 ,**, 罗怀勇,**, 李威涛, 郭建斌, 陈伟刚, 周小静, 黄莉, 刘念, 晏立英, 雷永, 廖伯寿, 姜慧芳,*中国农业科学院油料作物研究所 / 农业农村部油料作物生物学与遗传育种重点实验室, 湖北武汉 430062
,**, 罗怀勇,**, 李威涛, 郭建斌, 陈伟刚, 周小静, 黄莉, 刘念, 晏立英, 雷永, 廖伯寿, 姜慧芳,*中国农业科学院油料作物研究所 / 农业农村部油料作物生物学与遗传育种重点实验室, 湖北武汉 430062Genome-wide identification of peanut resistance genes and their response to Ralstonia solanacearum infection
ZHANG Huan,**, LUO Huai-Yong,**, LI Wei-Tao, GUO Jian-Bin, CHEN Wei-Gang, ZHOU Xiao-Jing, HUANG Li, LIU Nian, YAN Li-Ying, LEI Yong, LIAO Bo-Shou, JIANG Hui-Fang,*Oil Crops Research Institute, Chinese Academy of Agricultural Sciences / Key Laboratory of Biology and Genetic Improvement of Oil Crops, Ministry of Agriculture and Rural Affairs, Wuhan 430062, Hubei, China通讯作者: * 姜慧芳, E-mail:peanutlab@oilcrops.cn
第一联系人:
收稿日期:2020-12-8接受日期:2021-04-14网络出版日期:2021-06-15
| 基金资助: |
Corresponding authors: * E-mail:peanutlab@oilcrops.cn
First author contact:
Received:2020-12-8Accepted:2021-04-14Published online:2021-06-15
| Fund supported: |
作者简介 About authors
张欢,E-mail:18198335427@163.com;
罗怀勇,E-mail:huaiyongluo@caas.cn

摘要
花生是主要的油料作物之一, 在生产过程中受到多种病原微生物的危害。培育和选用抗病品种是防治病害最经济有效的途径之一, 而抗病基因是植物抵御病原微生物的重要基因。本文首次对花生抗病基因进行全基因组鉴定, 发掘抗病候选基因4156个, 其中RLK、RLP、NL、CNL、TNL这5种典型抗病基因分别有536、490、232、182和149个。抗病基因在染色体上分布不均匀, 多数抗病基因集中在B02染色体上。转录组测序发现, 抗病材料中特异表达的基因有111个, 感病材料中特异表达的基因有104个, 抗、感病材料均有表达的基因2216个、均不表达的有1725个。筛选出第1类响应青枯菌诱导的抗病基因5个, 第2类持续上调表达抗青枯病基因65个。qRT-PCR成功验证了1个抗病候选基因Arahy.5D95TJ。本文对花生抗病基因的鉴定分析, 为后续研究抗病基因功能与花生抗病的分子育种提供重要参考。
关键词:
Abstract
Peanut is one of the main oil crops, which is harmed by many pathogenic microorganisms during growth and development period. Breeding and selection of disease-resistant varieties is one of the most economical and effective ways to control disease, and disease resistance genes are important genes for plant resistance to pathogenic microorganisms. Here, the whole genome-wide identification of peanut disease resistance genes was carried out for the first time. A total of 4156 candidate disease resistance genes were identified. Among them, 536, 490, 232, 182, and 149 genes were RLK, RLP, NL, CNL, and TNL, respectively. The distribution of disease resistance genes was uneven on chromosomes, and most of them were concentrated on chromosome B02. Transcriptome profiling revealed that 111 genes were specifically expressed in resistant materials, 104 genes were specifically expressed in susceptible materials, 2216 genes were expressed in both resistant and susceptible materials, while 1725 genes were not expressed in both resistant and susceptible materials. Two kinds of differentiate expressed R genes were identified, including five genes in the first group responded to the infection of Ralstonia solanacearum at specific time and 65 genes in the second group which exhibited higher expressions in resistant cultivar than susceptible cultivar. A candidate gene Arahy.5D95TJ was successfully validated by qRT-PCR. In this study, the identification and analysis of peanut disease resistance genes provides the important reference for further research of their functions and molecular breeding of peanut disease resistance.
