科学院福清市渔溪肉羊试验基地提供,试验于2017年2—6月进行。选择2—3岁营养良好、体态匀称的空怀雌性福清山羊和努比亚黑山羊各5只。母羊发情鉴定以公羊试情为主,结合母羊外阴部观察。试情公羊每日早晚各试情1次(早9:00、晚21:00),母羊连续2次试情均接受试情公羊的爬跨,并在爬跨时静立不动即鉴定为发情。鉴定母羊发情后按照国家实验动物处理饲喂准则屠宰,取有成熟卵泡发育一侧的卵巢组织,置于-80℃备用。每个品种屠宰3只,福清山羊标记为F-1、F-2、F-3,努比亚黑山羊分别标记为N-4、N-5、N-6。1.2 RNA提取、转录组文库构建及测序
利用TaKaRa MiniBEST Universal RNA Extraction Kit (TaKaRa,大连宝生物)提取每个样品卵巢组织的总RNA,单个建池。为了保证数据准确性,采用Nanodrop检测RNA的纯度,Agilent 2100精确检测RNA的完整性。RNA样品检测合格后,送北京百迈克生物科技有限公司进行文库构建以及测序。文库构建完成后,对建成文库的质量进行检测。文库质量合格后,不同的文库利用Illumina HiSeq2500平台(Illumina,America),基于边合成边测序(sequencing by synthesis,SBS)技术进行高通量测序。1.3 转录组测序数据处理与分析
原始测序数据经过去除含有接头和低质量数据过滤后获得高质量测序数据。利用TopHat2[15]软件将获得的Clean reads比对到山羊参考基因组(ftp://ftp. ncbi.nlm.nih.gov/genomes/all/ GCF/001/704/415/GCF_ 001704415.1_ASM170441v1/GCF_001704415.1_ASM170441v1_genomic.fna.gz)[16],获取序列在参考基因组或基因上的位置信息,以及样品特有的序列特征信息。1.4 差异表达基因的筛选、功能注释和富集分析
参考JIANG等报道的数学模型[17],使用Cufflinks软件对转录本和基因的表达水平进行定量。采用FPKM作为衡量转录本或基因表达水平的指标[18]。皮尔逊相关系数r作为生物学重复相关性的评估指标[19]。以福清山羊为对照,利用DEseq进行2个品种间的差异表达分析[20]。将差异倍数≥2且错误发现率(False Discovery Rate,FDR)<0.01作为DEGs的筛选标准。绘制2个品种DEGs火山图并进行聚类分析。分别利用GO数据库[21]、COG数据库[22]和KEGG数据库[23]对DEGs进行生物信息学分析。
1.5 SNP/InDel分析
基于各样品reads与参考基因组序列的TopHat2比对结果,使用GATK软件[24]识别测序样品与参考基因组建的单碱基错配和插入缺失,识别潜在的SNP及InDel位点。进一步利用SnpEff软件[25]注释筛选到的变异(SNP、InDel)和预测变异造成的影响。1.6 荧光定量PCR验证
为了进一步验证高通量测序结果以及发现新转录本序列的准确性,以18S rRNA为内参基因,随机挑选6个 DEGs和2个新转录本,采用实时荧光定量PCR(qRT-PCR)验证基因表达量。内参基因引物参考文献设计[21],其余基因和新转录本引物根据测序序列信息由ABI公司的Primer Express Software v2.0设计,铂尚生物技术(上海)有限公司合成,基因名称及引物信息见表1。以1.2中各样品RNA池为模板,采用TAKARA PrimeScript? RT reagent Kit with gDNA Eraser(TaKaRa,大连宝生物)试剂盒逆转录后进行qRT-PCR扩增,扩增体系20μL:cDNA模板1μL,上、下游引物各1μL,2×SYBR? Select Master Mix 10μL,ddH2O7μL。qRT-PCR扩增程序:95℃预变性2 min;95℃变性10s,60℃退火10s,72℃延伸40s,50个循环;最后72℃延伸5min;4℃保存。采用2-ΔΔCt法计算基因的相对表达量。Table 1
