 ,1,**, 李书宇2,**, 詹杰鹏1, 李晏斌3, 师家勤,1,*, 王新发1, 王汉中1
,1,**, 李书宇2,**, 詹杰鹏1, 李晏斌3, 师家勤,1,*, 王新发1, 王汉中1Mapping and candidate gene analysis of silique number mutant in Brassica napus L.
ZHAO Gai-Hui,1,**, LI Shu-Yu2,**, ZHAN Jie-Peng1, LI Yan-Bin3, SHI Jia-Qin,1,*, WANG Xin-Fa1, WANG Han-Zhong1通讯作者: *师家勤, E-mail:shijiaqin@caas.cn
第一联系人:
收稿日期:2020-12-27接受日期:2021-04-14网络出版日期:2021-06-15
| 基金资助: |
Corresponding authors: *E-mail:shijiaqin@caas.cn
First author contact:
Received:2020-12-27Accepted:2021-04-14Published online:2021-06-15
| Fund supported: |
作者简介 About authors
E-mail:3468382960@qq.com

摘要
角果数是油菜单株产量重要的构成因子之一, 其优异等位基因的发掘和利用对产量的提高至关重要。油菜中已定位到上百个角果数QTL, 但大多数效应不大且不稳定, 难以进行精细定位或克隆。本研究前期发掘到一个油菜突变体(No.7931), 其花序顶端在分化出约十朵花后即停止生长, 因而成熟期角果极少。利用该少角果突变体和多角果品系No.73290构建F2分离群体, 从中挑选角果数极端单株各30株进行BSA-seq, 在C02染色体检测到3个关联区间: 0~1.1 Mb、4.7~6.2 Mb、11.5~12.4 Mb。该候选区间在油菜参考基因组DarmorV8.1中有522个注释基因, 存在SNP或Indel差异且有同源注释的基因235个。在花芽分化初期, 选取两亲本(No.73290和No.7931)的茎尖分生组织进行RNA-seq, 总共鉴定到8958个差异表达基因(DEGs)。这些DEGs显著富集于20个生物学通路, 包括碳代谢、翻译、氨基酸代谢(和花芽分化高度相关)等, 其中99个位于关联区间。结合基因功能注释以及序列和表达差异分析确定了9个候选基因(BnaC02g00490.1D2、BnaC02g01030.1D2、BnaC02g01120.1D2、BnaC02g00270.1D2、BnaC02g02670. 1D2、BnaC02g08680.1D2、BnaC02g08890.1D2、BnaC02g09480.1D2和BnaC02g10490.1D2), 它们主要参与花序分生组织特性的维持和花器官的发育。上述研究结果为后续油菜角果数基因的精细定位和克隆奠定了坚实的基础。
关键词:
Abstract
The silique number is one of the important components of yield per plant in oilseed rape (Brassica napus L.) and the exploitation and utilization of its excellent alleles are essential to increase yield. More than hundreds of silique number QTLs have been mapped in oilseed rape, but they are difficult to be fine-mapped or cloned because of their moderate and unstable effects. A oilseed rape mutant (No.7931) was detected in previous study and it had few siliques at mature stage due to the stop growth after differentiation about 10 flowers on the top of inflorescence. A F2 segregating population consisting of 3400 individuals was constructed using this mutant and another more-silique lines No.73290. Among them, we performed BSA-seq on 30 individuals with extreme more- or less-siliques and detected three associated intervals of 0-1.1 Mb, 4.7-6.2 Mb, and 11.5-12.4 Mb on the C02 chromosome. These genomic intervals contained a total of 522 annotated genes in the reference genome DarmorV8.1, among which 235 genes had functional annotation and SNP/InDel variation. At the early stage of flower bud differentiation, the shoot apical meristems of two parents were subjected to RNA-seq, and a total of 8958 differentially expressed genes (DEGs) were detected. These DEGs were significantly enriched into 20 pathways, including carbohydrate metabolism, translation, and amino acid metabolism (highly associated with flower bud differentiation) and so on, among which 99 were located in the associated intervals. By the integration of gene functional annotation as well as sequence and expression variation analysis, a total of nine candidate genes (BnaC02g00490.1D2, BnaC02g01030.1D2, BnaC02g01120.1D2, BnaC02g00270.1D2, BnaC02g02670.1D2, BnaC02g08680.1D2, BnaC02g08890.1D2, BnaC02g09480.1D2, and BnaC02g10490.1D2) were identified, which were mainly involved in the maintenance of inflorescence meristems and the regulation of flower development. The above results lay the foundation for the following fine-mapping and cloning of the silique number mutant gene in oilseed rape.