Keywords:
PDF (589KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
张欢, 罗怀勇, 李威涛, 郭建斌, 陈伟刚, 周小静, 黄莉, 刘念, 晏立英, 雷永, 廖伯寿, 姜慧芳. 花生全基因组抗病基因鉴定及其对青枯菌侵染的响应分析. 作物学报, 2021, 47(12): 2314-2323 DOI:10.3724/SP.J.1006.2021.04266
ZHANG Huan, LUO Huai-Yong, LI Wei-Tao, GUO Jian-Bin, CHEN Wei-Gang, ZHOU Xiao-Jing, HUANG Li, LIU Nian, YAN Li-Ying, LEI Yong, LIAO Bo-Shou, JIANG Hui-Fang.
植物抵抗外界微生物刺激而形成的系统称为植物固有免疫系统, 可分为2个层次。第1个层次是植物模式识别受体(pattern-recognition receptors, PRRS)识别病原相关的分子模式(pathogen-associated molecular patterns, PAMPs)触发免疫反应(PAMP-triggered immunity, PTI); 第2个层次是病原菌产生效应物抑制对PTI的基本免疫反应, 而植物抗病基因(resistance gene, R gene)通过编码靶向抗性蛋白直接或间接识别病原菌, 进而诱导效应器触发免疫(effector-triggered immunity, ETI)重建植物的抗性[1,2]。
目前植物中已经克隆了150多个抗病基因[3], 编码的蛋白质具有共同的结构域, 如卷曲螺旋(coiled coil, CC)、核苷酸结合区(nucleotide binding site, NBS)、Toll-白细胞介素区(toll-interleukin-1 receptor, TIR)、富含亮氨酸重复区(leucine-rich repeat, LRR)、激酶结构域(kinase, KIN) 和跨膜结构域(trans- membrane domain, TM)。胞质NBS-LRR基因分为2类: TNL (TIR-NBS-LRR)和CNL (CC-NBS-LRR), 分别拥有TIR或CC结构域[4]。如过表达TNL型抗病基因GmKR3增强大豆对多种病毒的抗性[5], 棉花GhTNL1抗病基因的表达减缓植物叶片黄化、萎蔫现象[6], 番茄CNL型抗细菌性斑点病基因SlNBRP1过表达增加其植株抗病性[7], 水稻中NL型Pi9抗病基因组成型表达抗稻瘟病[8]。包含Kinase和LRR结构域的跨膜受体蛋白, 如受体样蛋白(RLP)和受体样激酶(RLK)也参与其中。RLK型基因SlDALR1正调控番茄对Pst DC3000的抗性[9], RLP型基因TaRLP1.1参与小麦对小麦锈病的防御反应[10]。
花生是世界上重要的油料和经济作物, 广泛种植于热带和亚热带地区。中国是世界最大的花生生产国, 也是最大的花生消费国。据国家统计局统计, 2019年中国花生产量1751.96万吨, 占世界产量近40%。青枯病是我国花生的主要病害之一, 是由茄科雷尔式菌侵染引起的一种细菌性土传病害, 由于防治困难, 严重影响我国花生的品质和产量[11]。培育和种植抗青枯病花生品种是防治青枯病危害最为经济有效的途径。目前, 已获得的抗青枯病基因有: 拟南芥抗青枯病基因RRS1和ERECTA[12,13], 辣椒抗青枯病基因CaLRR-RLK1和CaLRR51[14,15], 花生中参与青枯病响应的基因AhRLK1和AhRRS5[16,17], 对于花生抗病基因的获得还需要开展更多的研究。
四倍体花生全基因组测序已在2019年完成[18,19], 为在全基因组水平研究花生抗病基因提供便利。目前尚没有关于花生全基因组抗病基因鉴定的相关研究, 本研究利用生物信息学分析鉴定了花生全基因组抗病基因数量及染色体分布, 结合抗、感病品种接种青枯病菌后转录组分析, 鉴定了与青枯病抗性有关的差异表达抗病基因, 旨在为克隆花生抗病基因以及抗病分子育种奠定基础。
1 材料与方法
1.1 花生材料和青枯病菌株
花生抗病品种远杂9102和感病品种徐州68-4由中国农业科学院油料作物研究所花生遗传育种团队提供。青枯病菌为茄科雷尔式菌(Ralstonia solanacearum)生理小种1, 生化型III, 分离自红安病害苗圃试验基地[20]。1.2 R基因的筛选和分类
根据植物抗病基因数据库(PRGdb;1.3 染色体分布
从花生数据库(1.4 花生材料接种青枯病菌和取样
参照Chen等[21]使用的方法进行青枯菌培养、花生幼苗培养及接种。接种后12、24、48、72、96 h分别对花生主根部位取样, 处理组标记为RT12/ ST12、RT24/ST24、RT48/ST48、RT72/ST72和RT96/ST96, 对照组标记为RC12/SC12、RC24/SC24、RC48/SC48、RC72/SC72和RC96/SC96。试验设3次重复, 样品立即冷冻于液氮中, 并在-80℃下保存。由武汉菲沙基因信息有限公司完成转录组测序。利用RSEM[22], 调用Bowtie2软件的比对结果进行统计, 得到每个样品比对到每个转录本上的Reads数目, 并对其进行FPKM[23] (Fragments Per Kilobase per Million bases)转换。根据抗感材料对照组和处理组(RC/RT/SC/ST)各时间点的3个生物学重复FPKM均值大于1, 将抗病基因的表达情况分为