表1
表1实时荧光定量PCR引物信息
Table 1
| 基因名称 Gene | 上游引物 Forward primer(5′-3′) | 下游引物 Reverse primer(5′-3′) | 产物长度 Product size(bp) |
|---|---|---|---|
| 18S rRNA | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG | 151 |
| New-Gene 4708 | CTCCAAATCGGGCAACACAG | TCCACAGTAATAATCGGCCTCG | 77 |
| New-Gene 1125 | ACAACAGTGTCAGCATCCGT | TGTTGGAGTCCCGTTATGGC | 136 |
| CREB3L4 | GGGCTCTTCGAAACAGCAGA | AGTCCCAGCTCCAGGAAAGA | 104 |
| THSD7B | AGTTACAAGGCGCATCACCA | TTTCGGCATGATGCAGGACT | 159 |
| CRABP1 | GTGCAGGAGCTTACCCACTT | CGGGTCCAGTAGGTTTTGGG | 93 |
| CTHRC1 | TTGGTGCTGGACTAGTGGAC | ATGCGAGACACCGAATTCCA | 100 |
新窗口打开|下载CSV
2 结果
2.1 测序数据评估及对比分析
经过文库质量检测和测序质量控制,各样品均符合测序要求。6个样品共得到46.68Gb Clean Data,各样品Clean Data 均达到7.24Gb以上,获得Clean reads 24 283 922—27 945 197条。测序数据碱基质量(Quality Score)Q30碱基百分比为91.15%—91.99%,碱基GC含量为50.98%—52.19%,各碱基质量稳定在20%—30%之间,低质量碱基比例小,测序质量好。表明文库构建质量和测序质量高,测序数据准确可靠,满足后续分析条件。将获得的Clean reads与参考山羊基因组进行对比分析,结果表明各样品的Clean reads与参考基因组的比对效率在84.62%—87.14%之间,比对到参考基因组唯一位置的Reads占Clean Reads的百分比为81.28%—84.35%,多处位置的百分比为2.30%—3.71%(表2),表明6个样本总RNA不存在污染。
Table 2
表2
表2测序数据与参考基因组的序列比对结果统计
Table 2
| 样品 Sample | 总Reads Total reads | 可定位的Reads Mapped reads | 唯一定位Reads Uniq mapped reads | 多点定位Reads Multiple mapped reads |
|---|---|---|---|---|
| F-1 | 48567844 | 41489885/85.43% | 39687502/81.72% | 1802383/3.71% |
| F-2 | 49910620 | 42262711/84.68% | 40565566/81.28% | 1697145/3.40% |
| F-3 | 49890394 | 42216928/84.62% | 40925490/82.03% | 1791438/3.59% |
| N-4 | 53482074 | 46603840/87.14% | 45110457/84.35% | 1293383/2.42% |
| N-5 | 53379622 | 46077636/86.32% | 44696951/83.73% | 1380685/2.59% |
| N-6 | 53967784 | 46261607/85.72% | 45017922/83.42% | 1243685/2.30% |
新窗口打开|下载CSV
2.2 DEGs的筛选、功能注释和富集分析
