 ,11
,11 2
3
Genome-Wide Association Study of Chlorophyll Content in Maize
SHI DaKun1, YAO TianLong1, LIU NanNan2, DENG Min3, DUAN HaiYang1, WANG LuLin1, WAN Jiong1, GAO JiongHao1, XIE HuiLing1, TANG JiHua1, ZHANG XueHai,11 2
3
通讯作者:
责任编辑: 李莉
收稿日期:2019-01-21接受日期:2019-02-28网络出版日期:2019-06-01
| 基金资助: |
Received:2019-01-21Accepted:2019-02-28Online:2019-06-01
作者简介 About authors
史大坤,E-mail:912320091@qq.com。
姚天茏,E-mail:yaotianlong0629@qq.com。

摘要
关键词:
Abstract
Keywords:
PDF (5697KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
史大坤, 姚天茏, 刘楠楠, 邓敏, 段海洋, 王路林, 万炯, 高炯浩, 谢惠玲, 汤继华, 张雪海. 玉米叶绿素含量的全基因组关联分析[J]. 中国农业科学, 2019, 52(11): 1839-1857 doi:10.3864/j.issn.0578-1752.2019.11.001
SHI DaKun, YAO TianLong, LIU NanNan, DENG Min, DUAN HaiYang, WANG LuLin, WAN Jiong, GAO JiongHao, XIE HuiLing, TANG JiHua, ZHANG XueHai.
0 引言
【研究意义】植物干物质的90%—95%通过光合作用产生,作物产量主要来源于叶片的光合产物[1]。而叶绿素含量是决定作物光合效率和生物量或产量的重要因素[2],也是高光效育种的重要育种目标之一。因此,对玉米叶绿素含量的遗传基础进行解析可为玉米高光效理想株型的设计育种提供理论支持。【前人研究进展】叶绿素是植物叶片的主要光合色素,也是植物光合作用的物质基础,已成为研究玉米生长特性及生理变化的重要指标[2]。孙红等[3]通过研究玉米生长期冠层反射光谱与叶绿素含量的关系,表明玉米拔节期和灌浆期冠层反射光谱与叶绿素含量呈正相关,从而认为叶绿素对玉米的生长至关重要。左宝玉等[4]研究表明玉米叶片的叶绿素含量为多基因控制的数量性状。文自翔等[5]通过亲本杂交建立的遗传群体结合连锁分析表明,QTL(quantitative trait locus)定位的准确性由亲本之间的遗传差异所决定,还发现一些位点在特定双亲间没有发生分离和重组,认为使用基于连锁分析的QTL定位存在一定的局限性。近年来,以连锁不平衡为基础的全基因组关联分析已被证明是一种解析复杂农艺性状遗传基础的强大工具[6,7,8]。随着测序技术发展、测序成本降低及统计模型改善,全基因组关联分析在作物QTL鉴定中被广泛应用[10]。ATWELL等[11]用约200份拟南芥材料,结合216 130个SNPs标记,对其开花期、抗病及发育相关的107个表型进行GWAS,鉴定到大量主效位点,其中一些位点内含已知基因,说明GWAS对植物复杂性状遗传基础的解析是可行的。THORNSBERRY等[12]首次将关联分析用于玉米开花期研究,成功发掘到Dwarf8并验证其功能。而在玉米中,较早的GWAS则是对553份自交系成熟籽粒脂肪酸含量的分析[13]。在水稻和拟南芥中,部分叶绿素含量相关的基因已被克隆,WANG等[14]通过对水稻剑叶期的剑叶SPAD(soil and plant analyzer development)值、叶绿素a、叶绿素b进行测定,并结合高密度标记对不同亚型的水稻群体进行GWAS,检测到多个叶绿素相关性状显著位点,并发现已知基因Ghd7也影响叶绿素含量;拟南芥AT5G18660编码二乙烯基叶绿素酯还原酶,该酶是二乙烯脱植基叶绿素a形成的关键酶,该基因突变会导致拟南芥叶片变黄[15]。在拟南芥发育初期,叶绿素合酶基因Ygl1的突变体使叶片呈黄绿色,植株生长缓慢,野生型的绿色程度显著高于该突变体[16]。【本研究切入点】在玉米中,叶绿素含量QTL鲜有报道,且对其自然变异遗传基础的了解较少。玉米具有丰富的遗传多样性及快速的连锁不平衡衰减等特性,是进行全基因组关联分析的理想材料[17]。玉米不同叶位叶片的叶绿素含量决定各叶位叶光合强度,进而导致各叶位叶对籽粒产量贡献不同,而棒三叶(穗位叶及穗位叶的上下两片叶)作为有效光合层,对玉米籽粒产量贡献最大[18]。因此,通过全基因组关联分析对玉米棒三叶叶绿素含量的遗传结构进行解析,为快速获取叶绿素含量相关的标记及候选基因尤为重要,同时也可为高光合效率玉米品种的改良提供理论支持。【拟解决的关键问题】本研究以538份玉米自交系构成的关联群体为研究对象,通过对其棒三叶叶绿素含量进行测定,并结合高密度SNP标记,用3种不同的统计模型(Q、K、Q+K)对叶绿素含量进行GWAS分析,随后结合最优模型的GWAS结果及eQTL分析,探索玉米叶绿素含量的遗传基础,为玉米高光效的理想株型设计育种提供理论基础。1 材料与方法
1.1 试验材料与田间种植
所用关联群体为华中农业大学严建兵教授提供,由538份玉米自交系构成。该群体于2012年夏季种植于河南鹤壁(12年鹤壁)鹤壁农业科学院浚县试验站(浚县,北纬N35°41′51.37″,东经E114°18′22.96″),2017年冬季种植于海南三亚(17年三亚)河南农业大学南繁基地(三亚,北纬N18°22′55.49″,东经E108°58′32.47″),2018年春季种植于湖南长沙(18年长沙)湖南农业大学试验基地(长沙,北纬N28°12′40.27″,东经E113°16′59.29″),2018年夏季种植于河南原阳(18年原阳)国家2011计划河南农业大学现代农业科技园区(原阳,北纬N35°03′58.63″,东经E113°56′2.78″)及河南永城(18年永城)棉花原种场(永城,北纬N33°52′53.11″,东经E116°27′11.51″)。采用单行区种植,单次重复,行长3 m,株距0.25 m,行距0.65 m,种植密度约63 000株/hm2。1.2 叶绿素含量测定
为了准确测定玉米棒三叶的叶绿素含量,于授粉后5 d,使用手持SPAD仪(型号:SPAD-502 plus)对关联群体棒三叶(穗上叶(above the uppermost ear leaf)、穗位叶(uppermost ear leaf)、穗下叶(below the uppermost ear leaf))的SPAD值(叶绿素相对含量)进行测定。每行测定5株,对于每一株的棒三叶,选择每片叶的中间三分之一位置进行测定,重复3次(误差小于5%),3次测量均值作为该片叶的叶绿素含量;5株对应叶片的SPAD值的均值作为该自交系棒三叶对应叶片的叶绿素含量并用于后续的表型一般统计分析和全基因组关联分析。1.3 数据处理与分析
利用Excel 2016对每个环境每个基因型棒三叶叶绿素含量的异常值进行剔除,并获得平均值。利用R语言的corrplot函数对不同环境下叶绿素含量进行相关性分析[19]。利用Excel 2016对5个环境下棒三叶叶绿素含量进行无重复双因素方差分析。利用R语言的lme4包[20]的混合线性模型[Y =(1|LINE)+(1|ENV)+(1|LINE:ENV)]计算5个环境下每一个材料每一性状的最佳线性无偏预测(best linear unbiased prediction,Blup)值,基因型为随机因子,环境为固定因子,公式中,Y为性状数据,括号表示随机效应,“1|”表示分组,“:”表示互作,LINE表示所有材料,ENV表示环境。Blup值可减少不平衡数据造成的预测偏差,最终也用于一般统计分析及全基因组关联分析。广义遗传力的计算采用混合线性模型将基因型和环境作为随机效应,得到δ2 g和δ2 e的估计值。利用公式H2=δ2 g/(δ2 g+δ2 e/n)计算每个部位的叶绿素含量的遗传力,其中,δ2 g为遗传方差,δ2 e为协方差,n为环境数[21]。1.3.1 全基因组关联分析 所用基因型为YANG等[22]推算得到的覆盖玉米全基因组且最小等位基因频率≥0.05的558 629个SNP。考虑到很多标记间可能存在高度的连锁不平衡,利用GEC软件[23]计算该套标记的有效标记数(effective number,En),并以该有效标记数En作为判断性状-标记显著的依据(P≤1/En)。统计学功效(即检测到真实显著关联的能力)是进行全基因组关联分析的首要考虑因素。因此,在Tassel 3.0软件[24]中使用3种模型,即只控制亲缘关系的K模型、只控制群体结构的Q模型、同时控制群体结构和亲缘关系的Q+K模型对关联群体棒三叶叶绿素含量分别进行全基因组关联分析,通过3种模型Quantile-quantile(QQ)图的比较,选择最优模型并对其结果进行进一步解析。
前期已有研究用558 629个SNP对该关联群体的连锁不平衡(linkage disequilibrium,LD)衰减程度进行评价,结果表明,该群体的LD衰减距离为50 kb(R2=0.1)[25]。因此,定义一个显著SNP标记物理位置的上下游各50 kb的区间范围为一个位点。基于B73参考基因组(RefGen_v2),从玉米数据库MaizeGDB(http://www.maizegdb.org)中下载玉米全基因组基因列表并从列表中搜索每个显著位点内的所有候选基因。根据基因的功能注释及其在B73参考基因组中的表达部位及表达量,选择一个最可能的候选基因作为该位点的候选基因。
1.3.2 表达量QTL分析 利用RNA-seq技术深度测序368份关联群体自交系授粉后15 d的籽粒,每个自交系有50%以上的基因表达且至少有10个reads可用,共获得了28 850个基因的表达量[26],通过eQTL定位鉴定调控基因表达的位点,方法同表型的全基因组关联分析。
2 结果
2.1 玉米棒三叶叶绿素含量表型数据分析
通过对该关联群体叶绿素含量进行分析,发现多个环境的穗位叶叶绿素含量均高于穗上叶和穗下叶。同时发现除鹤壁穗下叶叶绿素含量的偏度值绝对值大于1外,其余环境棒三叶叶绿素含量偏度值的绝对值均小于1(表1),表明不同叶片的叶绿素含量均遵从正态分布,为典型的数量性状。Table 1
表1
表1关联群体不同环境下玉米棒三叶叶绿素含量的描述统计分析
Table 1
| 环境 Environment | 性状 Traits | 变异范围 Range | 均值 Mean | 标准差 sd. | 偏度 Ske. | 峰度 Kur. |
|---|---|---|---|---|---|---|
| 2012年鹤壁Hebi, 2012 | 穗上叶Above the uppermost ear leaf | 26.25—67.85 | 52.98 | 6.91 | -0.87 | 1.51 |
| 穗位叶Uppermost ear leaf | 22.25—68.60 | 52.82 | 7.17 | -0.97 | 1.69 | |
| 穗下叶Below the uppermost ear leaf | 18.25—66.20 | 52.92 | 7.58 | -1.08 | 1.92 | |
| 2017年三亚Sanya, 2017 | 穗上叶Above the uppermost ear leaf | 29.25—68.45 | 50.97 | 6.39 | -0.46 | 0.90 |
| 穗位叶Uppermost ear leaf | 25.83—70.45 | 51.08 | 6.37 | -0.43 | 1.16 | |
| 穗下叶Below the uppermost ear leaf | 25.65—69.60 | 51.03 | 6.67 | -0.48 | 0.92 | |
| 2018年长沙Changsha, 2018 | 穗上叶Above the uppermost ear leaf | 27.90—82.93 | 53.84 | 6.20 | -0.26 | 3.08 |