Keywords:
PDF (10417KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
赵改会, 李书宇, 詹杰鹏, 李晏斌, 师家勤, 王新发, 王汉中. 甘蓝型油菜角果数突变体基因的定位及候选基因分析. 作物学报, 2022, 48(1): 27-39 DOI:10.3724/SP.J.1006.2022.04281
ZHAO Gai-Hui, LI Shu-Yu, ZHAN Jie-Peng, LI Yan-Bin, SHI Jia-Qin, WANG Xin-Fa, WANG Han-Zhong.
油菜是我国最大的油料作物, 大力发展油菜生产对保障我国油料供给安全具有十分重要的战略意义[1]。据国家统计局(
本实验室Li等[8]从种质资源中筛选出19份角果数极端品系进行花器官数目和结角率的考察和比较, 估算出两者对角果数的相对贡献率分别为75.2%和24.8%, 认为油菜角果数的变异主要决定于花器官数目的差异。虽然油菜中尚无角果数基因克隆的报道, 但在模式植物和作物中已鉴定了一批调控花器官数目的基因[19], 这些基因大多数参与到WUS-CLV3的反馈调节途径中。该途径在植物中比较保守, 具有调节分生组织大小的功能, 主要包括WUS、CLV1、CLV2/CRN、CLV3和BAM/RPK2等核心基因[20], 其中任何一个基因发生突变往往导致分生组织变大, 花器官数目增多。其中, 拟南芥WUS和水稻TAB1同源, 编码一个具有同源异型结构域(homeodomain)的转录因子, 主要参与腋分生组织干细胞活性的维持及其分化[21]; 拟南芥CLV1和水稻FON1、玉米TD1以及番茄FAB同源, 编码富含亮氨酸的受体激酶, 主要参与花分生组织特性的建立和维持[22,23,24]; 拟南芥CLV2和玉米FEA2同源, 编码富含亮氨酸重复的类受体蛋白, 主要参与调控分生组织和器官的发育[25]; 拟南芥CLV3和水稻FON2/FON4以及番茄FAS同源, 编码一个含有CLE功能域的干细胞特异的蛋白, 被认为是分泌肽激素的前体, 主要参与分生组织发育及其特性的维持[21,26]。
BSA-seq是将传统的集团分离分析(bulked segregant analysis)方法和现代测序技术相结合发展起来的一种比较新的高效快速的定位手段, 原先应用于质量性状, 随后又逐渐拓展到由主效基因控制的数量性状[27]。BSA-seq主要有基于简化基因组测序的BSA、基于重测序的BSA、基于转录组测序的BSR、基于芯片的BSA, 目前基于重测序的BSA仍被广泛应用[28]。RNA-seq是基于第二代测序的技术, 该技术主要应用于表达水平的分析和特定时期显著富集的差异表达基因分析[29]。RNA-seq能够深入揭示组织发育的分子机理, 并为候选基因的筛选提供可靠的依据。目前将BSA-seq和RNA-seq结合鉴定候选基因的报道很多。Gao等[30]利用BSA-seq和RNA-seq相结合方式鉴定了调控水稻紫叶的隐性基因Plr4, 为阐明花青素合成途径的分子机制奠定了基础; Sunggil等[31]通过BSA和RNA-seq的联合应用, 确定了洋葱育性恢复的候选基因AcPMS1。
本研究前期对油菜种质资源进行了广泛的筛选, 获得了少角果突变体No.7931和角果数极多品系No.73290[8], 两者角果数差异形成的原因是花芽分化产生的花蕾和花器官数目的差异。本研究利用这2个材料为亲本构建了F2分离群体, 从中挑选角果数极端单株进行BSA-seq, 实现对角果数基因的定位, 同时结合亲本花芽分化初期茎尖分生组织RNA-seq筛选候选基因, 旨在为后续精细定位和克隆奠定基础。
1 材料与方法
1.1 试验材料
甘蓝型油菜多角果品系No.73290为本实验室多年的自交种, 少角果突变体No.7931是田间筛选的自然变异材料, 利用No.7931为母本与No.73290杂交, 杂种F1代自交后获得F2代分离群体。从F2分离群体中挑选30株多角果植株和30株少角果植株分别构建多角果和少角果DNA混合池, 利用该混合池进行少角果突变体基因的初定位。油菜花芽分化初期取油菜多角果品系No.73290和少角果突变体No.7931的茎尖分生组织进行转录组测序, 每个样本取20个顶端分生组织进行混合, 设置3个生物学重复, 共计6个样本。1.2 油菜花期和成熟期性状调查
经过多年的油菜花期和成熟期性状调查, 评价油菜少角果突变体材料的标准为油菜主花序和一次分枝的顶端生长发育受阻, 且出现无效角果。开花期提前, 主枝和一次分枝花序出现顶生花, 但植株高度无显著差异, F2分离群体中表现为少角果突变体的材料基本无明显主枝, 二级分支和三级分支增多。2018年夏季将亲本No.73290和No.7931杂交的F1代种植在中国农业科学院油料作物研究所青海基地(36°67'41.91′′N, 101°75'66.38′′E), 2018年秋季将F1代收获的种子种植在中国农业科学院油料作物研究所武昌基地(30°35'1.12′′N, 114°20'21.70′′E), 花期和成熟期分别观察油菜表型。
1.3 顶端分生组织(SAM)显微观察