抗感病材料中均表达、仅在抗病材料中表达、仅在感病材料中表达、抗感病材料中均不表达4类。使用DESeq2[24]对5个时间点RT-vs-RC、RT-vs-ST和RT-vs-SC三个组分别进行差异表达分析, 筛选阈值为FDR (false discovery rate) < 0.05, log2 FC (fold change (condition 2/condition 1) for a gene) >1或log2 FC < -1。使用TBtools对RT-vs-RC、RT-vs-ST和RT-vs-SC 3个组差异表达上调的R基因各个时间点作韦恩分析, 筛选出的差异表达基因使用Microsoft Excel作折线图和RStudio[25]制作热图。
1.5 抗病基因qRT-PCR分析
以RNA-seq测序的根组织RNA为模板, 采用诺唯赞HiScript II Q RT SuperMix for qPCR试剂盒, 反转录合成cDNA。用Primer 5.0软件设计引物, 采用诺唯赞ChamQ Universal SYBR qPCR Master Mix试剂盒进行qRT-PCR试验, 反应体系(20 μL)包含2×ChamQ Universal SYBR qPCR Master Mix 10 μL、上下游引物(10 μmol L-1) 各0.4 μL、cDNA 2 μL、ddH2O 7.2 μL。反应程序为95℃预变性30 s, 95℃变性10 s, 60℃退火30 s, 72°C延伸20 s, 40个循环。每个反应进行3次重复, 花生内参基因Actin的引物为actin-F: 5°-TAAGAACAATGTTGCCATACAGA-3°, actin-R: 5°-GTTGCCTTGGATTATGAGC-3°。按照2-ΔΔCT计算基因相对表达量。2 结果与分析
2.1 花生R基因的鉴定和染色体分布
在花生Tifrunner参考基因组[18]中共筛选得到4156个抗病候选基因, 分为TNL、CNL、RLK、RLP等18个类别(表1)。在典型的5类抗病基因中, 基因数目较多的是RLK型(536个)和RLP型(490个), 占总抗病候选基因数目的12.90%和11.79%, 其次是NL型(232个)和CNL类抗病基因(182个), 占抗病候选基因总数的5.58%和4.38%, TNL型抗病基因(149个)占抗病候选基因总数的3.59%。非典型的13个抗病类型中, 仅含Kinase保守结构域的KIN型抗病候选基因数目为1714个, 占总抗病候选基因数目的41.24%; 其余12个类别候选基因含有NBS/CC/TIR/TM/LRR/Kinase等保守结构域的不同组合形式, 占比0.05%~5.32%, 共853个。抗病候选基因分布于花生20条染色体上(图1), 在染色体B02上的抗病基因数量最多, 为334个, A01和A06上抗病基因数量最少, 为137个。Table 1
表1
表1花生R基因的数量和分类
Table 1
| 类别 Type | 包含结构域 Domain contained | 基因数 Gene number | 占R基因比例 Proportion in R gene (%) |
|---|---|---|---|
| RLK | TM, LRR, Kinase | 536 | 12.90 |
| RLP | TM, LRR | 490 | 11.79 |
| CNL | CC, TM, NBS, LRR | 182 | 4.38 |
| TNL | TM, TIR, NBS, LRR | 149 | 3.59 |
| KIN | TM, Kinase | 1714 | 41.24 |
| NL | TM, NBS, LRR | 232 | 5.58 |
| CK | CC, TM, Kinase | 221 | 5.32 |
| N | TM, NBS | 146 | 3.51 |
| CN | CC, TM, NBS | 136 | 3.27 |
| CTNL | CC, TM, TIR, NBS, LRR | 77 | 1.85 |
| L | LRR | 76 | 1.83 |
| T | TM, TIR | 65 | 1.57 |
| TN | TM, TIR, NBS | 61 | 1.47 |
| CL | CC, TM, LRR | 28 | 0.67 |
| CLK | CC, TM, LRR, Kinase | 20 | 0.48 |
| CNT | CC, TM, NBS, TIR | 15 | 0.36 |
| CT | CC, TM, TIR | 6 | 0.14 |
| TRAN | TM | 2 | 0.05 |
| 总计Total | 4156 |
新窗口打开|下载CSV
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1花生R基因在染色体上的分布
绿色代表在抗病和感病材料中都表达; 红色是只在抗病材料中表达的抗病基因; 紫色是只在感病材料中表达的抗病基因; 黑色代表抗病基因在抗感材料中都不表达。
Fig. 1Distribution of R genes on peanut chromosomes
Expressed in both resistant and susceptible materials are marked by green; only expressed in resistant materials are marked by red; only expressed in susceptible materials are marked by purple; not expressed in resistant and susceptible materials are marked by black.