| 穗位叶Uppermost ear leaf | 29.47—82.95 | 53.83 | 6.10 | -0.08 | 2.98 | |
| 穗下叶Below the uppermost ear leaf | 22.88—95.28 | 53.93 | 7.44 | -0.09 | 4.59 | |
| 2018年原阳Yuanyang, 2018 | 穗上叶Above the uppermost ear leaf | 30.80—63.35 | 48.05 | 5.74 | -0.48 | 0.97 |
| 穗位叶Uppermost ear leaf | 30.32—61.99 | 47.88 | 5.80 | -0.39 | 0.84 | |
| 穗下叶Below the uppermost ear leaf | 29.73—60.64 | 47.97 | 6.13 | -0.33 | 0.52 | |
| 2018年永城Yongcheng, 2018 | 穗上叶Above the uppermost ear leaf | 40.92—69.59 | 54.67 | 6.61 | 0.00 | 0.00 |
| 穗位叶Uppermost ear leaf | 38.30—68.48 | 54.86 | 6.74 | -0.20 | 0.37 | |
| 穗下叶Below the uppermost ear leaf | 37.85—69.15 | 54.99 | 6.96 | -0.03 | -0.04 | |
| 综合Blup | 穗上叶Above the uppermost ear leaf | 43.46—58.68 | 52.51 | 3.16 | -0.57 | 1.02 |
| 穗位叶Uppermost ear leaf | 40.45—60.49 | 52.57 | 3.61 | -0.55 | 0.78 | |
| 穗下叶Below the uppermost ear leaf | 40.20—64.44 | 52.55 | 4.13 | -0.50 | 0.64 |
新窗口打开|下载CSV
2.2 玉米棒三叶叶绿素含量的相关性分析、方差分析及遗传力
通过对不同环境玉米棒三叶叶绿素含量的相关性分析,发现不同环境的棒三叶(穗上叶、穗位叶、穗下叶)叶绿素含量间均呈正相关(图1),并且发现各环境下棒三叶叶绿素含量间的相关系数,大多在0.6以上,推测棒三叶叶绿素的生物合成调控存在着相互协同促进作用。图1
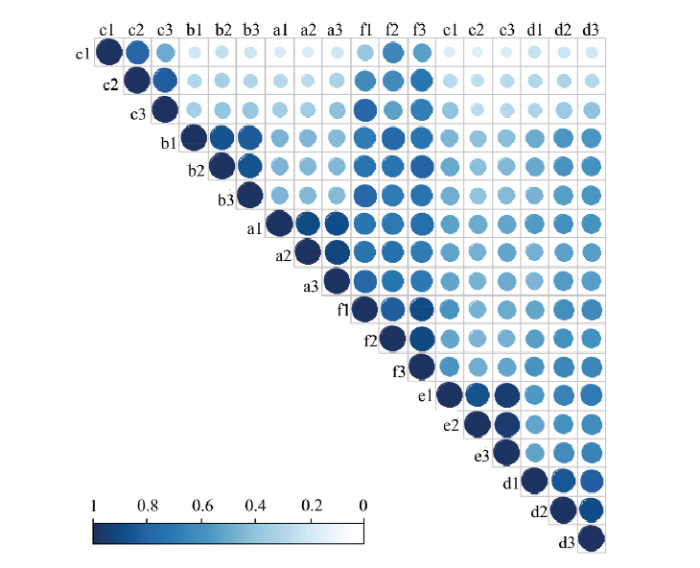 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图16个环境关联群体棒三叶叶绿素含量的相关性分析
a、b、c、d、e、f分别表示2012年鹤壁、2017年三亚、2018年长沙、2018年永城、2018年原阳及Blup等6个环境。字母后面的数字1—3分别代表穗上叶、穗位叶、穗下叶。下同
Fig. 1Correlation coefficients for chlorophyll content in maize three leaves of association population under six environments
a, b, c, d, e, f: Stands for Hebi in 2012, Sanya in 2017, Changsha in 2018, Yongcheng in 2018, Yuanyang in 2018 and Blup value, respectively. The number followed character, 1-3: Stands for above the uppermost ear leaf, uppermost ear leaf and below the uppermost ear leaf, respectively. The same as below
方差分析表明关联群体棒三叶叶绿素含量的基因型效应、环境效应、基因型与环境互作效应均达极显著水平(表2),说明棒三叶不同叶片的叶绿素含量存在显著的遗传变异,且受环境影响较大;若在环境影响较大的不同环境间检测到相同的位点或基因,则说明这些位点或基因可以稳定遗传。此外,穗上叶、穗位叶和穗下叶的叶绿素含量的遗传力分别为0.66、0.66和0.67。
Table 2
表2
表25个环境玉米棒三叶叶绿素含量的方差分析
Table 2
| 性状 Trait | 变异来源 Source | 离均差平方和 Type sum of squares | 自由度 Df | 均方 Mean square | F值 F-value |
|---|---|---|---|---|---|
| 穗上叶 Above the uppermost ear leaf | 环境E | 62052.294 | 4 | 15513.073 | 1202.047** |
| 基因型G | 51835.582 | 537 | 96.528 | 7.479** | |
| 基因型×环境 G×E | 122215.029 | 2148 | 56.897 | 4.408** | |
| 误差Error | 138863.674 | 10760 | 12.906 | ||
| 总变异Total | 374966.579 | 13449 | |||
| 穗位叶 Uppermost ear leaf | 环境E | 74363.802 | 4 | 18590.951 | 1466.621** |
| 基因型G | 56009.168 | 537 | 104.300 | 8.228** | |
| 基因型×环境 G×E | 126440.437 | 2148 | 58.864 | 4.643** | |
| 误差Error | 136394.238 | 10760 | 12.676 | ||
| 总变异Total | 393207.645 | 13449 | |||
| 穗下叶 Below the uppermost ear leaf | 环境E | 101559.963 | 4 | 25389.991 | 1822.287** |
| 基因型G | 59378.695 | 537 | 110.575 | 7.936** | |
| 基因型×环境 G×E | 135210.636 | 2148 | 62.947 | 4.518** | |
| 误差Error | 149919.461 | 10760 | 13.933 | ||
| 总变异Total | 446068.755 | 13449 |
新窗口打开|下载CSV
2.3 玉米棒三叶叶绿素含量的全基因组关联分析
2.3.1 阈值确定 利用GEC软件计算558 629个SNPs的有效标记数(En),结果表明,En为250 345,软件建议的P=3.99×10-6(1/En),该值作为关联分析的显著阈值。2.3.2 关联分析结果 对6个环境下的棒三叶叶绿素含量的GWAS结果进行分析,通过比较3种模型的QQ图(图2),针对不同模型对假阳性的控制来看,Q模型(图中红点)最差,K模型(图中黑点)和Q+K模型(图中蓝点)优于Q模型。从对假阴性的控制效果看,Q+K模型比K模型稍微过于严格。因此,选用表现相对好的K模型的GWAS结果用于后续的位点挖掘和基因预测。基于K模型,在P≤3.99×10-6下,6个环境一共检测到29个SNPs与棒三叶叶绿素含量显著关联,涉及到18个位点(图3;表3)。图2
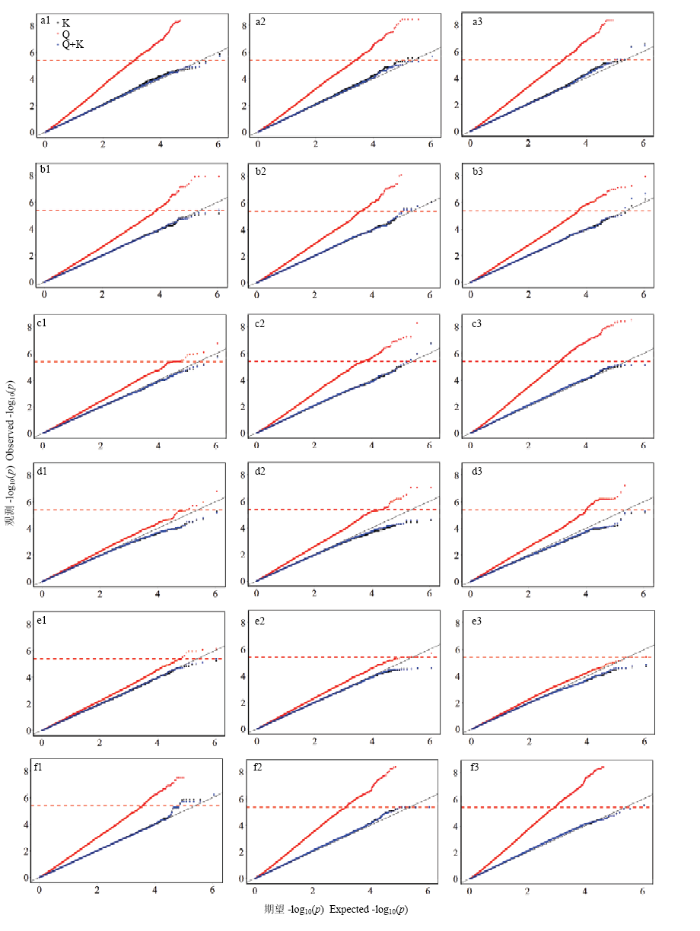 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图26个环境下棒三叶叶绿素含量3种模型(Q、K、Q+K)的QQ图
Fig. 2Quantile-quantile (QQ) plots resulting from GWAS using three models for chlorophyll content of maize three ear leaves at six environments
图3
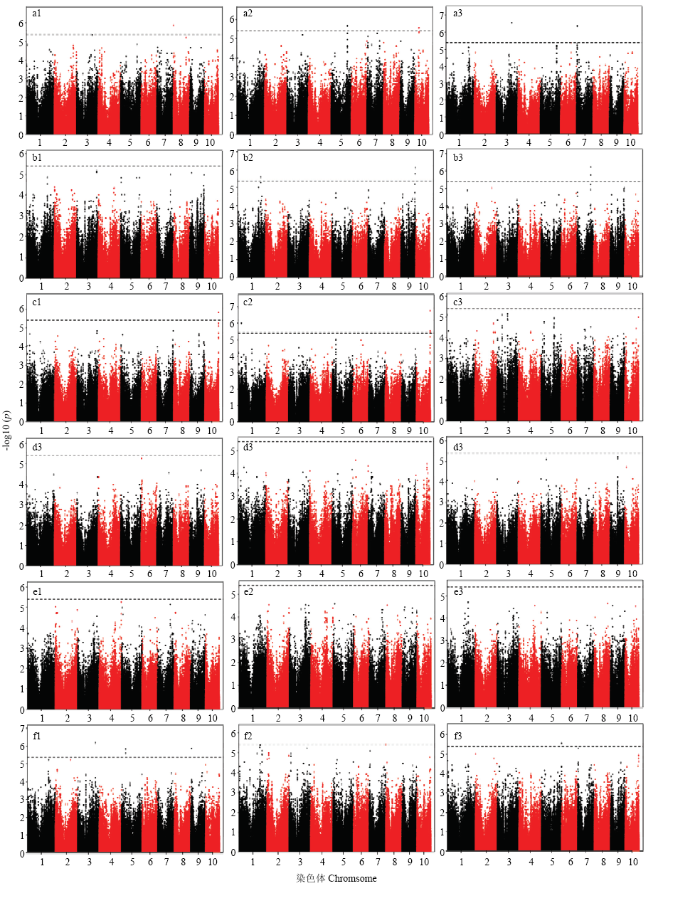 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3棒三叶叶绿素含量关联分析的曼哈顿图
Fig. 3The Manhattan plots of chlorophyll content of maize three ear leaves at six environments
Table 3
表3
表3玉米棒三叶叶绿素含量候选基因及功能注释
Table 3
| 位点a Loci | 染色 Chr. | 环境 Enviro. | 性状 Trait | 峰值SNP Peak SNP | 物理位置b Posi. (bp) | P值c P value | 贡献率d R2(%) | 候选基因e Candidate gene | 功能注释 Annotation |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 2018年长沙 Changsha, 2018 | 穗位叶 Uppermost ear leaf | Chr.1.S_33879839 | 33879839 | 1.05E-06 | 5.89 | GRMZM2G110023 | ATP结合亚基CLPT1叶绿体 ATP-dependent Clp protease ATP-binding subunit CLPT1 chloroplastic |
| GRMZM2G110004 | NA | ||||||||
| GRMZM2G035552 | ACR8 | ||||||||
| GRMZM2G035461 | 自噬蛋白9Autophagy protein 9 | ||||||||
| 2 | 1 | 2017年三亚 Sanya, 2017 | 穗位叶 Uppermost ear leaf | Chr.1.S_249417295 | 249417295 | 2.65E-06 | 5.43 | GRMZM2G031771 | NA |
| GRMZM2G031788 | NA | ||||||||
| GRMZM2G058057 | 同源线粒体导入受体亚基 Mitochondrial import receptor subunit TOM5 homolog TOM5 | ||||||||
| GRMZM2G058227 | 甘油磷酰二元酸酯磷酸二酯酶Glycerophosphoryl diester phosphodiesterase | ||||||||
| GRMZM2G058244 | UDP -葡萄糖6脱氢酶UDP-glucose 6-dehydrogenase | ||||||||
| GRMZM2G172512 | 未知Unknown | ||||||||
| GRMZM2G172584 | 未知Unknown | ||||||||
| 2 | 1 | 2017年三亚 Sanya, 2017 | 穗位叶 Uppermost ear leaf | Chr.1.S_249417300 | 249417300 | 2.65E-06 | 5.43 | GRMZM2G031771 | NA |
| GRMZM2G031788 | NA | ||||||||
| GRMZM2G058057 | TOM5同源线粒体导入受体亚基 Mitochondrial import receptor subunit TOM5 homolog | ||||||||
| GRMZM2G058227 | 甘油磷酰二元酸酯磷酸二酯酶Glycerophosphoryl diester phosphodiesterase | ||||||||
| GRMZM2G058244 | UDP -葡萄糖6脱氢酶UDP-glucose 6-dehydrogenase | ||||||||
| GRMZM2G172512 | 未知Unknown | ||||||||
| GRMZM2G172584 | 未知Unknown | ||||||||