2.2.1 DEGs的筛选 通过定位到参考基因组区域或基因外显子区的Reads的计数来估计基因(转录本)的表达水平,基因(转录本)的真实表达水平与Reads计数、基因(转录本)的长度和测序深度成正相关。采用FPKM作为衡量转录本或基因表达水平的指标,计算得到了6个样本18 996个表达基因的FPKM值。为了保证测序数据的可重复性以及可靠性,分析了6个样品之间转录本表达水平的相关性。皮尔逊相关系数的平方(r2)越接近于1,说明2个样品的表达模式相似度越高。Encode计划建议r2大于0.92,福清山羊3个样品的r2为0.926—0.935,努比亚黑山羊3个样品的r2为0.928—0.947,表明2个品种内部样品之间表达模式的相似度较高,重复性好,试验结果可信度高。
根据差异基因筛选标准,以福清山羊为对照,获得了努比亚黑山羊和福清山羊发情期卵巢组织的DEGs 149个,其中上调基因53个,下调基因96个,图2为DEGs火山图。进一步根据DEGs在2个品种卵巢组织的差异表达值和文献检索,初步认为输卵管素基因(oviductin,OVN)、类固醇合成快速调节蛋白基因(steroidogenic acute regulatory protein,STAR)、早期生长应答1基因(early growth response 1,EGR1)可作为福清山羊繁殖性能的候选基因。
图1
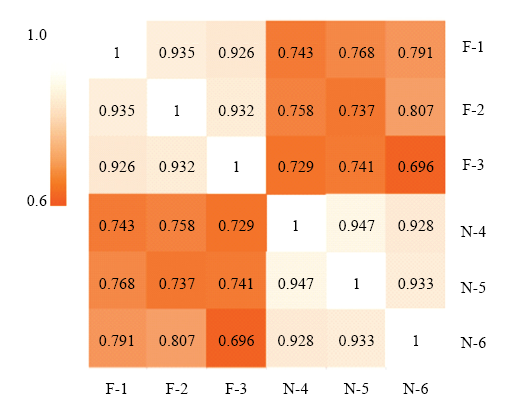 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1样品的表达量相关性热图
Fig. 1Correlation heat map of gene expression level in 6 samples
图2
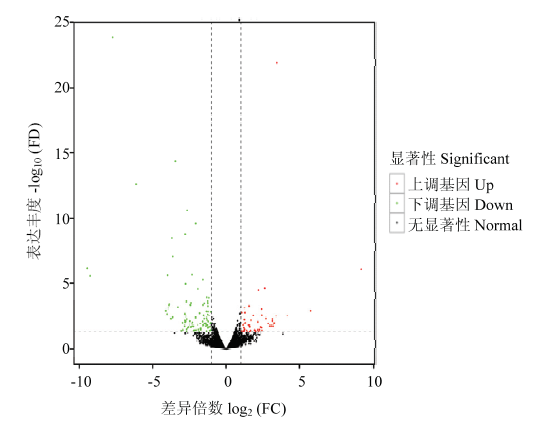 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2差异表达基因火山图
图中绿点表示表达量下调的基因,红点表示表达量上调的基因,黑点表示无显著性差异基因
Fig. 2Volcano plot of differentially expressed genes
Green dots are significantly down-regulated genes. Red dots are significantly up-regulated genes. Black dots are genes without differentially expression
2.2.2 DEGs的GO功能富集 GO注释系统是一个有向无环图,包含生物学过程(biological process, BP),细胞组分(cellular component, CC)和分子功能(molecular function, MF)3个主要分支。筛选出的149个DEGs中的108个基因能够被GO数据库注释(图3)。被注释的DEGs分别参与了BP、CC和MF的22、19和20个功能亚分类。在BP分类中,细胞过程、单一的生物过程和生物调节参与的DEGs最多,与繁殖相关联的繁殖过程和繁殖的DEGs也被注释。在CC分类中,参与细胞部分;2细胞;3细胞器的DEGs最多。在MF分类中,DEGs在结合中所占比例最高,其次为催化活性及分子传感器活性。
图3
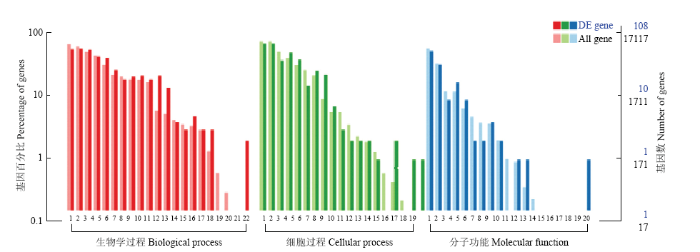 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3差异表达基因GO注释