从苗期五叶期(花芽分化初期)开始, 对亲本突变体材料(No.7931)和正常野生型材料进行连续取样。选取长势一致的植株3~5株, 首先用解剖刀将植株从根部切除, 摘除顶端分生组织周围大的叶片, 在普通体式显微镜下, 用解剖针剥去生长在顶端分生组织周围的嫩叶。利用体式显微镜(NikonSMZ25)进行连续显微观察。1.4 混池测序
1.4.1 极端混合池的构建和DNA测序 根据F2代分离群体的花期表型鉴定结果, 在No.73290× No.7931组合的F2群体中分别选取30株极端材料, 每株材料取0.2 g叶片提取基因组DNA, 并利用琼脂糖凝胶电泳检测其质量, 将检测合格的样品分别等量混合构建油菜多角果混合池和油菜少角果混合池, 连同亲本No.73290一起送未来组生物科技有限公司进行DNA测序。1.4.2 变异检测和候选区间的确定 对Illumina HiSeq Xten测序所得的数据进行质控(Quality Control, QC)。质控后, 将过滤得到的clean reads比对到参考基因组DarmorV8.1, 使用samtools工具[32]和GATK软件[33]检测SNP、InDel。将检测得到的SNP和InDel利用GATK进行过滤和筛选。
利用SNP-index值确定候选区间, SNP-index值等于亲本型等位基因的测序深度比总深度。为排除不可靠标记的影响, 进行SNP过滤: (1) 过滤掉亲本、混池任意一个有缺失的位点; (2) 过滤掉亲本为杂合的位点; (3) 过滤掉任一混池测序深度<20的位点; (4) 过滤掉2个子代混池基因型相同且为纯合的位点。以测序亲本No.73290的实际基因型为参考, 分别计算2个混池的SNP-index及总的Δ(SNP-index), 通过相应的划窗(步长100 kb窗口大小1 Mb)进行拟合; 分别对每条染色体作图, 最后确定99%的置信区间为候选区域。
1.5 RNA测序
1.5.1 总RNA提取和RNA测序 为得到可靠的数据, 在油菜花芽分化的初期(花原基形成阶段)取No.73290和No.7931的茎尖分生组织各20株进行混合, 设置3个生物学重复, 共6个样本。使用RNeasy Plant Minit Kit试剂盒提取样本RNA, 并使用NanoDrop1000分光光度计和琼脂糖凝胶电泳检测RNA的浓度和质量。将检测合格的样本送上海欧易生物科技公司测序。1.5.2 基因表达分析 首先将质控得到的clean reads比对到油菜的参考基因组(
1.5.3 差异表达基因的功能注释与分类 将得到的差异表达基因与参考基因组进行比对, 得到所有差异表达基因的CDS序列。利用差异表达基因的CDS序列与拟南芥的序列进行Blastn分析, 将E值设为1e-8, 将匹配度最高的拟南芥的基因进行差异表达基因的功能注释, 使用在线软件(
1.5.4 qRT-PCR验证RNA测序结果 为验证测序数据的准确性, 本研究随机选取了14个差异表达基因进行qRT-PCR验证, 使用在线软件Primer3.0设计引物, 并由北京擎科生物科技公司合成。使用PrimeScript RT reagent Kit with gDNA Eraser 反转录试剂盒将转录组测序的RNA样品反转录成cDNA, 选择BraActin作为内参基因。每个基因在试验组(No.7931)和对照组(No.73290)中设置3个生物学重复, 以2-ΔΔCt算法计算基因的差异表达倍数, 并以log2 Fold Change展示试验结果。
1.6 候选基因鉴定
本研究采取基因功能注释为主, 同时结合序列和表达差异的方法筛选候选基因。首先从已发表的文章和作物网站(2 结果与分析
2.1 油菜少角果突变体No.7931和多角果品系No.73290花期和成熟期表型观察
从外形上来看, 少角果突变体No.7931最显著的特征是: 花序轴的顶端发育成一朵顶生花或若干小花苞粘连在一起, 无明显的主序, 二次分枝和三次分枝增多(图1)。相比于多角果品系No.73290, 少角果突变体No.7931的一次花序的花器官和角果数都要少很多, 而且花期更早, 但株高相差不大(表1)。因此, No.7931和No.73290角果数的差异是由于两者花器官数目的差异。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1油菜少角果突变体No.7931和多角果品系No.73290花期和成熟期花序的观察
A1: No.7931花期; A2: No.73290花期; B1: No.7931成熟期; B2: No.73290成熟期。
Fig. 1Observation on the inflorescences at flowering and mature stages of the rapeseed oligocyte mutant No.7931 and polycygocyte stain No.73290
A1: No.7931 at flowering stage; 2: No.73290 at flowering stage; B1: No.7931 at mature stage; B2: No.73290 at mature stage.