| 2 | 1 | 2017年三亚 Sanya, 2017 | 穗位叶 Uppermost ear leaf | Chr.1.S_249417301 | 249417301 | 2.65E-06 | 5.43 | GRMZM2G031771 | NA |
| GRMZM2G031788 | NA | ||||||||
| GRMZM2G058057 | TOM5同源线粒体导入受体亚基 Mitochondrial import receptor subunit TOM5 homolog | ||||||||
| GRMZM2G058227 | 甘油磷酰二元酸酯磷酸二酯酶Glycerophosphoryl diester phosphodiesterase | ||||||||
| GRMZM2G058244 | UDP -葡萄糖6脱氢酶UDP-glucose 6-dehydrogenase | ||||||||
| 位点a Loci | 染色 Chr. | 环境 Enviro. | 性状 Trait | 峰值SNP Peak SNP | 物理位置b Posi. (bp) | P值c P value | 贡献率d R2(%) | 候选基因e Candidate gene | 功能注释 Annotation |
| GRMZM2G172512 | 未知Unknown | ||||||||
| GRMZM2G172584 | 未知Unknown | ||||||||
| 3 | 3 | 2012年鹤壁 Hebi, 2012 | 穗下叶Below the uppermost ear leaf | Chr.3.S_167437401 | 167437401 | 2.88E-07 | 6.52 | GRMZM2G701895 | NA |
| GRMZM2G044055 | 甲酸精蛋白1Formin-like protein 1 | ||||||||
| GRMZM2G044092 | 葡萄糖苷酶42Beta-glucosidase 42 | ||||||||
| GRMZM2G118362 | 含五肽复合物的线粒体蛋白 Pentatricopeptide repeat-containing protein mitochondrial | ||||||||
| 4 | 3 | 综合Blup | 穗上叶Above the uppermost ear leaf | PZE-103136749 | 192318356 | 6.48E-07 | 5.10 | GRMZM2G038953 | 54 kD蛋白3信号识别粒子Signal recognition particle 54 kD protein 3 |
| 5 | 5 | 综合Blup | 穗上叶Above the uppermost ear leaf | PZE-105049606 | 41552842 | 1.46E-06 | 4.80 | GRMZM2G007185 | β13-半乳糖基转移酶4 Probable beta-13-galactosyltransferase 4 |
| 5 | 5 | 综合Blup | 穗上叶Above the uppermost ear leaf | PZE-105049607 | 41552873 | 2.58E-06 | 4.53 | GRMZM2G007185 | β13-半乳糖基转移酶4 Probable beta-13-galactosyltransferase 4 |
| 6 | 5 | 2012年鹤壁 Hebi, 2012 | 穗位叶 Uppermost ear leaf | Chr.5.S_174139937 | 174139937 | 2.23E-06 | 5.36 | GRMZM5G836353 | 单氧酶1 Monooxygenase 1 |
| GRMZM2G339523 | 单氧酶1 Monooxygenase 1 | ||||||||
| GRMZM2G087169 | 单氧酶1 Monooxygenase 1 | ||||||||
| GRMZM2G087068 | 类Cmc1细胞色素c氧化酶生物发生蛋白 Cytochrome c oxidase biogenesis protein Cmc1-like | ||||||||
| GRMZM2G388099 | 未知Unknown | ||||||||
| GRMZM2G087040 | 乙烯反应转录因子ERF021Ethylene-responsive transcription factor ERF021 | ||||||||
| GRMZM2G086946 | TPX2(靶向蛋白为Xklp2)蛋白家族 TPX2 (targeting protein for Xklp2) protein family | ||||||||
| 7 | 5 | 综合Blup | 穗下叶Below the uppermost ear leaf | Chr.5.S_216586758 | 216586758 | 2.75E-06 | 5.03 | GRMZM2G014116 | 26S蛋白酶体非ATP酶调节亚基 26S proteasome non-ATPase regulatory subunit 12 homolog B |
| GRMZM2G014793 | O-岩藻糖转移酶家族蛋白O-fucosyltransferase family protein | ||||||||
| GRMZM2G015090 | 植物细胞内ras -group相关LRR蛋白 Plant intracellular Ras-group-related LRR protein 9 | ||||||||
| GRMZM2G145061 | 脯氨酰4羟化酶亚基α2 Prolyl 4-hydroxylase alpha-2 subunit | ||||||||
| GRMZM2G145107 | 蔗糖转运2 Sucrose transporter2 | ||||||||
| GRMZM2G145128 | 含有重复PPR的线粒体蛋白Pentatricopeptide repeat-containing protein | ||||||||
| 位点a Loci | 染色 Chr. | 环境 Enviro. | 性状 Trait | 峰值SNP Peak SNP | 物理位置b Posi. (bp) | P值c P value | 贡献率d R2(%) | 候选基因e Candidate gene | 功能注释 Annotation |
| GRMZM2G145133 | 未知Unknown | ||||||||
| GRMZM2G445944 | 转录因子TCP21Transcription factor TCP21 | ||||||||
| GRMZM2G445958 | 复制蛋白a32 kD亚基A Replication protein A 32 kD subunit A | ||||||||
| GRMZM2G145473 | 未知Unknown | ||||||||
| 8 | 7 | 2012年鹤壁 Hebi, 2012 | 穗下叶Below the uppermost ear leaf | Chr.7.S_9204823 | 9204823 | 4.39E-07 | 6.37 | GRMZM2G704349 | Pumilio同源3 Pumilio homolog 3 |
| GRMZM2G046776 | Pumilio同源3 Pumilio homolog 3 | ||||||||
| GRMZM2G046750 | 脂质转运蛋白Lipid-transfer protein 种子贮藏2S白蛋白超家族蛋白Seed storage 2S albumin superfamily protein | ||||||||
| GRMZM2G046529 | 叶绿体中蛋白质脂肪酸输出1 Protein FATTY ACID EXPORT 1 chloroplastic | ||||||||
| GRMZM2G004301 | Ⅳ型肌醇多磷酸酶Type Ⅳ inositol polyphosphate | ||||||||
| 9 | 7 | 2017年三亚 Sanya, 2017 | 穗下叶Below the uppermost ear leaf | Chr.7.S_145681493 | 145681493 | 1.72E-06 | 5.54 | GRMZM2G168913 | RNA聚合酶Ⅱ的中介体亚单位8 Mediator of RNA polymerase Ⅱtranscription subunit 8 |
| GRMZM2G168858 | NADPH-细胞色素P450还原酶NADPH--cytochrome P450 reductase 2 | ||||||||
| GRMZM2G357688 | 锌指蛋白2 Zinc finger protein 2 | ||||||||
| GRMZM2G436511 | BTB/POZ蛋白域 BTB/POZ domain-containing protein | ||||||||
| GRMZM2G135651 | 环核苷酸封闭通道Cyclic nucleotide-gated ion channel 14 | ||||||||
| 9 | 7 | 2017年三亚 Sanya, 2017 | 穗下叶Below the uppermost ear leaf | Chr.7.S_145682189 | 145682189 | 5.85E-07 | 6.07 | GRMZM2G168913 | RNA聚合酶Ⅱ的中介体亚单位8 Mediator of RNA polymeraseⅡ transcription subunit 8 |
| GRMZM2G168858 | NADPH-细胞色素P450还原酶2 NADPH--cytochrome P450 reductase 2 | ||||||||
| GRMZM2G357688 | 锌指蛋白2Zinc finger protein 2 | ||||||||
| GRMZM2G436511 | BTB/POZ蛋白域BTB/POZ domain-containing protein | ||||||||
| GRMZM2G135651 | 环核苷酸封闭通道Cyclic nucleotide-gated ion channel 14 | ||||||||
| 10 | 8 | 2012年鹤壁 Hebi, 2012 | 穗上叶Above the uppermost ear leaf | Chr.8.S_1454124 | 1454124 | 1.32E-06 | 5.55 | GRMZM2G378906 | 外被体蛋白β亚基Coatomer subunit beta |
| GRMZM2G110368 | 未知Unknown | ||||||||
| 11 | 8 | 综合Blup | 穗位叶 Uppermost ear leaf | Chr.8.S_1454124 | 1454124 | 3.95E-06 | 4.70 | GRMZM2G378906 | 外被体蛋白β亚基Coatomer subunit beta |
| GRMZM2G110368 | 未知Unknown | ||||||||
| 位点a Loci | 染色 Chr. | 环境 Enviro. | 性状 Trait | 峰值SNP Peak SNP | 物理位置b Posi. (bp) | P值c P value | 贡献率d R2(%) | 候选基因e Candidate gene | 功能注释 Annotation |
| 12 | 9 | 综合Blup | 穗上叶Above the uppermost ear leaf | Chr.9.S_9638369 | 9638369 | 1.32E-06 | 4.86 | GRMZM2G159402 | 假定锌指蛋白Putative zinc finger protein |
| GRMZM2G033130 | DUF1296结构域蛋白家族 Putative DUF1296 domain containing family protein | ||||||||