生物学过程Biological process:1细胞过程 Cellular process;2单一的生物过程 Single-organism process;3生物调节 Biological regulation;4代谢过程 Metabolic process;5刺激反应 Response to stimulus;6多细胞生物过程 Multicellular organismal process;7信号 Signaling;8定位 Localization;9发育过程 Developmental process;10组织或生物起源细胞组件 Cellular component organization or biogenesis;11免疫系统过程 Immune system process;12多生物体过程 Multi-organism process;13繁殖过程Reproductive process;14移动Locomotion;15生物粘附Biological adhesion;16繁殖Reproductive;17生长Growth;18有节奏的过程Rhythmic process;19激素分泌 Hormone secretion;20生物相Biological phase;21细胞杀伤 Cell killing;22细胞聚集Cell aggregation
Fig. 3GO annotation of DEGs
细胞过程Cellular process:1细胞部分 Cell part;2细胞 Cell;3细胞器 Organelle;4膜 Membrane;5膜部分 Membrane part;6细胞器部分Organelle part;7高分子复合物 Macromolecular complex;8胞外区Extracellular region;9膜包围内腔Membrane-enclosed lumen;10细胞外区域部分Extracellular region part;11细胞连接Cell junction;12细胞外基质Extracellular matrix;13突触Synapse;14突出部分Synapse part;15细胞外基质部分 Extracellular matrix part;16胶原三聚体Collagen trimer;17细胞核Nucleoid;18病毒体Virion;19病毒体部分Virion part
分子功能Molecular function:1结合 Binding;2催化活性 Catalytic activity;3分子传感器活性Molecular transducer activity;4受体活性Receptor activity;5运输活性Transporter activity;6核酸结合转录因子活性Nucleic acid binding transcription factor activity;7酶调节活性Enzyme regulator activity;8结构分子活性Structural molecule activity;9蛋白结合转录因子活性Protein binding transcription factor activity;10鸟嘌呤核苷酸交换因子活性Guanyl- nucleotide exchange factor activity;11电子载体活性Electron carrier activity;12抗氧化活性Antioxidant activity;13通道调节活性Channel regulator activity;17金属伴侣活性Metallochaperone activity;18排斥活动Chemorepellent activity;19蛋白标签Protein tag ;20成形素活性 Morphogen activity
2.2.3 差异表达基因的COG分类 利用COG数据库可以对基因产物进行直系同源分类。筛选出的149个DEGs中,有30个DEGs能够被COG数据库注释,分类统计结果见图4。其中一般的功能预测基因数目最多,其次为脂质运输与代谢。
图4
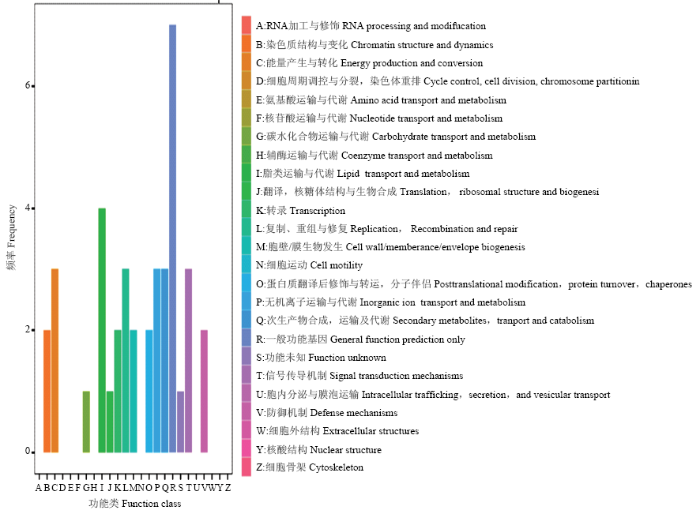 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4差异表达基因COG注释分类统计图
Fig. 4COG annotation classification statistics of DEGs