Table 1
表1
表1No.7931和No.73290表型数据统计分析
Table 1
| 亲本 Parent | 主花序花器官数目 FNm | 主花序角果数 SNm | 株高 PH |
|---|---|---|---|
| No.7931 | 8.33±1.24 | 7.67±0.94 | 154.3±2.06 |
| No.73290 | 199.3±1.24 | 93.67±2.49 | 150.4±1.57 |
| t检验值t-test value | 5.45E-09 | 6.91E-07 | 0.0170 |
新窗口打开|下载CSV
2.2 油菜少角果突变体No.7931花芽分化的显微观察
为进一步解析No.7931花器官数目少的原因, 从花芽分化初期开始, 对该突变体和多角果材料No.73290的花芽分化过程进行连续显微观察。以最外围花苞所处发育阶段为划分标准, 主要分4个阶段进行比.较: 第1个阶段, 花原基分化(图2-A1, A2); 第2个阶段为花萼分化(图2-B1, B2); 第3个阶段为花瓣原基分化, 蕾轴伸长(图2-C1, C2); 第4个阶段为现蕾早期(图2-D1, D2)。在花芽分化前期两种类型的材料无明显差异, 花瓣原基分化至现蕾早期, No.7931分化的花器官彼此不能正常分开, 粘连在一起。因此, 显微观察的结果表明, No.7931花器官数目少的原因是花芽分化过程出现异常, 较早就终止了。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2油菜角果数突变体的显微观察
A、B、C、D分别代表花原基分化、花萼分化、花瓣原基分化(蕾轴伸长)与现蕾早期4个时期; 1、2分别代表No.7931和No.73290。
Fig. 2Microscopic observation of silique number mutant in Brassica napus L.
A, B, C, and D represent the four stages of flower primordium differentiation, calyx differentiation, petal primordium differentiation (bud axis elongation), and early budding, respectively. 1, 2 represent No.7931 and No.73290, respectively.
2.3 油菜少角果突变体基因的BSA定位
2.3.1 测序数据的统计分析 对突变体混池(F2A)、多角果混池(F2B)以及亲本多角果品系(No.73290)进行测序, 测序深度分别为69.42、62.71和109.15 G。过滤后碱基数产量共有288 G, 测序数据质量较高(Q30>94.40%), GC含量在36.70%~ 36.83%之间。利用BWA软件将过滤后的reads比对到油菜Darmor V8.1参考基因组, 比对结果经samtools去除重复, 其中F2A、F2B和No.73290特异性比对的百分比分别为88.65%、88.97%和88.88%。整体测序和比对结果良好, 可用于后续分析。2.3.2 油菜角果数候选区间的鉴定 将2个池观测到的SNP均计算出SNP-index值, 然后将2个值相减后得到Δ(SNP-index), 将Δ(SNP-index)对应到SNP所在染色体位置作图, Δ(SNP-index)比较大的区域即可作为QTL的候选区域。分析2个混池的ΔSNP-index, 用于少角果突变体基因的定位。分析结果表明, 候选区间定位在C2染色体3个区段: 0~1.1 Mb、4.7~6.2 Mb、11.5~12.4 Mb (图3)。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3候选区间峰图
横坐标表示Δ(SNP-index)在染色体上的位置, 纵坐标表示Δ(SNP-index)的数值。
Fig. 3Candidate interval peak map
The abscissa represents the position of the Δ(SNP-index), and the ordinate represents the value of the Δ(SNP-index).