| 12 | 9 | 综合Blup | 穗上叶Above the uppermost ear leaf | Chr.9.S_9639904 | 9639904 | 1.32E-06 | 4.86 | GRMZM2G159402 | 假定锌指蛋白Putative zinc finger protein |
| GRMZM2G033130 | DUF1296结构域蛋白家族Putative DUF1296 domain containing family protein | ||||||||
| 12 | 9 | 综合Blup | 穗上叶Above the uppermost ear leaf | chr9.S_9642472 | 9642472 | 1.32E-06 | 4.86 | GRMZM2G159402 | 假定锌指蛋白Putative zinc finger protein |
| GRMZM2G033130 | DUF1296结构域蛋白家族Putative DUF1296 domain containing family protein | ||||||||
| 12 | 9 | 综合Blup | 穗上叶Above the uppermost ear leaf | Chr.9.S_9643507 | 9643507 | 1.32E-06 | 4.86 | GRMZM2G159402 | 假定锌指蛋白Putative zinc finger protein |
| GRMZM2G033130 | DUF1296结构域蛋白家族Putative DUF1296 domain containing family protein | ||||||||
| 12 | 9 | 综合Blup | 穗上叶Above the uppermost ear leaf | Chr.9.S_9644832 | 9644832 | 1.32E-06 | 4.86 | GRMZM2G159402 | 假定锌指蛋白Putative zinc finger protein |
| GRMZM2G033130 | DUF1296结构域蛋白家族Putative DUF1296 domain containing family protein | ||||||||
| 13 | 9 | 2017年三亚 Sanya, 2017 | 穗位叶 Uppermost ear leaf | Chr.9.S_151327393 | 151327393 | 1.72E-06 | 6.01 | GRMZM2G178787 | 叶绿体蛋白激酶Protein kinase 2B chloroplastic |
| GRMZM2G178826 | 脱氢奎尼酸合酶3-dehydroquinate synthase | ||||||||
| GRMZM2G479529 | 未知Unknown | ||||||||
| GRMZM2G178847 | 未知Unknown | ||||||||
| GRMZM2G178859 | 原血红素Ⅸ法氏转移酶Protoheme Ⅸ farnesyltransferase | ||||||||
| GRMZM2G178880 | 甘露聚糖合酶7 Probable mannan synthase 7 | ||||||||
| GRMZM2G178916 | 含有重复PPR的线粒体蛋白 Pentatricopeptide repeat-containing protein mitochondrial | ||||||||
| GRMZM2G479581 | 未知Unknown | ||||||||
| GRMZM2G178945 | O-岩藻糖转移酶家族蛋白O-fucosyltransferase family protein | ||||||||
| GRMZM2G178960 | 核酮糖磷酸3-表异构酶Ribulose-phosphate 3-epimerase | ||||||||
| GRMZM2G057950 | 核糖体蛋白L32含有蛋白质的异构体1%3B核糖体蛋白L32,含有蛋白质的异构体 2Ribosomal protein L32 containing protein isoform 1%3B, Ribosomal protein L32 containing protein isoform 2 | ||||||||
| 位点a Loci | 染色 Chr. | 环境 Enviro. | 性状 Trait | 峰值SNP Peak SNP | 物理位置b Posi. (bp) | P值c P value | 贡献率d R2(%) | 候选基因e Candidate gene | 功能注释 Annotation |
| GRMZM2G058037 | 类HVA22蛋白Ⅰ HVA22-like proteinⅠ | ||||||||
| GRMZM2G361256 | ATP结合盒(ABC)蛋白家族3 ABC transporter C family member 3 | ||||||||
| 14 | 9 | 2017年三亚 Sanya, 2017 | 穗位叶 Uppermost ear leaf | Chr.9.S_153737236 | 153737236 | 7.91E-07 | 6.59 | GRMZM2G382537 | 未知Unknown |
| GRMZM2G382534 | 羟化酶5 Hydroxylase5 | ||||||||
| GRMZM2G060564 | PLAC8 家族蛋白PLAC8 family protein | ||||||||
| GRMZM2G060507 | 假定类结转录因子家族蛋白Putative knotted-like transcription factor family protein | ||||||||
| GRMZM2G064005 | Protein FIZZY-RELATED 1 FIZZY-RELATED蛋白 | ||||||||
| GRMZM2G063931 | 假定泛素连接酶酶家族,E2泛素连接酶 Putative ubiquitin-conjugating enzyme family%3B, ubiquitin-conjugating enzyme E2 | ||||||||
| 15 | 10 | 2012年鹤壁 Hebi, 2012 | 穗位叶 Uppermost ear leaf | Chr.10.S_45404195 | 45404195 | 2.81E-06 | 4.76 | NO | - |
| 16 | 10 | 2012年鹤壁 Hebi, 2012 | 穗位叶 Uppermost ear leaf | Chr.10.S_46780569 | 46780569 | 2.76E-06 | 4.75 | GRMZM2G135800 | NA |
| GRMZM2G371316 | 未知Unknown | ||||||||
| GRMZM2G371345 | 黄酮醇3-O-葡糖基转移酶Flavonol 3-O-glucosyltransferase | ||||||||
| 16 | 10 | 2012年鹤壁 Hebi, 2012 | 穗位叶 Uppermost ear leaf | Chr.10.S_46780652 | 46780652 | 2.76E-06 | 4.75 | GRMZM2G135800 | NA |
| GRMZM2G371316 | 未知Unknown | ||||||||
| GRMZM2G371345 | 黄酮醇3-O-葡糖基转移酶Flavonol 3-O-glucosyltransferase | ||||||||
| 17 | 10 | 2018年长沙 Changsha, 2018 | 穗位叶 Uppermost ear leaf | Chr.10.S_148643242 | 148643242 | 3.04E-06 | 5.30 | GRMZM2G074787 | DNA错配修复蛋白MSH3 DNA mismatch repair protein MSH3 |
| GRMZM2G074773 | 核转录因子Y亚基C-4 Nuclear transcription factor Y subunit C-4 | ||||||||
| GRMZM2G074759 | 酰基活化酶3 Acyl activating enzyme3 | ||||||||
| GRMZM2G074754 | C2钙/脂结合植物磷脂酰转移酶家族蛋白 C2 calcium/lipid-binding plant phosphoribosyltransferase family protein | ||||||||
| GRMZM2G074718 | 表达蛋白Expressed protein | ||||||||
| GRMZM2G074107 | DNA结合蛋白DNA binding protein | ||||||||
| 位点a Loci | 染色 Chr. | 环境 Enviro. | 性状 Trait | 峰值SNP Peak SNP | 物理位置b Posi. (bp) | P值c P value | 贡献率d R2(%) | 候选基因e Candidate gene | 功能注释 Annotation |
| 17 | 10 | 2018年长沙 Changsha, 2018 | 穗位叶 Uppermost ear leaf | Chr.10.S_148643247 | 148643247 | 3.87E-06 | 5.27 | GRMZM2G074787 | DNA错配修复蛋白MSH3 DNA mismatch repair protein MSH3 |
| GRMZM2G074773 | 核转录因子Y亚基C-4 Nuclear transcription factor Y subunit C-4 | ||||||||
| GRMZM2G074759 | 酰基活化酶3 Acyl activating enzyme3 | ||||||||
| GRMZM2G074754 | C2钙/脂结合植物磷脂酰转移酶家族蛋白 C2 calcium/lipid-binding plant phosphoribosyltransferase family protein | ||||||||
| GRMZM2G074718 | 表达蛋白Expressed protein | ||||||||
| GRMZM2G074107 | DNA结合蛋白DNA binding protein | ||||||||
| 17 | 10 | 2018年长沙 Changsha, 2018 | 穗位叶 Uppermost ear leaf | Chr.10.S_148643249 | 148643249 | 1.78E-07 | 6.71 | GRMZM2G074787 | DNA错配修复蛋白MSH3 DNA mismatch repair protein MSH3 |
| GRMZM2G074773 | 核转录因子Y亚基C-4 Nuclear transcription factor Y subunit C-4 | ||||||||
| GRMZM2G074759 | 酰基活化酶3 Acyl activating enzyme3 | ||||||||
| GRMZM2G074754 | C2钙/脂结合植物磷脂酰转移酶家族蛋白 C2 calcium/lipid-binding plant phosphoribosyltransferase family protein | ||||||||
| GRMZM2G074718 | 表达蛋白Expressed protein | ||||||||
| GRMZM2G074107 | DNA结合蛋白DNA binding protein | ||||||||
| 18 | 10 | 2018年长沙 Changsha, 2018 | 穗上叶Above the uppermost ear leaf | Chr.10.S_148643249 | 148643249 | 1.58E-06 | 5.80 | GRMZM2G074787 | DNA错配修复蛋白MSH3 DNA mismatch repair protein MSH3 |