2.2.4 差异表达基因的KEGG注释 经过分析,可被注释到KEGG数据库的91个DEGs参与了多个代谢通路,将差异表达基因KEGG的注释结果按照KEGG中通路类型进行分类,分类图见图5。差异表达基因参与最多的是人类疾病相关的通路,其次是生物系统相关通路和环境信息处理相关通路。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5差异表达基因显著富集的KEGG通路
Fig. 5List of KEGG pathway for DEGs
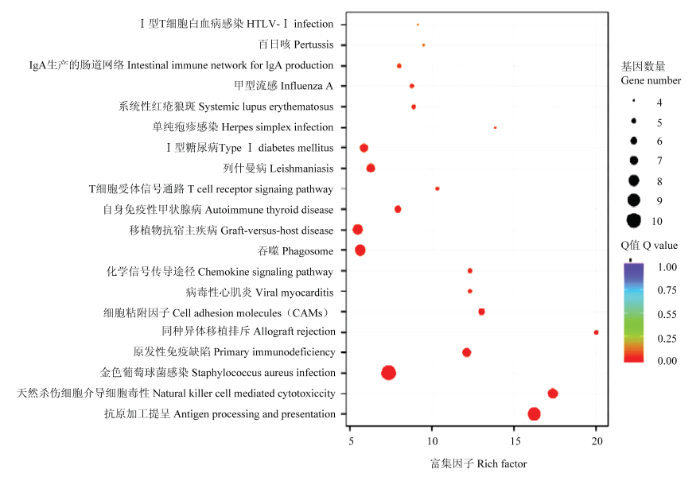
以KEGG数据库总通路为单位,应用超几何检验,找出与整个基因组背景相比,在DEGs中显著性富集的通路,KEGG通路分析表明DEGs共富集到21条信号通路中,6条通路显著富集(P<0.05)。图6为DEGs的KEGG通路富集分析结果,图中呈现了显著性Q值最小的20个通路,其中自身免疫性甲状腺病、同种异体移植排斥、吞噬体、病毒性心肌炎、哮喘5个代谢通路的富集显著性最可靠,推测其相关的DGEs可能与山羊繁殖性能相关。
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6差异表达基因KEGG通路富集散点图
Fig. 6Enriched scatter map of DEGs KEGG pathway
2.3 新基因分析
使用Cufflinks软件对Mapped Reads进行拼接,并与参考基因组注释信息进行比较,寻找原来未被注释的转录区,发掘该物种的新转录本和新基因,补充和完善参考基因组注释信息。过滤掉编码的肽链过短(少于50个氨基酸残基)或只包含单个外显子的序列,共发掘1 506个新基因(转录本),其中25个在2个品种中差异表达。2.4 SNP/InDel分析
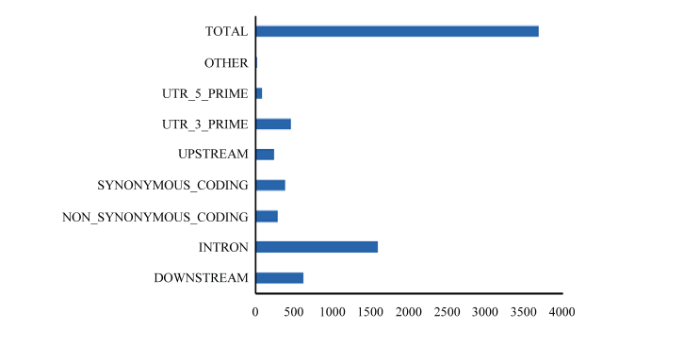
经过与参考基因组序列的对比,已知的124个DEGs中发现了3 689个SNP/InDel位点,其中6个DEGs未发现突变(图7)。位于内含子区域(INTRON)的SNP/InDel位点最多,占基因序列突变总量的43.2%,其次为下游序列(DOWNSTREAM),占比16.8%。而引起基因编码序列非同义突变(NON- SYNONYMOUS-CODING)的SNP/InDel位点占比7.8%。图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7差异表达基因的SNP类型统计
Fig. 7The number of different SNP/InDel effects of DEGs
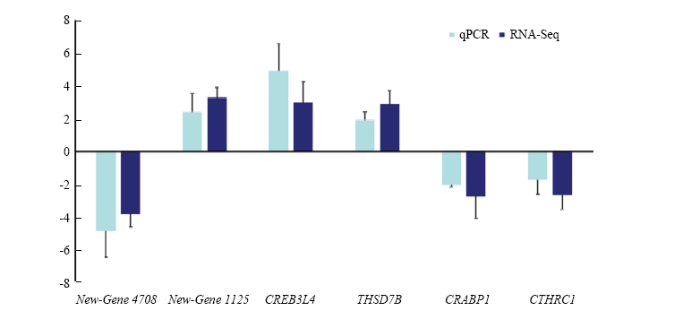
2.5 qRT-PCR验证测序
6个基因(转录本)的 qRT-PCR结果见图8,选择的目的基因在2个品种个体间表达变化模式与转录组测序结果一致,表明本研究利用转录组测序获得差异表达基因以及新基因(转录本)序列数据较为准确。图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8测序结果的qRT-PCR验证
Fig. 8Validation of sequencing results by qRT-PCR
3 讨论
随着高通量测序技术的发展和广泛应用,动物遗传学从以前候选基因、单一性状为主的微观研究转向以物种进化构架内的全基因组水平、多性状、组学化的宏观研究,利用高通量测序技术研究畜禽主要经济性状取得了较大的研究进展[26]。卵巢是雌性动物重要的繁殖器官,具有提供卵细胞和产生类固醇激素的功能,常用于发情和排卵等繁殖生理过程的研究。罗庆苗等[8]利用DGE技术研究了小尾寒羊高繁殖性状相关基因,但筛选的DEGs直接与已知动物繁殖性状相关基因极少,DI等[9,10]在不同品种研究中也得到了类似结果,与本研究结果一致,并且不同品种筛选出的与繁殖性状相关的DEGs也不尽相同。兰道亮等[27]认为部分DEGs可能涉及卵巢的相关功能及活动,但缺乏现有试验验证;同时可能也与器官功能的复杂性和多样性有关[8,28]。