2.2 抗病候选基因对青枯病侵染的响应
在接种青枯菌后5个时间点中, 抗病材料远杂9102对照组(RC)共2383个抗病候选基因表达, 处理组(RT)有2337个表达; 在感病材料徐州68-4对照组(SC)共2304个候选基因表达, 处理组(ST)有2325个表达。抗感材料中表达的抗病候选基因数目基本一致。抗病材料(RC+RT)中特异表达的基因有111个, 感病材料(SC+ST)中特异表达的有104个, 抗病、感病材料均有表达的基因2216个, 抗、感病材料中均不表达的有1725个。根据FPKM值将所有抗病基因分为5组(图2), 从图2可看出, FPKM值在1~5的抗病候选基因数量最多, 占总表达基因数目的42.6%~46.8%; FPKM值在50以上最少, 基因数量在1.8%~2.3%, 植物体内大部分的抗病基因转录水平较低。图2
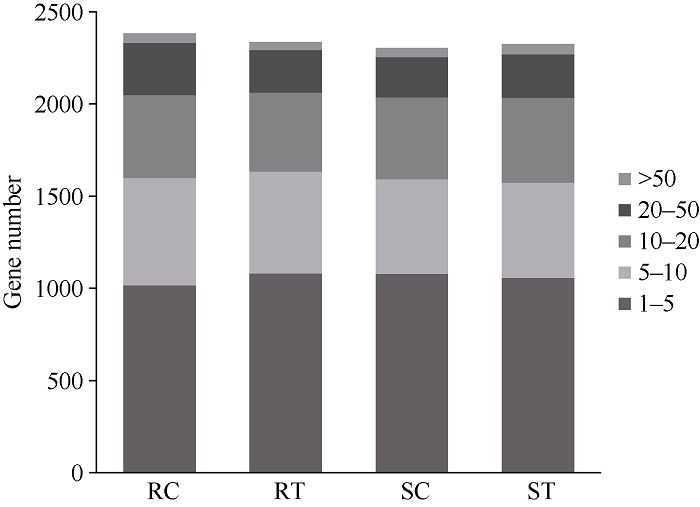 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2花生R基因FPKM值分布范围
RC: 抗病材料对照; RT: 抗病材料处理; SC: 感病材料对照; ST: 感病材料处理。
Fig. 2Distribution range of FPKM values of R genes in peanut
RC: control of resistant material; RT: treatment of resistant material; SC: control of susceptible material; ST: treatment of susceptible material.