2.4 油菜少角果突变体SAM的转录组分析
2.4.1 RNA-seq数据统计 本研究对6个样本进行有参转录组测序, 共获得39.63 G的CleanData, 各样本的有效数据量分布在6.48~6.72 G, Q30碱基分布在94.35%~94.74%之间, 平均GC含量为47.54%, 测序质量良好, 可以用于后续分析。将reads比对到参考基因组上, 得到各个样本的基因组比对情况, 总比对率为95.23%~96.46%, 比对到多个位置上的比率为8.32%~8.56%, 特异性比对率为86.69%~87.97%, 与参考序列比对结果良好, 可以用于生物信息学分析。2.4.2 差异表达基因分析 将显著性检验P<0.05且差异倍数FC>2的基因作为差异表达基因(DEGs)。本研究在No.73290和No.7931的茎尖分生组织中筛选到了8958个DEGs。其中4769个DEGs表达上调, 4189个DEGs表达下调。并根据基因的表达情况获得样本间的相关系数, 重复间相关系数均在0.998以上, 重复间差异很小, 数据可靠(附图1)。
附图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图1样本间相关系数热图
Fig. S1Heat map of correlation coefficient between samples
2.4.3 qRT-PCR验证RNA-seq结果分析 对挑选的6个上调差异表达基因和8个下调差异表达基因进行实时荧光定量PCR (qRT-PCR)试验(附表1)表明, 所选的14个差异表达基因在qRT-PCR与RNA-seq的数据中上调和下调的趋势一致(图4), 说明RNA-seq的结果可靠。但2种检测方法得到的表达差异倍数数值有差异, 与已发表的相关研究一致[36], 属正常现象。
Table S1
附表1
附表1qRT-PCR引物
Table S1
| 基因 Gene | 正向引物 Forward primer (5°-3°) | 反向引物 Reverse primer (5°-3°) |
|---|---|---|
| BnaA02G0314300ZS | CCGACACGCTTCAGAAGGTC | AGAGATCCCGCTTCGACTCC |
| BnaC03G0027000ZS | TGAGAACACGCCGGTCAAAG | CCTCGCGTTCTCTCTCTCCA |
| BnaC03G0100100ZS | CTGATGGCCGTAGAGCATGC | AGCCATCTCCTGTTGTTGCA |
| BnaC03G0091000ZS | TCCCACTCTACCCTGCACTG | GACGTTGTTCACATTCGCGC |
| BnaC02G0283700ZS | ACAACATGGCCCTGGAAACTG | CTTGGAGGCGGATGATCGTT |
| BnaC02G0139700ZS | CGGCGGAGGTACAGACATTT | TCGTCATGATGAGCCTCTCCT |
| BnaC02G0175400ZS | GCGCCTCGTATCCATTCTCG | CCTGAAGTTGTGGCGAGCTT |
| BnaA01G0077800ZS | CCACCGTCATGTCTTCCTTCC | ACGAGTTGGAAGTGTGCGTT |
| BnaA01G0061200ZS | GGGAACGGCTTAGATGGTGC | TTGCTAGTACCAGGGCTGCT |
| BnaC02G0013900ZS | CATTGGCTCGTCATGAACATC | TTCACGAGTGTTGAACTGATCC |
| BnaC02G0159100ZS | CTGTCACTGGAAACCACCCG | TCAATCGACCATGGCAAGCA |
| BnaA01G0009100ZS | TCTGCTCTGAACGCGACCAA | ACCAGCCAAAGAACCAGGGT |
| BnaA01G0037200ZS | TCCTCAACTGTGCCGACATGT | AAAGCCGTCGTCAATCTCGC |
| BnaC05G0266700ZS | AGACTACGTGAAGCAGCCGA | CTCCAGCTTCCGACCAGTCT |