Ovn具有促进受精和早期胚胎发育的作用[29],张译夫等[30]研究表明其还在卵母细胞成熟和排卵过程中具有重要的生物学作用。Ovn的合成受卵巢类固醇激素的调控[31],STAR编码的蛋白通过增强胆固醇转化为孕烯醇酮在类固醇激素合成的急性调节中起着关键作用[32],罗庆苗等[8]研究认为STAR的显著下调可导致FecBBFecBB小尾寒羊性激素合成能力下降而提高繁殖力。本研究发现,与福清山羊对比,努比亚黑山羊发情期卵巢组织的Ovn表达量显著上调,而STAR表达量显著下调,很可能是造成2品种繁殖性能差异的关键因素之一。EGR1涉及不同组织多种基因的反式激活及功能调控,在牛的排卵前卵泡中EGR1具有促性腺激素依赖性的诱导表达[33],吴阳升等[34]研究表明EGR1参与绵羊卵泡早期发育功能,本研究中该基因显著上调表达,可作为福清山羊繁殖性能候选基因进行进一步验证研究。此外,其他DEGs(包括新转录本)与繁殖性能相关的确切功能及作用尚不明确,有待进一步的实验验证。
由于针对不同羊品种RNA-Seq筛选到的DEGs存在差异,对应差异表达基因KEGG显著富集到的代谢通路也不尽相同[9,10,11]。罗庆苗等[8]在小尾寒羊中富集到细胞内吞过程、Notch信号通路和嘧啶代谢3个调控通路,LING等[3]在安徽白山羊繁殖力差异研究中富集到细胞外基质受体相互作用、粘着斑和肌动蛋白细胞骨架调节等11条信号通路,本研究富集到自身免疫性甲状腺病、同种异体移植排斥、吞噬体、病毒性心肌炎、哮喘等免疫和人类疾病的代谢通路,与兰道亮等[27,35-36]的研究结果相似,MIAO等[35]认为疾病相关通路的富集表明代谢、免疫等途径可能调控家畜繁殖力,兰道亮等[27]则认为DEGs富集的疾病通路虽然源于疾病发生的生理过程,但这些通路中的部分基因可能涉及卵巢的相关功能及活动。表明畜禽繁殖性能形成的分子机理其调控机制十分复杂,造成不同品种间高繁殖力差异的调控基因可能存在差异。
繁殖性状是羊的重要经济性状,而繁殖性状是受多基因控制的,遗传力低,传统的选择方法对其遗传进展的改良非常缓慢,利用显著影响羊繁殖性能的分子标记进行辅助选择能提高选育效果[29]。但研究表明分子标记具有品种特异性,因此在不同品种中筛选出更丰富、更适用的分子标记是当前的重要工作之一[3]。RNA-Seq高通量转录组技术在完善基因结构及挖掘新转录本及新基因方面具有强大的优势[27]。本研究发掘了1 506个新基因(转录本),其中25个为2个品种的差异表达基因。经过进一步与参考基因组序列的对比,在已知的124个DEGs中发现了3689个SNP/InDel位点,引起基因编码序列非同义突变(NON-SYNONYMOUS-CODING)的SNP/InDel位点占比7.8%,验证这些潜在影响福清山羊繁殖性能的分子标记、评价其在福清山羊群体中的遗传效应,筛选出控制福清山羊繁殖性状的主效基因(分子标记)是下一步研究的关键。
4 结论
本研究通过对DRI指数存在明显差异的福清山羊和努比亚黑山羊的发情期卵巢组织进行RNA-Seq分析,在转录组水平上筛选出了可能与羊繁殖性能的149个DGEs,并发掘了1 506个新基因(转录本)。初步认为OVN、STAR、EGR1在福清山羊和努比亚黑山羊发情期卵巢组织中的差异表达可能是造成2品种繁殖力差异的关键因素之一,可作为福清山羊繁殖性能的候选基因进行深入验证研究。该研究为揭示福清山羊繁殖性能形成的分子机制提供了线索,为开发和利用畜禽优良性状分子标记提供了参考依据。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
[本文引用: 4]
[本文引用: 1]
DOI:10.1038/nrg2484 [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 5]
URL [本文引用: 5]
[本文引用: 3]
[本文引用: 3]
URL [本文引用: 2]
URL [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
.