2.3 差异表达R基因的鉴定
在接种青枯菌12 h的样品中, 通过对RT12-vs- RC12、RT12-vs-ST12和RT12-vs-SC12 3个组差异表达基因的分析, 鉴定出2类差异表达R基因: 第1类是3个在抗病材料接种青枯菌后诱导表达的基因, 在RT12的表达量显著高于RC12、ST12和SC12, 推测其受到青枯菌侵染的诱导表达(图3-a~c); 第2类是33个在抗病材料中持续上调表达的差异基因, 在RT12和RC12中的表达量差异不显著, 但是显著高于ST12和SC12 (图3-f)。通过类似的方法, 在接种青枯菌24、48、72和96 h后, 鉴定到第1类差异表达R基因分别有1、0、1和0个(图3-d, e); 第2类差异表达R基因有42、41、39和28个, 在抗病材料中差异不显著, 但显著高于感病材料(图3-g~j)。图3
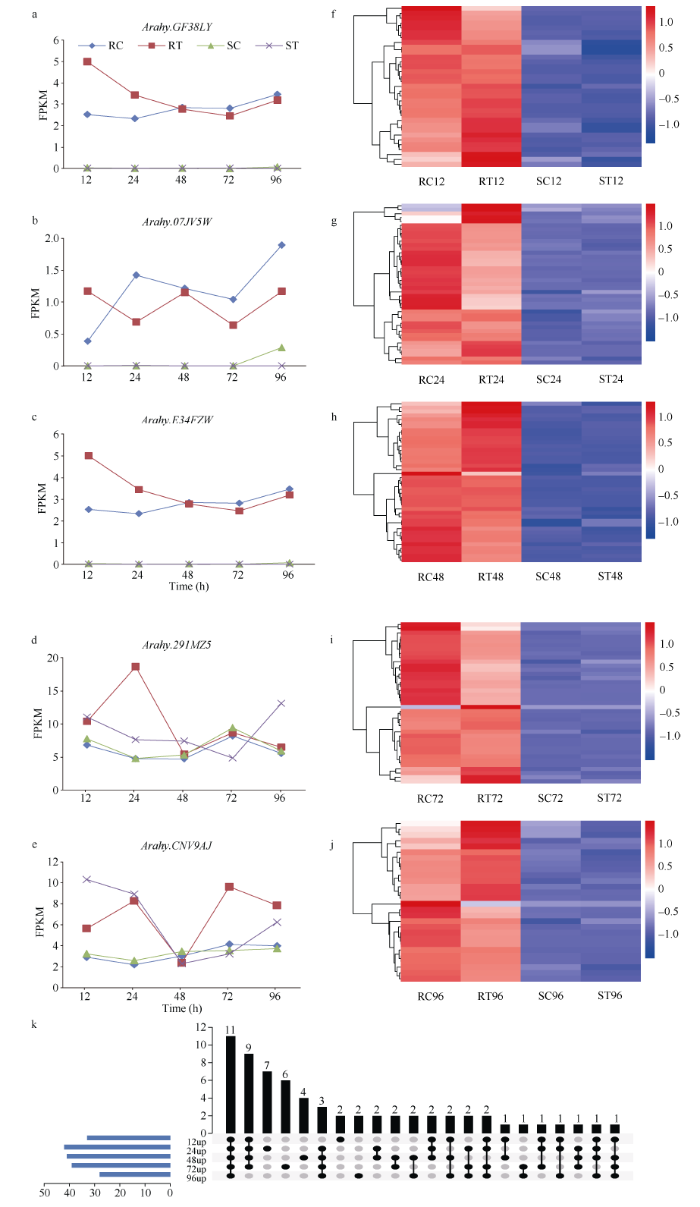 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3花生接种青枯菌后差异表达的2类R基因
RC: 抗病材料对照; RT: 抗病材料处理; SC: 感病材料对照; ST: 感病材料处理。RT12、RT24、RT48、RT72和RT96分别表示抗病材料接种青枯菌后12、24、48、72和96 h的样本; RC12、RC24、RC48、RC72和RC96分别表示抗病材料对照组(清水处理) 12、24、48、72和96 h的样本; ST12、ST24、ST48、ST72和ST96分别表示感病材料接种青枯菌后12、24、48、72和96 h的样本; SC12、SC24、SC48、SC72和SC96分别表示感病材料对照组(清水处理) 12、24、48、72和96 h的样本。
Fig. 3Two classes of differentially expressed R genes infected by Ralstonia solanacearum in peanut
RC: control of resistant material; RT: treatment of resistant material; SC: control of susceptible material; ST: treatment of susceptible material. RT12, RT 24, RT 48, RT 72, and RT 96 represented the resistant materials collected at 12, 24, 48, 72, and 96 hours post inoculated with Ralstonia solanacearum (treatment group); RC12, RC24, RC48, RC72, and RC 96 represented the resistant materials collected at 12, 24, 48, 72, and 96 hours post inoculated with water (control group); ST12, ST24, ST48, ST72, and ST 96 represented the susceptible materials collected at 12, 24, 48, 72, and 96 hours post inoculated with Ralstonia solanacearum (treatment group); SC12, SC24, SC48, SC72, and SC96 represented the susceptible materials collected at 12, 24, 48, 72, and 96 hours post inoculated with water (control group). FPKM: fragments per kilobase per million bases.