| GRMZM2G074773 | 核转录因子Y亚基C-4 Nuclear transcription factor Y subunit C-4 | ||||||||
| GRMZM2G074759 | 酰基活化酶3 Acyl activating enzyme3 | ||||||||
| GRMZM2G074754 | C2钙/脂结合植物磷脂酰转移酶家族蛋白 C2 calcium/lipid-binding plant phosphoribosyltransferase family protein | ||||||||
| GRMZM2G074718 | 表达蛋白Expressed protein | ||||||||
| GRMZM2G074107 | DNA结合蛋白DNA binding protein |
新窗口打开|下载CSV
对其他几个环境来说,在鹤壁,共鉴定到7个显著SNP,涉及到6个位点,分别是位于第8染色体与穗上叶叶绿素有关的1个位点,解释5.55%的表型变异;位于第5和第10染色体与穗位叶叶绿素有关的3个位点,分别解释5.36%、4.76%和4.75%的表型变异;其余2个与穗位叶叶绿素有关,位于第3和第7染色体,可分别解释6.52%和6.37%的表型变异。
在三亚,共检测到7个显著SNP(4个位点),其中1个位于第7染色体与穗下叶叶绿素相关的位点,解释6.07%的表型变异;另外3个位于玉米第1和第9染色体,与穗位叶叶绿素有关,解释的表型变异分别为5.43%、6.01%和6.59%。
在长沙,检测到5个SNP-性状关联,这5个SNP落在3个位点内,其中位于第1和第10染色体的2个与穗位叶叶绿素显著相关的位点,能分别解释5.89%和6.71%的表型变异;位于第10染色体上与穗上叶叶绿素含量显著相关的位点,能解释5.80%的表型变异。
在5个环境综合(Blup)条件下,共检测到10个SNP,涉及到5个位点。其中有3个位点与穗上叶叶绿素含量显著相关,分别位于第3、第5和第9染色体,解释的表型变异分别为5.10%、4.80%和4.86%;1个位于玉米第8染色体与穗位叶叶绿素有关的位点,可解释4.70%的表型变异,此位点在鹤壁也被检测到,为后续的进一步研究奠定了基础;此外,还有1个位于第5染色体,与穗下叶叶绿素含量显著关联,可解释5.03%的表型变异。利用线性回归模型计算了穗上叶、穗位叶与穗下叶叶绿素含量检测到的位点的总表型变异,分别可解释18.5%、26.2%和11.4%的表型变异;但仍有部分变异未能被解释,由此造成的缺失遗传力(missing heritability)可能因为存在微效位点或上位性互作导致,当前GWAS方法难以检测。
鉴定的18个位点,其中10号与11号位点共定位于第8染色体1.40—1.50 Mb区间内,分别解释5.55%和4.70%的表型变异,该区间内共有2个基因(表3)。17号和18号位点共定位于第10染色体148.59—148.69 Mb区间,分别解释6.71%和5.08%的表型变异,该区间内共有6个基因(表3)。共定位结果表明,这两个位点受环境影响较小,可在不同环境下稳定遗传;此外,2号位点可同时被长沙环境下的穗上叶与穗位叶叶绿素含量检测到(表3)。遗憾的是,原阳和永城均未检测到显著位点(3.99×10-6)(图3和表3)。表达量QTL分析(eQTL)发现76个候选基因中,85.5%的基因具有eQTL(65/76),表型与表达量之间的相关性分析发现,11.8%(9/76)的基因与对应表型显著相关(P<0.05),说明这些基因可能是通过表达量的变化来调控表型变异(数据未列出)。
2.3.3 候选基因确定 由于该群体的连锁不平衡衰减距离是50 kb(R2=0.1),因此,在峰值SNP的上下游各50 kb,即100 kb范围内搜索候选基因。一共有76个候选基因落在这18个位点内,其中60个具有功能注释(表3),根据基因功能注释及其在B73参考基因组中的表达部位及表达量,选择一个最可能的候选基因作为该位点的候选基因(表3)。
3 讨论
统计功效是GWAS首要考虑因素,对玉米来说,受群体结构影响,不同性状对不同模型的敏感程度不同。YANG等[27]研究表明,使用最优统计模型,可增加GWAS的统计功效。此外,刘坤等[28]使用3种模型(Q、K、Q+K)对玉米株高(plant height,PH)、穗位高(ear height,EH)、EH/PH、总叶片数(leaf number,LN)、穗上叶片数(leaf number above ear,LNAN)、LNAN/LN等6个性状进行GWAS,发现Q模型对假阳性的控制不好,Q+K模型对假阳性的过于严格,而K模型表现最优;ZHAO等[29]使用3种相同模型对玉米籽粒、穗轴、苞叶、茎杆、叶片等5个组织的砷含量进行GWAS,发现K和Q+K模型对假阳性的控制过于严格,最终选择表现较好的Q模型的结果进行解析。本研究中,Q模型的表现最差,K模型和Q+K模型相当,但Q+K模型过于严格(图2),因此,选择K模型结果对玉米棒三叶叶绿素含量的遗传基础进行解析。赵永萍[30]通过对玉米单交种先玉335的棒三叶进行剪叶处理,探索玉米各部位叶片对百粒重、果穗干重、产量的影响,发现棒三叶的缺失会造成产量的明显下降,证明棒三叶对产量贡献至关重要。陈永欣等[31]对玉米棒三叶相关性状进行分析,得出棒三叶各叶片面积差异不大,在光合作用中发挥的作用近乎相同。本研究所测的棒三叶叶绿素含量相差不大,表明棒三叶在光合作用中可能发挥相似作用,与该结果吻合。唐海涛等[32]对65个玉米杂交种棒三叶的叶长、叶宽、叶肉厚度、叶面积、叶绿素含量、叶向值、光合速率、叶片干重和比叶重等性状进行分析,发现穗上叶、穗位叶、穗下叶间存在一定的相关性,且棒三叶比叶重(单位叶面积的叶片重量从小到大依次为穗位叶、穗上叶、穗下叶)和叶绿素含量(从小到大依次为穗上叶、穗位叶、穗下叶)呈现一定变化规律。左宝玉等[4]对玉米不同叶位叶片进行叶绿体超微结构观察及叶绿素含量进行测定,发现叶绿体的光合膜系统是叶绿素含量最高的部位,并且发现叶绿素含量和光合速率和产量之间呈显著正相关[33,34]。苏云松等[35]研究发现马铃薯叶片叶绿素含量(SPAD值)在正常日照条件下与马铃薯产量呈显著正相关,认为SPAD值的大小会直接影响马铃薯产量。王康等[36]研究表明夏玉米生育期的SPAD值与其产量呈正相关。以上研究均表明叶绿素含量对产量有重要贡献。
在前人研究中,拟南芥中发现的VIPP1,将其进行过表达可显著抵抗氧化胁迫,可以修复nyc1突变体中受损的叶绿体膜结构,保护叶绿体膜,使叶绿素免受进一步降解[37]。在水稻中,与野生型相比,过表达Gc植株的叶绿素a、叶绿素b和叶绿素总量分别比野生型增加5%、100%和25%,光合速率、生物量和产量相对增加20%、17%和16%[38]。作为Gc的上位基因,DE1表达的增加导致叶绿素含量显著增加,叶片叶色变为深绿色,光合速率和产量均显著提高[39]。在玉米中,也有叶绿素含量定位相关报道,王爱玉等[40]通过玉米A150-3-2和Mo17为亲本构建的189个F2单株为作图群体,分别在喇叭口期和开花期对叶绿素a,叶绿素b,叶绿素c和总叶绿素含量进行测定,共检测到32个与叶绿素含量相关的QTL。刘宗华等[41]以优良玉米杂交种农大108的203个F2:3家系为材料,利用复合区间作图法,在施氮和不施氮情况下,对玉米不同发育时期的叶绿素含量进行了QTL分析,认为叶绿素含量的QTL表达存在时空差异,其中2个QTL(qchl4和qchl5b)可以在整个生育期被检测到,是生长发育必需的QTL。方永丰等[42]对已定位的玉米叶绿素QTL进行整合,利用荟萃分析(meta-analysis)发现多个共定位热区,并在这些热点区内挖掘与玉米叶绿素含量相关的候选基因。以上研究仅停留在连锁分析层面,需要构建作图群体,费时费力,且定位精度较低。随着测序技术的发展和测序成本的降低,关联分析在植物中被广泛使用,因其定位精度可达单基因水平,已成为挖掘植物基因的重要工具。
本研究对538份玉米自交系的棒三叶叶绿素含量(SPAD值)进行测定,结合55万个SNPs标记,通过全基因组关联分析,检测到29个与棒三叶叶绿素含量显著关联的SNP,涉及18个位点,共有76个候选基因落在这18个位点内,其中60个候选基因有功能注释,16个功能未知。已注释基因主要编码转运蛋白,结合蛋白,还原酶等,涉及功能包括能量代谢、生物合成调节及物质输送代谢等途径。18个位点中,有2个位点(第10和第11位点)在不同环境下共定位(表3),表明该位点受环境影响较小,在不同环境可稳定遗传。此外,另2个位点(第17和第18位点)在相同环境不同叶片被检测到(表3),说明这两个叶片的叶绿素含量有共同的遗传机制。此外还发现一些参与叶绿素合成及分解途径的基因。
3.1 共定位结果及候选基因分析
对定位结果进行分析发现2012年鹤壁穗上叶有关的第10位点和Blup条件下穗位叶相关的第11位点共定位,该位点内共有2个基因,分别为GRMZM2G378906和GRMZM2G110368。其中GRMZM2G110368功能未知,而GRMZM2G378906编码外被体β蛋白亚基(coatomer subunit beta),该基因仅在人类中有过描述,当敲除外被体β蛋白亚基时可使人类前列腺癌细胞凋亡速率升高,从而降低癌细胞的存活率[43]。但在植物中外被体β蛋白亚基的研究未见报道,因此,其作用机理尚不明确,推测其在玉米中也可能与细胞凋亡有关。位于第10染色体上的与2018年长沙穗位叶相关的第17位点和2018年长沙穗上叶相关的第18位点共定位,位点内存在6个基因,GRMZM2G074787(DNA mismatch repair protein MSH3)、GRMZM2G074773(Nuclear transcription factor Y subunit C-4)、GRMZM2G074759(acyl activating enzyme3,AAE3)、GRMZM2G074754(C2 calcium/lipid-binding plant phosphoribosyltransferase family protein)、GRMZM2G074718(Expressed protein)和GRMZM2G074107(DNA binding protein)。GRMZM2G074759编码一种与AAE3高度相似的酰基活化酶。opaque 7的不透明表型即是由该酶减少所引起的α-玉米醇溶蛋白的浓度降低导致,通过氨基酸和代谢物的分析表明,该基因可能通过提高α-酮戊二酸(ALA)和草酰乙酸含量进而影响氨基酸生物合成,将该基因转入水稻中,同样会出现不透明胚乳表型。该基因的克隆为谷物种子贮藏蛋白含量的遗传调控提供了有效靶点[44]。也有研究表明,ALA的合成会促使叶绿素含量升高[45,46,47]。结合前人研究认为基因GRMZM2G074759在参与调控玉米棒三叶叶绿素含量的同时也参与调控玉米籽粒赖氨酸的合成,当该基因过表达时叶绿素含量及籽粒中赖氨酸含量均升高。
3.2 叶绿素代谢相关候选基因
通过eQTL分析,发现2个参与叶绿素代谢的基因(GRMZM2G178859和GRMZM2G168858)。叶绿素属于四吡咯物质,当四吡咯物质不与蛋白结合时,会产生对植物有害的自由基。为了保护植物免受自由基的伤害,可以通过调节ALA的合成来阻止四吡咯物质的积累[45]。PONTOPPIDAN等[46]证明谷氨酰-tRNA还原酶(Glu TR)在ALA合成中起拮抗作用,而血红素参与Glu TR活性的调节。在拟南芥中,可通过降低血红素加氧酶活性积累血红素,进而降低Glu TR活性[47]。本研究鉴定到的位于第9染色体的基因GRMZM2G178859编码原血红素Ⅸ法氏转移酶,该酶可增加植物中血红素量积累,进而降低Glu TR活性,促进ALA的合成,从而促进叶绿素的合成。此外,7号染色体上检测到的编码NADPH-细胞色素P450还原酶2(CPR2)的基因GRMZM2G168858参与叶绿素代谢。细胞色素P450(CYPs)是一种血红素结合蛋白,细胞色素P450接受电子后才能发挥活性。CPR2是细胞色素P450的主要电子供体,CPR2的存在使得细胞中血红素积累,谷氨酰-tRNA还原酶(Glu TR)活性降低,促使ALA合成,叶绿素合成增加[48]。并且,GRMZM2G168858具有CPR功能,由于P450家族中的酶或基因功能多样,在玉米中并未明确其作用机理,仅在其他植物中有报道,表明CPR是一种将电子从NADPH转移到细胞色素P450酶的关键酶。在杉木中已明确CPR基因表达和酶活性的调控机制,通过Northern blot发现CPR在种子成熟前与萌发过程中的表达均受发育调控,而在种子和幼苗组织的子叶、胚根和大配子体中存在差异[49],推测其在玉米中具有类似功能,即CPR在玉米籽粒成熟前调控籽粒大小。4 结论