| BraActin | CTGGAATTGCTGACCGTATGAG | ATCTGTTGGAAAGTGCTGAGGG |
新窗口打开|下载CSV
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4转录组测序与实时荧光定量PCR结果的比较
Fig. 4Comparison of the relative expression abundance measured by qRT-PCR and RNA-seq
2.4.4 差异表达基因的富集分析 对有功能注释的8047个DEGs进行GO富集分析, 图5展示了具体的结果。在生物过程方面, 最富集的组为对生物或者非生物刺激的反应和发育过程, 其次为转录和电子传递; 此外显著富集的组为抗逆反应、信号转导、细胞组织和生物发生、转运、DNA或RNA代谢等。在分子功能方面, 最富集的组为转录因子活性、蛋白结合和核酸结合, 其次为转运活性、转运分子活性和核苷酸结合; 此外显著富集的分子功能为水解酶活性、DNA或RNA结合、转移酶活性、激酶活性、受体结合等。在细胞组份方面, 最富集的组为细胞壁和细胞质液, 其次为ER和核糖体; 此外, 显著富集的还有细胞骨架、高尔基体、叶绿体、质体等。同时对这8047个DEGs进行功能分类(图6), 显著富集的有34组。值得注意的是, 发酵作用和多胺代谢为最富集的组。此外大多数富集的组与能量合成转运、新陈代谢、生长发育、植物逆境等有关, 例如糖酵解、乙醛酸循环、三羧酸循环(TCA)、转导、氨基酸代谢、核苷酸代谢、激素代谢、发育、植物逆境等。表明能量供给、养分吸收和同化、激素水平和信号转导在油菜茎尖分生组织的发育过程中发挥重要作用, 影响花器官数目。
图5
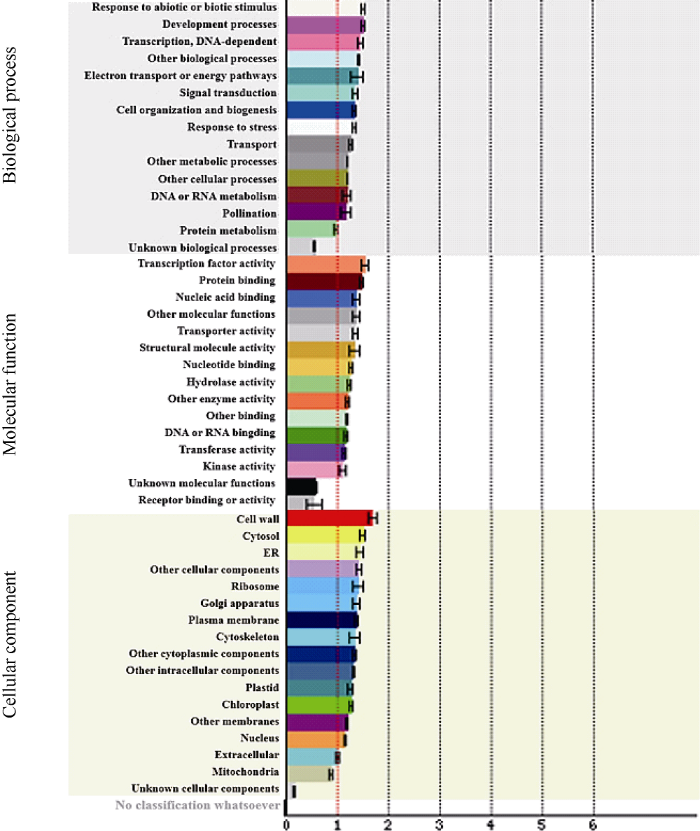 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5差异表达基因GO富集分析
Fig. 5GO enrichment analysis of differentially expressed genes
图6
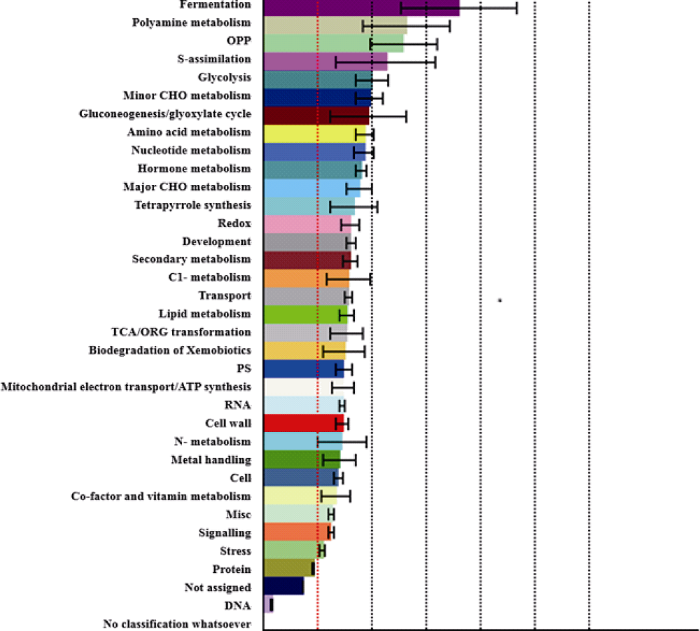 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6差异表达基因的功能分类
Fig. 6Functional classification of differentially expressed genes
2.4.5 差异基因KEGG富集分析 为进一步明确DEGs参与的生物学路径, 对其中有功能注释的8047个进行KEGG分析, 富集到的生物学路径主要被划分为5个大类, 分别为细胞过程、环境信息过程、遗传信息过程、代谢、有机系统, 共富集到了3052个DEGs (图7)。其中, 代谢类富集到了1785个DEGs, 占富集到通路基因的58.23%, 说明代谢在No.73290和No.7931茎尖分生组织发育差异中发挥重要作用。另外, 最富集的3个生物学路径为碳代谢(469个基因)、翻译(327个基因)和氨基酸代谢(255个基因), 其中2个都属于代谢类。碳代谢是植物体内最重要的基础代谢, 包括光合产物的合成、降解与转化, 也包括呼吸过程中的糖酵解、三羧酸循环、戊糖磷酸途径、乙醇酸氧化途径以及乙醛酸循环等。因此, KEGG分析的结果表明, 这些DEGs参与的生物学路径和油菜花芽分化密切相关。
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7差异表达基因的KEGG分析
Fig. 7KEGG analysis of differentially expressed genes
2.5 油菜少角果突变体候选基因