URL [本文引用: 1]
URL [本文引用: 1]
.
URL [本文引用: 1]
DOI:10.1186/gb-2013-14-4-r36URL [本文引用: 1]
[本文引用: 1]
DOI:10.1093/bioinformatics/btp113URL [本文引用: 1]
DOI:10.12688/f1000researchURL [本文引用: 1]
DOI:10.1186/1471-2164-13-524URL [本文引用: 1]
DOI:10.1093/bioinformatics/btp612URL [本文引用: 1]
[本文引用: 2]
.
[本文引用: 1]
DOI:10.1093/nar/gkh063URL [本文引用: 1]
DOI:10.1101/gr.107524.110URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 4]
URL [本文引用: 4]
URL [本文引用: 1]
URL [本文引用: 1]
DOI:10.1530/rep.0.1230355URL [本文引用: 2]
URLMagsci [本文引用: 1]

输卵管蛋白(oviductin, Ovn)具有促进受精和早期胚胎发育的作用。本研究旨在克隆牦牛(Bos grunniens)输卵管蛋白基因(Ovn)的完整开放阅读框序列,并从基因和蛋白水平分析其在发情周期的表达差异。本研究RT-PCR克隆牦牛输卵管蛋白cDNA,并采用实时荧光定量PCR(quantitative Real-time PCR, qRT-PCR)和免疫组织化学方法分别分析Ovn在卵泡期前期阶段(卵泡直径3~6 mm)、卵泡期后期阶段(卵泡直径>6 mm)、黄体期前期阶段(可见一个或多个血体)和黄体期后期阶段(可见一个或多个黄体)输卵管伞部和壶腹部的表达差异。成功克隆出牦牛Ovn的cDNA序列(GenBank登录号: KF303541),其包含的完整开放阅读框由1 443个核苷酸组成,共编码480个氨基酸。序列分析显示,牦牛输卵管蛋白cDNA序列与黄牛(Bos taurus)和印度水牛(Bubalus bubalis)具有很高的同源性,Ovn的氨基酸序列进化树表明,牦牛输卵管蛋白与黄牛的同源性最高,达到99.1%,与水牛、绵羊(Ovis aries)和冠毛猕猴(Macaca radiate)的同源性依次为93.7%、88.1%和79.8%。qRT-PCR结果显示,卵泡期后期阶段输卵管蛋白mRNA的表达最高,黄体期前期和黄体期后期阶段表达依次减少,卵泡期前期阶段相对表达量最低,而壶腹部和伞部的表达差异不显著。蛋白免疫标记在卵泡期后期和黄体期前期阶段较明显,其他阶段则减弱。本研究得到了包含完整开放阅读框的牦牛Ovn的cDNA序列,检测出发情周期中其卵泡期后期阶段输卵管表达最高,表明其在卵母细胞成熟和排卵过程中具有重要的生物学作用,为进一步研究其在提高受精率和早期胚胎发育率中的作用机制提供了一定的理论基础。
URLMagsci [本文引用: 1]
输卵管蛋白(oviductin, Ovn)具有促进受精和早期胚胎发育的作用。本研究旨在克隆牦牛(Bos grunniens)输卵管蛋白基因(Ovn)的完整开放阅读框序列,并从基因和蛋白水平分析其在发情周期的表达差异。本研究RT-PCR克隆牦牛输卵管蛋白cDNA,并采用实时荧光定量PCR(quantitative Real-time PCR, qRT-PCR)和免疫组织化学方法分别分析Ovn在卵泡期前期阶段(卵泡直径3~6 mm)、卵泡期后期阶段(卵泡直径>6 mm)、黄体期前期阶段(可见一个或多个血体)和黄体期后期阶段(可见一个或多个黄体)输卵管伞部和壶腹部的表达差异。成功克隆出牦牛Ovn的cDNA序列(GenBank登录号: KF303541),其包含的完整开放阅读框由1 443个核苷酸组成,共编码480个氨基酸。