基于K模型的GWAS结果,共检测到29个与棒三叶叶绿素含量显著关联的SNP,涉及到18个位点,其中2个位点可以在2个环境或2个组织中被共定位。18个位点内共有76个基因,其中85.5%的候选基因具有eQTL(65/76),11.8%的候选基因与对应表型显著相关(P<0.05)。此外,共有76个候选基因落在这些位点内,其中60个基因具有功能注释并讨论了共定位以及与叶绿素代谢相关的基因。致谢:
本研究所用关联群体材料及基因型由华中农业大学作物遗传改良国家重点实验室严建兵教授提供,在此表示感谢。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.13430/j.cnki.jpgr.2014.04.003 [本文引用: 1]

以6个籼型杂交稻三系不育系和5个恢复系按不完全双列杂交设计配制的30个杂交稻组合及其亲本品种为材料,对其光合性状进行了测定和分析,结果表明:(1)杂交稻组合的光合特性存在显著或极显著的组合间遗传差异,光合特性的遗传变异主要来自基因的非加性效应;(2)胞间CO2浓度、蒸腾速率、叶绿素a、叶绿素b和类胡萝卜素含量受不育系的影响大于恢复系,而气孔导度、叶绿素a+b含量受恢复系的影响大于不育系;(3)杂交稻光合性状的广义遗传力均大于狭义遗传力,各性状主要受基因互作及环境的影响。狭义遗传力的大小依次为叶绿素b、叶绿素a+b、叶绿素a、气孔导度、胞间CO2浓度、类胡萝卜素和蒸腾速率,这些性状具有中等遗传力;(4)9个光合性状杂交稻F1表型值与父母本一般配合力效应值之和的相关系数均达极显著水平。因此,可以根据父母本一般配合力效应值之和来预测杂交稻组合光合性状的表现,有利于高效选育高光效杂交稻组合。
DOI:10.13430/j.cnki.jpgr.2014.04.003 [本文引用: 1]
以6个籼型杂交稻三系不育系和5个恢复系按不完全双列杂交设计配制的30个杂交稻组合及其亲本品种为材料,对其光合性状进行了测定和分析,结果表明:(1)杂交稻组合的光合特性存在显著或极显著的组合间遗传差异,光合特性的遗传变异主要来自基因的非加性效应;(2)胞间CO2浓度、蒸腾速率、叶绿素a、叶绿素b和类胡萝卜素含量受不育系的影响大于恢复系,而气孔导度、叶绿素a+b含量受恢复系的影响大于不育系;(3)杂交稻光合性状的广义遗传力均大于狭义遗传力,各性状主要受基因互作及环境的影响。狭义遗传力的大小依次为叶绿素b、叶绿素a+b、叶绿素a、气孔导度、胞间CO2浓度、类胡萝卜素和蒸腾速率,这些性状具有中等遗传力;(4)9个光合性状杂交稻F1表型值与父母本一般配合力效应值之和的相关系数均达极显著水平。因此,可以根据父母本一般配合力效应值之和来预测杂交稻组合光合性状的表现,有利于高效选育高光效杂交稻组合。
[本文引用: 2]
[本文引用: 2]
URL [本文引用: 1]
分析了在正常、 偏低、 偏高等不同施氮水平下, 玉米生长期冠层反射光谱与叶绿素含量的相关关系, 结果表明玉米叶绿素含量检测的敏感期为拔节期和喇叭口期。 正常施氮水平下玉米冠层光谱反射率与叶绿素含量相关关系高于其他施氮水平, 即r正常>r偏高>r偏低。 整个生长期由苗期开始二者相关系数绝对值满足先上升后下降, 于开花吐丝期达最低后回升的趋势, 其中玉米拔节期和灌浆期冠层反射光谱与叶绿素含量呈正相关。 选取558, 667, 714和912 nm, 分别对玉米拔节期和喇叭口期建立了MLR和PLSR检测模型, 经比较, 虽然PLSR模型复相关系数较MLR模型有所降低, 但模型鲁棒性得到增强。 分析拔节期和喇叭口期各种植被指数与叶绿素含量的相关关系, 表明DVI优于其他指数, 且拔节期DVI与叶绿素含量呈二项式相关, 喇叭口期二者呈指数相关。
URL [本文引用: 1]
分析了在正常、 偏低、 偏高等不同施氮水平下, 玉米生长期冠层反射光谱与叶绿素含量的相关关系, 结果表明玉米叶绿素含量检测的敏感期为拔节期和喇叭口期。 正常施氮水平下玉米冠层光谱反射率与叶绿素含量相关关系高于其他施氮水平, 即r正常>r偏高>r偏低。 整个生长期由苗期开始二者相关系数绝对值满足先上升后下降, 于开花吐丝期达最低后回升的趋势, 其中玉米拔节期和灌浆期冠层反射光谱与叶绿素含量呈正相关。 选取558, 667, 714和912 nm, 分别对玉米拔节期和喇叭口期建立了MLR和PLSR检测模型, 经比较, 虽然PLSR模型复相关系数较MLR模型有所降低, 但模型鲁棒性得到增强。 分析拔节期和喇叭口期各种植被指数与叶绿素含量的相关关系, 表明DVI优于其他指数, 且拔节期DVI与叶绿素含量呈二项式相关, 喇叭口期二者呈指数相关。
Magsci [本文引用: 2]
对玉米不同层次的叶片进行电镜观察和叶绿素含量测定,结果发现:叶绿体的光合膜系随叶位上升而愈加发达和增高,直至果穗叶达到顶峰的趋势。邻近果穗叶的叶片中的叶绿体,也具有较为发达的基粒和基质类囊体膜。而着生部位最高的顶生叶,其叶绿体中的光合膜系却不及果穗叶和邻近果穗叶的发达。但顶生叶中维管束鞘细胞的叶绿体内充
Magsci [本文引用: 2]
对玉米不同层次的叶片进行电镜观察和叶绿素含量测定,结果发现:叶绿体的光合膜系随叶位上升而愈加发达和增高,直至果穗叶达到顶峰的趋势。邻近果穗叶的叶片中的叶绿体,也具有较为发达的基粒和基质类囊体膜。而着生部位最高的顶生叶,其叶绿体中的光合膜系却不及果穗叶和邻近果穗叶的发达。但顶生叶中维管束鞘细胞的叶绿体内充
DOI:10.3724/SP.J.1006.2008.01339Magsci [本文引用: 1]
在前文研究已检出与农艺品质性状显著关联的SSR位点的基础上, 本文进一步对与性状关联位点的等位变异作解析, 通过将携带某等位变异的所有材料表型均值与携带无效等位基因( allele)材料表型均值做比较, 估计等位变异的潜在表型效应增量(减量), 进一步利用该信息估计位点增效(减效)等位变异的平均效应, 鉴别出一批农艺品质性状优异位点、等位变异及携带优异等位变异的载体材料。发现在栽培及野生种质中检出的优异等位变异有同、有异、有互补性。发现关联位点正、负效应等位变异均值间有差异, 可根据育种目标性状选择要求, 选取适合的位点及相应等位变异。同一标记位点可与多性状关联, 其等位变异在不同性状间各有其表型效应的方向和大小; 等位变异在相关性状效应上方向、大小的异同解释了性状间正、负相关的遗传原因。关联作图得到的信息可以弥补家系连锁法QTL定位信息的不足, 并直接利用等位变异信息进行亲本选拔、组合选配及后代等位条带辅助选择以提高育种成效。
DOI:10.3724/SP.J.1006.2008.01339Magsci [本文引用: 1]
在前文研究已检出与农艺品质性状显著关联的SSR位点的基础上, 本文进一步对与性状关联位点的等位变异作解析, 通过将携带某等位变异的所有材料表型均值与携带无效等位基因( allele)材料表型均值做比较, 估计等位变异的潜在表型效应增量(减量), 进一步利用该信息估计位点增效(减效)等位变异的平均效应, 鉴别出一批农艺品质性状优异位点、等位变异及携带优异等位变异的载体材料。发现在栽培及野生种质中检出的优异等位变异有同、有异、有互补性。发现关联位点正、负效应等位变异均值间有差异, 可根据育种目标性状选择要求, 选取适合的位点及相应等位变异。同一标记位点可与多性状关联, 其等位变异在不同性状间各有其表型效应的方向和大小; 等位变异在相关性状效应上方向、大小的异同解释了性状间正、负相关的遗传原因。关联作图得到的信息可以弥补家系连锁法QTL定位信息的不足, 并直接利用等位变异信息进行亲本选拔、组合选配及后代等位条带辅助选择以提高育种成效。
DOI:10.3724/SP.J.1006.2012.00962Magsci [本文引用: 1]
为揭示小麦自然群体干旱胁迫条件下旗叶叶绿素含量的变化, 筛选相关标记的优异等位变异, 以262份小麦种质资源组成的自然群体为材料, 分别种植在北京的2个试验地点, 均设雨养和灌溉处理, 于开花期和灌浆期检测旗叶叶绿素含量。以分布于21条染色体的169个SSR标记检测所有材料的基因型, 利用STRUCTURE 2.3.2软件分析群体结构, 用TASSEL软件的MLM (mixed linear model)方法对小麦自然群体的旗叶叶绿素含量进行关联分析。在此基础上, 将携带某等位变异的所有材料表型均值与携带无效等位基因( allele)材料表型均值比较, 估计等位变异的表型效应, 鉴别优异等位变异。共检测到2048个等位变异, 每位点2~37个等位变异, 平均12个。每位点的标记多态性信息量(PIC)为0.008~0.936, 平均0.628。在22个标记位点共检测出40个(次)与旗叶叶绿素含量极显著的关联, 其中11个标记位点有2次以上的关联, <em>Xwmc419-1B</em>和<em>Xgwm501-2B</em>分别有3次关联。在<em>Xcfa2123-7A</em>、<em>Xgwm232- 1D</em>和<em>Xgwm429-2B</em>位点分别检测到效应值大于4.0的等位变异。
DOI:10.3724/SP.J.1006.2012.00962Magsci [本文引用: 1]
为揭示小麦自然群体干旱胁迫条件下旗叶叶绿素含量的变化, 筛选相关标记的优异等位变异, 以262份小麦种质资源组成的自然群体为材料, 分别种植在北京的2个试验地点, 均设雨养和灌溉处理, 于开花期和灌浆期检测旗叶叶绿素含量。以分布于21条染色体的169个SSR标记检测所有材料的基因型, 利用STRUCTURE 2.3.2软件分析群体结构, 用TASSEL软件的MLM (mixed linear model)方法对小麦自然群体的旗叶叶绿素含量进行关联分析。在此基础上, 将携带某等位变异的所有材料表型均值与携带无效等位基因( allele)材料表型均值比较, 估计等位变异的表型效应, 鉴别优异等位变异。共检测到2048个等位变异, 每位点2~37个等位变异, 平均12个。每位点的标记多态性信息量(PIC)为0.008~0.936, 平均0.628。在22个标记位点共检测出40个(次)与旗叶叶绿素含量极显著的关联, 其中11个标记位点有2次以上的关联, <em>Xwmc419-1B</em>和<em>Xgwm501-2B</em>分别有3次关联。在<em>Xcfa2123-7A</em>、<em>Xgwm232- 1D</em>和<em>Xgwm429-2B</em>位点分别检测到效应值大于4.0的等位变异。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.molp.2015.02.014URLPMID:25747843 [本文引用: 1]
Forty-six significant association loci for chlorophyll content variation in rice were identified by GWAS and the validity of the GWAS signals was demonstrated by linkage mapping. Ghd7 and NAL1 were found to be two major loci, and the enhanced expression of Ghd7 decreased the chlorophyll content, mainly through down-regulating the expression of genes involved in the biosynthesis of chlorophyll and chloroplast. The loci or candidate genes identified would help fine-tune the antenna size of canopies.