在BSA定位的基础上, 结合显微观察(明确是花芽分化过程出现问题)、基因功能注释以及序列和表达差异(分别来自亲本DNA和RNA测序)的结果, 鉴定候选基因。BSA定位获得的3个关联区间在Darmor V8.1参考基因组中共有522 (231/224/67)个基因, 其中259 (147/111/0)个存在SNP或InDel差异, 235个(141/94/0)有功能注释。另外, 3个候选区间内有99 (67/28/4)个基因存在表达差异, 95 (65/26/4)个在拟南芥中有功能注释。最终, 3个关联区间内总共鉴定了9个与花芽分化相关的候选基因(表2), 其中1个存在序列和表达差异(BnaC02g00490.1D2), 7个仅存在序列差异(BnaC02g00830.1D2、BnaC02g01570.1D2、BnaC02g01970.1D2、BnaC02g00270.1D2、BnaC02g08680.1D2、BnaC02g08890.1D2和BnaC02g09480.1D2), 2个仅存在表达差异(BnaC02g02670.1D2、BnaC02g10490.1D2), 2个无序列和表达差异(BnaC02g01030.1D2、BnaC02g01120.1D2)。Table 2
表2
表2关联区间内的候选基因预测
Table 2
| 油菜基因编号 B. napus ID | 拟南芥 基因名 Name of A. thaliana | 拟南芥基因 编号 A. thaliana ID | 位置Position (Mb) | 基因注释 Gene annotation | 序列差异 Sequence difference | 差异表达基因 Differential expressed genes |
|---|---|---|---|---|---|---|
| BnaC02g00490.1D2 | FLC | AT5g10140 | 0.208 | K盒区和MADS转录因子家族蛋白 K-box region and MADS-box transcription factor family protein | 有Yes | 是No |
| BnaC02g01030.1D2 | TPR1 | AT1G80490 | 0.465 | TPL相关的基因1 TOPLESS-related 1 | 无No | 否No |
| BnaC02g01120.1D2 | AGAL2 | AT5G08370 | 0.493 | α-半乳糖苷酶2 Alpha-galactosidase 2 | 无No | 否No |
| BnaC02g00270.1D2 | AIL6 | AT5G10510 | 0.996 | 类ANT基因6 AINTEGUMENTA-LIKE 6 | 有Yes | 否No |
| BnaC02g02670.1D2 | TFL1 | AT5G03840 | 1.10 | 磷脂酰乙醇胺结合蛋白 Phosphatidylethanolamine-binding-protein | 无No | 是Yes |
| BnaC02g08680.1D2 | COL1 | AT5G15850 | 4.83 | 类CO基因1 CONSTANS-like 1 | 有Yes | 否No |
| BnaC02g08890.1D2 | ELF9 | AT5G16260 | 4.96 | RNA结合(RRM/RBD/RNP基序)家族蛋白RNA binding (RRM/RBD/RNP motifs) family protein | 有Yes | 否No |
| BnaC02g09480.1D2 | AIM1 | AT4G29010 | 5.23 | 烯酰辅酶A水合酶/异构酶家族 Enoyl-CoA hydratase/isomerase family | 有Yes | 否No |
| BnaC02g10490.1D2 | CHR17 | AT5G18620 | 5.97 | 染色质重塑因子17 Chromatin remodeling factor 17 | 无No | 是Yes |
新窗口打开|下载CSV
3 讨论
3.1 油菜少角果突变体No.7931的表型特征