序列分析显示,牦牛输卵管蛋白cDNA序列与黄牛(Bos taurus)和印度水牛(Bubalus bubalis)具有很高的同源性,Ovn的氨基酸序列进化树表明,牦牛输卵管蛋白与黄牛的同源性最高,达到99.1%,与水牛、绵羊(Ovis aries)和冠毛猕猴(Macaca radiate)的同源性依次为93.7%、88.1%和79.8%。qRT-PCR结果显示,卵泡期后期阶段输卵管蛋白mRNA的表达最高,黄体期前期和黄体期后期阶段表达依次减少,卵泡期前期阶段相对表达量最低,而壶腹部和伞部的表达差异不显著。蛋白免疫标记在卵泡期后期和黄体期前期阶段较明显,其他阶段则减弱。本研究得到了包含完整开放阅读框的牦牛Ovn的cDNA序列,检测出发情周期中其卵泡期后期阶段输卵管表达最高,表明其在卵母细胞成熟和排卵过程中具有重要的生物学作用,为进一步研究其在提高受精率和早期胚胎发育率中的作用机制提供了一定的理论基础。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
URL [本文引用: 1]
DOI:10.1016/j.mce.2014.12.023URL [本文引用: 2]
DOI:10.11843/j.issn.0366-6964.2016.07.004URLMagsci [本文引用: 1]
<p>本研究通过构建和分析小尾寒羊间情期和发情期卵巢组织microRNA(miRNA)表达谱,筛选两个时期差异表达的microRNAs,为研究microRNA调控小尾寒羊繁殖过程提供相应的理论基础。利用活体手术法在发情期(第1天)和间情期(第13天)分别采集一侧卵巢。从卵巢中提取总RNA,利用illumina Hiseq2000测序平台获取RNA数据,对表达谱和差异表达microRNAs进行生物信息学分析。实时荧光定量PCR(qRT-PCR)验证差异表达的microRNAs在小尾寒羊卵巢中表达水平。结果,成功地构建出小尾寒羊间情期和发情期microRNA的表达谱,oar-miR-99a和oar-miR-143分别是间情期和发情期表达量最高的microRNA,并筛选出在两个时期间3个显著差异表达的microRNAs,分别为oar-miR-200a、oar-miR-200b和oar-miR-200c。利用qRT-PCR对随机选择的2个显著差异表达的microRNAs进行验证,其表达水平和RNA-Seq分析结果一致。microRNA表达谱为后续的绵羊卵巢microRNA研究提供更详尽的信息。结合靶基因预测及通路富集分析,推测差异表达的microRNAs是通过代谢和免疫途径调控卵巢周期性活动。</p>
DOI:10.11843/j.issn.0366-6964.2016.07.004URLMagsci [本文引用: 1]
<p>本研究通过构建和分析小尾寒羊间情期和发情期卵巢组织microRNA(miRNA)表达谱,筛选两个时期差异表达的microRNAs,为研究microRNA调控小尾寒羊繁殖过程提供相应的理论基础。利用活体手术法在发情期(第1天)和间情期(第13天)分别采集一侧卵巢。从卵巢中提取总RNA,利用illumina Hiseq2000测序平台获取RNA数据,对表达谱和差异表达microRNAs进行生物信息学分析。实时荧光定量PCR(qRT-PCR)验证差异表达的microRNAs在小尾寒羊卵巢中表达水平。结果,成功地构建出小尾寒羊间情期和发情期microRNA的表达谱,oar-miR-99a和oar-miR-143分别是间情期和发情期表达量最高的microRNA,并筛选出在两个时期间3个显著差异表达的microRNAs,分别为oar-miR-200a、oar-miR-200b和oar-miR-200c。利用qRT-PCR对随机选择的2个显著差异表达的microRNAs进行验证,其表达水平和RNA-Seq分析结果一致。microRNA表达谱为后续的绵羊卵巢microRNA研究提供更详尽的信息。结合靶基因预测及通路富集分析,推测差异表达的microRNAs是通过代谢和免疫途径调控卵巢周期性活动。</p>

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}