DOI:10.1105/tpc.104.027276URL [本文引用: 1]
DOI:10.1104/pp.107.100321URL [本文引用: 1]
Magsci [本文引用: 1]
<p>鉴于混合群体的连锁不平衡分析在实现致病突变基因高解析度定位研究中的重要理论意义与应用前景, 为准确确定群体的混合比例, 进而实现基因高解析度定位中遗传重组率的精确估计, 以无选择、无突变的无限随机交配群体为对象, 分析了利用单倍型频率估计群体混合比例的误差, 建议采用随群体进化相对稳定的基因频率来估计群体的混合比例; 提出了利用混合群体连锁不平衡值的非线性部分随群体进化的衰减速率来估计基因间重组率的理论与方法. 理论研究和模拟实验结果表明, 初始群体在相关座位2的基因频率差异愈大, 混合比例愈接近0.5, 则混合群体连锁不平衡值非线性部分的作用愈明显, 这样的混合群体在基因高解析度定位研究中的应用价值也愈大.</p>
Magsci [本文引用: 1]
<p>鉴于混合群体的连锁不平衡分析在实现致病突变基因高解析度定位研究中的重要理论意义与应用前景, 为准确确定群体的混合比例, 进而实现基因高解析度定位中遗传重组率的精确估计, 以无选择、无突变的无限随机交配群体为对象, 分析了利用单倍型频率估计群体混合比例的误差, 建议采用随群体进化相对稳定的基因频率来估计群体的混合比例; 提出了利用混合群体连锁不平衡值的非线性部分随群体进化的衰减速率来估计基因间重组率的理论与方法. 理论研究和模拟实验结果表明, 初始群体在相关座位2的基因频率差异愈大, 混合比例愈接近0.5, 则混合群体连锁不平衡值非线性部分的作用愈明显, 这样的混合群体在基因高解析度定位研究中的应用价值也愈大.</p>
[本文引用: 1]
在玉米抽雄后剪去玉米植株不同层次和不同叶位叶片,观察各层次和各叶位叶对玉米籽粒产量的效应。在去叶同时测定各层次和各叶位叶的光合强度、叶绿素含量,并向各叶位叶喂以<SUP>32</SUP>P,观察其向果穗运输的情况。前后经过四年的实验,发现玉米抽雄后不同层次叶片对籽粒产量的效应是不同的,其作用大小顺序是中部叶片>上部
[本文引用: 1]
在玉米抽雄后剪去玉米植株不同层次和不同叶位叶片,观察各层次和各叶位叶对玉米籽粒产量的效应。在去叶同时测定各层次和各叶位叶的光合强度、叶绿素含量,并向各叶位叶喂以<SUP>32</SUP>P,观察其向果穗运输的情况。前后经过四年的实验,发现玉米抽雄后不同层次叶片对籽粒产量的效应是不同的,其作用大小顺序是中部叶片>上部
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/nph.13814URL [本文引用: 1]
DOI:10.1371/journal.pgen.1004573URLPMID:4161304 [本文引用: 1]
Association mapping is a powerful approach for dissecting the genetic architecture of complex quantitative traits using high-density SNP markers in maize. Here, we expanded our association panel size from 368 to 513 inbred lines with 0.5 million high quality SNPs using a two-step data-imputation method which combines identity by descent (IBD) based projection and k-nearest neighbor (KNN) algorithm. Genome-wide association studies (GWAS) were carried out for 17 agronomic traits with a panel of 513 inbred lines applying both mixed linear model (MLM) and a new method, the Anderson-Darling (A-D) test. Ten loci for five traits were identified using the MLM method at the Bonferroni-corrected threshold 61log10 (P) >5.74 (α66=661). Many loci ranging from one to 34 loci (107 loci for plant height) were identified for 17 traits using the A-D test at the Bonferroni-corrected threshold 61log10 (P) >7.05 (α66=660.05) using 556809 SNPs. Many known loci and new candidate loci were only observed by the A-D test, a few of which were also detected in independent linkage analysis. This study indicates that combining IBD based projection and KNN algorithm is an efficient imputation method for inferring large missing genotype segments. In addition, we showed that the A-D test is a useful complement for GWAS analysis of complex quantitative traits. Especially for traits with abnormal phenotype distribution, controlled by moderate effect loci or rare variations, the A-D test balances false positives and statistical power. The candidate SNPs and associated genes also provide a rich resource for maize genetics and breeding. Genotype imputation has been used widely in the analysis of genome-wide association studies (GWAS) to boost power and fine-map associations. We developed a two-step data imputation method to meet the challenge of large proportion missing genotypes. GWAS have uncovered an extensive genetic architecture of complex quantitative traits using high-density SNP markers in maize in the past few years. Here, GWAS were carried out for 17 agronomic traits with a panel of 513 inbred lines applying both mixed linear model and a new method, the Anderson-Darling (A-D) test. We intend to show that the A-D test is a complement to current GWAS methods, especially for complex quantitative traits controlled by moderate effect loci or rare variations and with abnormal phenotype distribution. In addition, the traits associated QTL identified here provide a rich resource for maize genetics and breeding.
DOI:10.1007/s00439-011-1118-2URLPMID:22143225 [本文引用: 1]
Abstract) for the adjustment of multiple testing, but current methods of calculation for are limited in accuracy or computational speed. Here, we report a more robust and fast method to calculate . Applying this efficient method [implemented in a free software tool named Genetic type 1 error calculator (GEC)], we systematically examined the , and the corresponding -value thresholds required to control the genome-wide type 1 error rate at 0.05, for 13 Illumina or Affymetrix genotyping arrays, as well as for HapMap Project and 1000 Genomes Project datasets which are widely used in genotype imputation as reference panels. Our results suggested the use of a -value threshold of ~10 as the criterion for genome-wide significance for early commercial genotyping arrays, but slightly more stringent -value thresholds ~502×0210 for current or merged commercial genotyping arrays, ~10 for all common SNPs in the 1000 Genomes Project dataset and ~502×0210 for the common SNPs only within genes.
DOI:10.1093/bioinformatics/btm308URL [本文引用: 1]
DOI:10.1038/ng.2484PMID:23242369 [本文引用: 1]
Maize kernel oil is a valuable source of nutrition. Here we extensively examine the genetic architecture of maize oil biosynthesis in a genome-wide association study using 1.03 million SNPs characterized in 368 maize inbred lines, including 'high-oil' lines. We identified 74 loci significantly associated with kernel oil concentration and fatty acid composition (P < 1.8 x 10(-6)), which we subsequently examined using expression quantitative trait loci (QTL) mapping, linkage mapping and coexpression analysis. More than half of the identified loci localized in mapped QTL intervals, and one-third of the candidate genes were annotated as enzymes in the oil metabolic pathway. The 26 loci associated with oil concentration could explain up to 83% of the phenotypic variation using a simple additive model. Our results provide insights into the genetic basis of oil biosynthesis in maize kernels and may facilitate marker-based breeding for oil quantity and quality.
DOI:10.1038/ncomms3832URLPMID:24343161 [本文引用: 1]
Abstract RNA sequencing can simultaneously identify exonic polymorphisms and quantitate gene expression. Here we report RNA sequencing of developing maize kernels from 368 inbred lines producing 25.8 billion reads and 3.6 million single-nucleotide polymorphisms. Both the MaizeSNP50 BeadChip and the Sequenom MassArray iPLEX platforms confirm a subset of high-quality SNPs. Of these SNPs, we have mapped 931,484 to gene regions with a mean density of 40.3 SNPs per gene. The genome-wide association study identifies 16,408 expression quantitative trait loci. A two-step approach defines 95.1% of the eQTLs to a 10-kb region, and 67.7% of them include a single gene. The establishment of relationships between eQTLs and their targets reveals a large-scale gene regulatory network, which include the regulation of 31 zein and 16 key kernel genes. These results contribute to our understanding of kernel development and to the improvement of maize yield and nutritional quality.
[本文引用: 1]
[本文引用: 1]
【目的】玉米株型性状与植株产量、光合效率、抗倒性等密切相关,是理想株型设计育种的基础,通过对玉米多个株型相关性状进行全基因组关联分析,构建玉米理想株型,为抗倒伏、耐密性的玉米新品种选育提供理论基础。【方法】以284份温带、亚热带和热带材料组成的关联群体为研究对象,在郑州与浚县2个环境下对玉米总叶片数(LN)、穗上叶片数(LNAN)、穗上叶片数与总叶片数比值(LNAN/LN)、株高(PH)、穗位高(EH)、穗位高与株高的比值(EH/PH)等6个玉米株型相关性状进行调查,借助覆盖玉米全基因组约56万个SNP标记,进行全基因组关联分析,以期为玉米新品种的选育提供理论依据。【结果】2个环境下,6个性状均表现为正态分布;大多数性状之间均存在高度正相关或负相关;方差分析表明这6个株型相关性状的基因型与环境以及基因型×环境的互作均达到显著水平。在选择最优关联分析模型时,发现Q模型假阳性较高,Q+K模型对假阳性的控制过于严格,而K模型的表现最好;在2个环境下,全基因组关联分析(K模型)共检测到56个显著SNP-性状关联(P≤3.99E-6),这些SNPs共涉及5个性状的17个位点,每个位点解释的表型变异从7.97%—10.56%不等。同时发现有4个位点能够在2个环境中同时被检测到,表明这4个位点受环境影响较小,在不同环境下可以稳定存在。通过对显著关联的SNP上下游各50 kb范围内候选基因进行搜索,共发现80个候选基因,其中42个具有功能注释。例如,与株高和穗位高显著相关的基因GRMZM2G161293编码一个具有乙酰葡糖转移酶活性的蛋白,该酶催化UDP-N-乙酰氨基葡萄糖生成糖过程中的N-乙酰葡糖氨基残基的转移,可能通过影响玉米籽粒可溶性糖含量进而影响产量,推测其为最可能的候选基因。【结论】K模型对假阳性的控制效果最好,基于K模型的GWAS结果,一共检测到17个株型性状相关的位点。
[本文引用: 1]
【目的】玉米株型性状与植株产量、光合效率、抗倒性等密切相关,是理想株型设计育种的基础,通过对玉米多个株型相关性状进行全基因组关联分析,构建玉米理想株型,为抗倒伏、耐密性的玉米新品种选育提供理论基础。【方法】以284份温带、亚热带和热带材料组成的关联群体为研究对象,在郑州与浚县2个环境下对玉米总叶片数(LN)、穗上叶片数(LNAN)、穗上叶片数与总叶片数比值(LNAN/LN)、株高(PH)、穗位高(EH)、穗位高与株高的比值(EH/PH)等6个玉米株型相关性状进行调查,借助覆盖玉米全基因组约56万个SNP标记,进行全基因组关联分析,以期为玉米新品种的选育提供理论依据。【结果】2个环境下,6个性状均表现为正态分布;大多数性状之间均存在高度正相关或负相关;方差分析表明这6个株型相关性状的基因型与环境以及基因型×环境的互作均达到显著水平。在选择最优关联分析模型时,发现Q模型假阳性较高,Q+K模型对假阳性的控制过于严格,而K模型的表现最好;在2个环境下,全基因组关联分析(K模型)共检测到56个显著SNP-性状关联(P≤3.99E-6),这些SNPs共涉及5个性状的17个位点,每个位点解释的表型变异从7.97%—10.56%不等。同时发现有4个位点能够在2个环境中同时被检测到,表明这4个位点受环境影响较小,在不同环境下可以稳定存在。通过对显著关联的SNP上下游各50 kb范围内候选基因进行搜索,共发现80个候选基因,其中42个具有功能注释。例如,与株高和穗位高显著相关的基因GRMZM2G161293编码一个具有乙酰葡糖转移酶活性的蛋白,该酶催化UDP-N-乙酰氨基葡萄糖生成糖过程中的N-乙酰葡糖氨基残基的转移,可能通过影响玉米籽粒可溶性糖含量进而影响产量,推测其为最可能的候选基因。【结论】K模型对假阳性的控制效果最好,基于K模型的GWAS结果,一共检测到17个株型性状相关的位点。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
对65个玉米杂交种棒三叶的叶长、叶宽、叶肉厚度、叶面积、叶绿素含量、叶向值、光合速率、叶片干重和比叶重等性状进行比较分析。结果表明:除比叶重和光合效率外,下位叶均超过上位叶和穗位叶,穗位叶又大于上位叶,呈现出形状上的金字塔结构。这种棒三叶的生长结构有利于透光,使光线较均匀地分布在棒三叶上,以提高玉米杂交种中部叶片的整体光合性能。穗位叶的比叶重和光合效率高于上位叶和下位叶,可能与穗位叶需提供玉米杂交种产量较多的光合产物有关。
[本文引用: 1]
对65个玉米杂交种棒三叶的叶长、叶宽、叶肉厚度、叶面积、叶绿素含量、叶向值、光合速率、叶片干重和比叶重等性状进行比较分析。结果表明:除比叶重和光合效率外,下位叶均超过上位叶和穗位叶,穗位叶又大于上位叶,呈现出形状上的金字塔结构。这种棒三叶的生长结构有利于透光,使光线较均匀地分布在棒三叶上,以提高玉米杂交种中部叶片的整体光合性能。穗位叶的比叶重和光合效率高于上位叶和下位叶,可能与穗位叶需提供玉米杂交种产量较多的光合产物有关。
[本文引用: 1]
[本文引用: 1]
Magsci [本文引用: 1]
水稻不同品种的叶绿素(a+b)含量及叶绿素 a/b 比值是不同的。叶绿素(a+b)含量生育初期较少,然后逐渐增多,到生育后期则下降。水稻各品种由苗期至乳熟期叶绿素(a+b)含量的变异系数为12~20%,而叶绿素 a/b比值的变异系数只有4~8%。剑叶的叶绿素 a/b 比值最大,越是下位叶越低。叶绿素(a+b)含量与净光合速率呈显著的正相关,其相
Magsci [本文引用: 1]
水稻不同品种的叶绿素(a+b)含量及叶绿素 a/b 比值是不同的。叶绿素(a+b)含量生育初期较少,然后逐渐增多,到生育后期则下降。水稻各品种由苗期至乳熟期叶绿素(a+b)含量的变异系数为12~20%,而叶绿素 a/b比值的变异系数只有4~8%。剑叶的叶绿素 a/b 比值最大,越是下位叶越低。叶绿素(a+b)含量与净光合速率呈显著的正相关,其相
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3389/fpls.2016.00533URL [本文引用: 1]
Chlorophylls (Chl) in photosynthetic apparatuses, along with other macromolecules in chloroplasts, are known to undergo degradation during leaf senescence. Several enzymes involved in Chl degradation, by which detoxification of Chl is safely implemented, have been identified. Chl degradation also occurs during embryogenesis and seedling development. Some genes encoding Chl degradation enzymes such as Chlbreductase (CBR) function during these developmental stages.Arabidopsismutants lacking CBR (NYC1 and NOL) have been reported to exhibit reduced seed storability, compromised germination, and cotyledon development. In this study, we examined aberrant cotyledon development and found that NYC1 is solely responsible for this phenotype. We inferred that oxidative damage of chloroplast membranes caused the aberrant cotyledon. To test the inference, we attempted to trans-complementnyc1mutant with overexpressing VIPP1 protein that is unrelated to Chl degradation but which supports chloroplast membrane integrity. VIPP1 expression actually complemented the aberrant cotyledon ofnyc1, whereas stay-green phenotype during leaf senescence remained. The swollen chloroplasts observed in unfixed cotyledons ofnyc1, which are characteristics of chloroplasts receiving envelope membrane damage, were recovered by overexpressing VIPP1. These results suggest that chloroplast membranes are a target for oxidative damage caused by the impairment in Chl degradation. Trans-complementation ofnyc1with VIPP1 also suggests that VIPP1 is useful for protecting chloroplasts against oxidative stress.