在成熟期, 少角果突变体No.7931的主序和大部分一次分枝花序都很短(个别甚至退化了), 其上的角果只有10个左右, 明显少于油菜种质资源中花序发育正常的少角果材料[8]。因为角果数决定于花器官数目和结角率的乘积, 为明确No.7931角果少的原因, 进一步对其花器官数目和结角率进行考察。结果表明, No.7931的结角率基本正常, 而花器官数目远少于油菜种质资源中的少花材料(≈70)[8],因此其角果少的原因是花器官少。花器官数目决定于花芽分化过程, 对No.7931花芽分化全程(分为4个时期)进行连续显微观察表明, 花芽分化早期, No.7931的顶端分生组织和花原基暂没有发现明显异常, 但在中间阶段分化出的花芽出现粘连, 开花期花序顶部形成一个由几朵发育不完整的花期簇拥而成的“顶生花”, 进而终止分化转变成有限花序, 随后一次花序变短或退化而二次花序增多。另外, No.7931的开花期和成熟期提前, 株高降低, 这些特征都与油菜中发现的其他几个有限花序突变体(如di1、BjSdt1、Bnsdt1)以及拟南芥TFL1基因突变体类似[37,38,39,40]。然而No.7931突变体定位在C2染色体, 而di1定位在A8~A10和C8~C9五条染色体, BjSdt1定位在B5染色体, Bnsdt1定位在A10染色体, 因此No.7931应该是一个新的油菜有限花序突变体。3.2 油菜少角果突变体No.7931的定位区间和角果数QTL的位置关系
本研究利用少角果突变体No.7931和角果数极多品系No.73290的F2分离群体进行BSA-seq, 将目标基因定位在C02染色体的3个区间: 0~1.1 Mb、4.7~6.2 Mb、11.5~12.4 Mb (对应DarmorV8)。有意思的是, Radoev等[14]利用‘Express617’和‘R53’构建的DH群体在C02染色体5.9 Mb之前的位置也定位到了1个单位面积角果数QTL-Sil/dm2_N12b; Shi等[17]利用多角果品系No.73290和测序品种中双11构建的F2:3家系群体在C02染色体的1.3~3.7 Mb定位到了1个主序角果数QTL-qPN.C02-1。因此, 少角果突变体No.7931的定位区间和油菜C2染色体上这2个角果数QTL的物理位置重叠, 可能对应相同的目的基因。由于3个候选区间间距较小, 也可能为一个大的候选区间, 后续研究中可以针对这3个区段分别构建近等基因系, 对上述可能性进行验证。3.3 油菜少角果突变体No.7931的候选基因
本研究基于油菜少角果突变体No.7931花芽分化全程的显微观察和BSA定位的结果, 结合基因功能注释以及序列和表达差异分析, 共筛选到9个与花发育或者花器官数目相关的候选基因, 它们应该是后续功能验证的重点。转录因子和转录共调节因子(如WUS、STM)在茎尖分生组织的发育过程中具有重要作用, 本研究筛选到的BnaC02g00490.1D2、BnaC02g01030.1D2、BnaC02g00270.1D2为转录因子基因, 而BnaC02g02670.1D2为转录共调节因子基因。在本研究中, BnaC02g00490.1D2、BnaC02g02670.1D2存在表达量差异, 而BnaC02g00270.1D2存在序列差异。其中, BnaC02g00490.1D2在拟南芥中的同源基因是FLC, 该基因是春化途径中抑制开花的关键因子(功能突变会使开花提前), 参与调控花分生组织的确定性并负调控花的发育(
BnaC02g01120.1D2在拟南芥中的同源基因是AGAL2, 是糖苷水解酶(GH27)家族成员, 参与碳代谢过程, 正向调控花发育(
BnaC02g08680.1D2在拟南芥中的同源基因为COL1 (与开花基因CONSTANS同源), 通过光周期途径正调控开花时间[45], 同时也在花序分生组织和花原基中表达, 参与调控花的发育(
4 结论
本研究对油菜少角果突变体No.7931进行表型观察发现其少角果的原因是花器官数目少。进一步显微观察明确其花器官数目少的原因是花芽分化中期出现异常而较早停止生长, 导致一次花序变短、退化而二次花序增多, 且开花提前。利用少角果突变体No.7931和多角果品系No.73290构建的F2群体进行BSA-seq, 定位到了位于C02染色体的3个候选区间: 0~1.1 Mb、4.7~6.2 Mb、11.5~12.4 Mb。在花芽分化初期, 选取两亲本SAM进行RNA-seq, 共鉴定到8958个DEGs, 功能分类和KEGG分析结果暗示它们和花芽分化密切相关。结合基因功能注释以及序列和表达差异分析在3个关联区间内筛选到9个与花器官数目或者花发育相关的候选基因。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 6]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}