DOI:10.1016/j.jplph.2006.11.006URL [本文引用: 1]
DOI:10.1016/j.plantsci.2013.06.003URL [本文引用: 1]
DOI:DOI: 10.3724/SP.J.1005.2008.01083Magsci [本文引用: 1]
为了探讨玉米叶绿素含量的遗传规律, 以A150-3-2×Mo17杂交组配的189个F2单株作为作图群体, 构建了具有112个标记位点的玉米分子遗传图谱, 于喇叭口期和开花期分别进行了玉米叶绿素a含量(chla), 叶绿素b含量(chlb), 其他叶绿素含量(chlc)和叶绿素总含量(chlz)4个性状的测定, 并进行QTL分析。在喇叭口期和开花期共检测到32个QTL, 分布在除第6和10染色体以外的其他染色体上。在喇叭口期检测到24个QTL, 分布于第1、2、3、5、7、8和9染色体上, 叶绿素a、叶绿素b、其他叶绿素和叶绿素总含量各检测到6个QTL, 在同一区间内检测到的4个性状的QTL之间的距离在0~2 cM之间。喇叭口期检测到控制叶绿素a、叶绿素b、其他叶绿素和叶绿素总含量的4个主效QTL位于第5染色体上的umc1098~bnlg557区间, 分别可解释表型变异的11.63%、10.3%、10.77%和11.51%。开花期检测到8个QTL, 分布于第4和5染色体上。其中叶绿素a、叶绿素b、其他叶绿素和叶绿素总含量各2个QTL。标记umc1098和bnlg557之间同时存在控制喇叭口期4个叶绿素含量性状的QTL和开花期控制叶绿素a和叶绿素b的QTL。标记umc2308和bnlg386之间只存在控制开花期4个叶绿素含量性状的QTL。
DOI:DOI: 10.3724/SP.J.1005.2008.01083Magsci [本文引用: 1]
为了探讨玉米叶绿素含量的遗传规律, 以A150-3-2×Mo17杂交组配的189个F2单株作为作图群体, 构建了具有112个标记位点的玉米分子遗传图谱, 于喇叭口期和开花期分别进行了玉米叶绿素a含量(chla), 叶绿素b含量(chlb), 其他叶绿素含量(chlc)和叶绿素总含量(chlz)4个性状的测定, 并进行QTL分析。在喇叭口期和开花期共检测到32个QTL, 分布在除第6和10染色体以外的其他染色体上。在喇叭口期检测到24个QTL, 分布于第1、2、3、5、7、8和9染色体上, 叶绿素a、叶绿素b、其他叶绿素和叶绿素总含量各检测到6个QTL, 在同一区间内检测到的4个性状的QTL之间的距离在0~2 cM之间。喇叭口期检测到控制叶绿素a、叶绿素b、其他叶绿素和叶绿素总含量的4个主效QTL位于第5染色体上的umc1098~bnlg557区间, 分别可解释表型变异的11.63%、10.3%、10.77%和11.51%。开花期检测到8个QTL, 分布于第4和5染色体上。其中叶绿素a、叶绿素b、其他叶绿素和叶绿素总含量各2个QTL。标记umc1098和bnlg557之间同时存在控制喇叭口期4个叶绿素含量性状的QTL和开花期控制叶绿素a和叶绿素b的QTL。标记umc2308和bnlg386之间只存在控制开花期4个叶绿素含量性状的QTL。
DOI:10.11674/zwyf.2008.0505Magsci [本文引用: 1]
以优良玉米杂交种农大108的203个F2:3家系为材料,构建了包含189个SSR标记的遗传连锁图谱,利用复合区间作图法,在施氮(+N)和不施氮(-N)情况下,对玉米不同发育时期的叶绿素含量进行了QTL分析。结果表明:在-N和+N条件下,亲本许178的5个时期叶绿素(SPAD)均值分别为54.12和55.76,比黄C分别高1.80和2.40;而F<SUB>2:3</SUB>家系的SPAD均值分别达55.6和58.32,高于双亲的中亲值;同时,在+N情况下,叶绿素含量变异范围相对较小,说明氮胁迫对玉米叶绿素含量的变化具有一定影响。-N和+N情况下,在玉米喇叭口期、散粉期、灌浆初期、灌浆中期和灌浆后期分别检测到2、2、1、3、1个和2、2、2、7、1个叶绿素含量的非条件QTL,可分别解释对应时期叶绿素含量表型总变异的22.75%、22.93%、19.77%、49.87%、12.79%和16.95%、23.49%、19.38%、84.56%、13.38%。在-N和+N情况下,喇叭口期―散粉期、灌浆中期―灌浆后期和喇叭口期―散粉期、散粉期―灌浆初期各检测到1个条件QTL,可分别解释绿素含量表型变异的12.90%、25.51%和5.98%、26.67%。4)叶绿素含量的QTL表达存在时空性,其中<EM>qchl4</EM>和<EM>qchl5b</EM>在整个生育时期均能检测到,是玉米生长发育所必需的两个QTL。
DOI:10.11674/zwyf.2008.0505Magsci [本文引用: 1]
以优良玉米杂交种农大108的203个F2:3家系为材料,构建了包含189个SSR标记的遗传连锁图谱,利用复合区间作图法,在施氮(+N)和不施氮(-N)情况下,对玉米不同发育时期的叶绿素含量进行了QTL分析。结果表明:在-N和+N条件下,亲本许178的5个时期叶绿素(SPAD)均值分别为54.12和55.76,比黄C分别高1.80和2.40;而F<SUB>2:3</SUB>家系的SPAD均值分别达55.6和58.32,高于双亲的中亲值;同时,在+N情况下,叶绿素含量变异范围相对较小,说明氮胁迫对玉米叶绿素含量的变化具有一定影响。-N和+N情况下,在玉米喇叭口期、散粉期、灌浆初期、灌浆中期和灌浆后期分别检测到2、2、1、3、1个和2、2、2、7、1个叶绿素含量的非条件QTL,可分别解释对应时期叶绿素含量表型总变异的22.75%、22.93%、19.77%、49.87%、12.79%和16.95%、23.49%、19.38%、84.56%、13.38%。在-N和+N情况下,喇叭口期―散粉期、灌浆中期―灌浆后期和喇叭口期―散粉期、散粉期―灌浆初期各检测到1个条件QTL,可分别解释绿素含量表型变异的12.90%、25.51%和5.98%、26.67%。4)叶绿素含量的QTL表达存在时空性,其中<EM>qchl4</EM>和<EM>qchl5b</EM>在整个生育时期均能检测到,是玉米生长发育所必需的两个QTL。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1534/genetics.111.133967URL [本文引用: 1]
DOI:10.1073/pnas.221252798URLPMID:11606728 [本文引用: 2]
Tetrapyrroles such as chlorophylls and bacteriochlorophylls play a fundamental role in the energy absorption and transduction activities of photosynthetic organisms. Because of these molecules, however, photosynthetic organisms are also prone to photooxidative damage. They had to evolve highly efficient strategies to control tetrapyrrole biosynthesis and to prevent the accumulation of free intermediates that potentially are extremely destructive when illuminated. In higher plants, the metabolic flow of tetrapyrrole biosynthesis is regulated at the step of -aminolevulinic acid synthesis. This regulation previously has been attributed to feedback control of Glu tRNA reductase, the first enzyme committed to tetrapyrrole biosynthesis, by heme. With the recent discovery of chlorophyll intermediates acting as signals that control both nuclear gene activities and tetrapyrrole biosynthesis, it seems likely that heme is not the only regulator of this pathway. A genetic approach was used to identify additional factors involved in the control of tetrapyrrole biosynthesis. In Arabidopsis thaliana, we have found a negative regulator of tetrapyrrole biosynthesis, FLU, which operates independently of heme and seems to selectively affect only the Mg2+branch of tetrapyrrole biosynthesis. The identity of this protein was established by map-based cloning and sequencing the FLU gene. FLU is a nuclear-encoded plastid protein that, after import and processing, becomes tightly associated with plastid membranes. It is unrelated to any of the enzymes known to be involved in tetrapyrrole biosynthesis. Its predicted features suggest that FLU mediates its regulatory effect through interaction with enzymes involved in chlorophyll synthesis.
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.1023/A:1006425025702URLPMID:11117258 [本文引用: 1]
NADH -cytochrome P450 is a key enzyme that transfers electrons from NADPH to the cytochrome P450 family of enzymes. To begin to determine the regulation of CPR gene expression and enzyme activity in Douglas-fir a full-length cDNA was isolated from a seedling ZAP cDNA library and the ORF was used to develop a synthetic CPR-peptide-based antiserum. Northern blot analysis indicated CPR expression was regulated both developmentally prior to seed maturation and during germination, and differentially in the cotyledons, radicle and megagametophyte of seed and seedling tissues. The CPR-peptide antiserum detected a single CPR in seed and seedling microsomes analyzed by western blot of two-dimensional SDS-polyacrylamide gels. In microsomal extracts from whole seeds and seedlings, the amount of CPR protein remained constant while NADPH:cytochrome c reductase activity increased during stratification, germination and early seedling development. In contrast to cotyledons and megagametophyte, the level of CPR protein detected in radicles was higher than expected when compared to the amount of CPR transcript.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}