综合所有时间点, 第1类差异表达R基因只在对应的时间点特异上调表达。但是在第2类差异上调的65个抗病候选基因中, 有27个R基因在4个时间点及以上差异表达, 在5个时间点连续差异上调表达的R基因11个, 有9个R基因在12~72 h连续差异上调表达, 3个R基因在24~96 h连续差异上调表达, 在12、24、72、96 h差异上调表达的R基因2个, 在12、24、48、96 h差异上调表达1个, 12、48、72、96 h差异上调表达1个(图3-k)。
2.4 抗青枯病QTL区间候选基因表达差异分析及qRT-PCR验证
前期在远杂9102和徐州68-4构建的RIL群体中, 通过QTL分析鉴定到1个主效抗青枯病QTL qBWRB02.1 [26]。通过对其候选区间分子标记的比对发现, 该QTL位于Tifrunner B02染色体4.05~6.12 Mb区间内, 该区间包含13个抗病候选基因, 其中6个候选基因在接种青枯菌后表达, 7个不表达。在表达的6个基因中, 有3个包含在上述鉴定到的第2 类差异表达基因中, 其表达情况如图4-a所示。针对Arahy.5D95TJ设计了特异荧光定量引物, F: 5°-TGCAAGGTACAATAAGGAGACAGG-3°, R: 5°- AATACTAGCCTCCAATAAGCATCC-3°。qRT-PCR结果显示该基因的表达趋势(图4-b)与转录组测序结果(图4-c)基本一致。图4
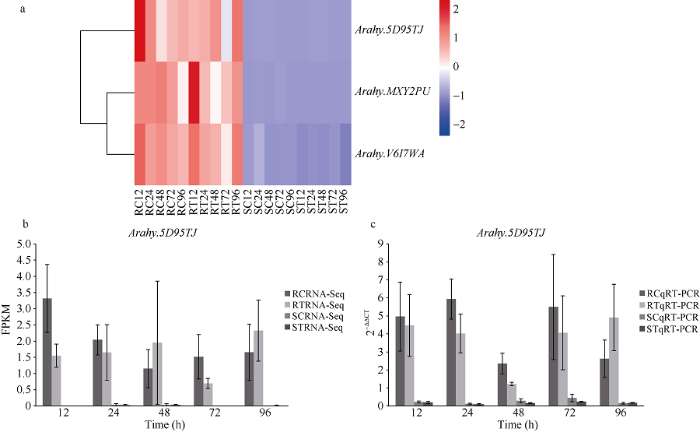 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4主效抗青枯病QTL区间差异表达R基因及验证
缩写同
Fig. 4Differentially expressed R genes in the confidential interval of the major QTL for resistance to bactrierl wilt
Abbreviations are the same as those given in
3 讨论
抗病基因在植物抵抗细菌、真菌、病毒等病原体过程具有非常重要的作用[27]。本研究根据保守结构域在花生参考基因组中鉴定出4156个抗病候选基因, 将其分为18类抗病分型。豆科植物木豆、菜豆、大豆中分别鉴定到289、337和465个NBS-LRR类抗病基因[28], 各占全基因组基因数目的0.5%、1.1%和1.9% [29,30,31]。本研究发现, 典型抗病基因NBS-LRR类641个, 占花生全基因组数目的0.8%, 推测抗病基因数目与基因组大小没有直接的联系。单子叶植物谷子、玉米、水稻各有NBS-LRR类家族基因数目为411、21和545个, 但是没有发现TNL型抗病基因[32,33,34]。在双子叶植物三裂叶薯、黑籽南瓜、番茄、葡萄、鹰嘴豆中CNL型抗病基因数目大于TNL型[35,36,37,38,39], 而向日葵、大豆、拟南芥等植物中TNL型抗病基因数目大于CNL型[40,41,42], 本研究鉴定出花生抗病基因CNL型182个, TNL型149个(CNL/TNL约1.2倍), 这说明双子叶植物中CNL型和TNL型抗病基因可能有不同的进化模式。
在植物中, RLK和RLP富含植物激素信号和植物病原菌互作途径[43], RLK类抗病基因作为胁迫表达相关基因的一部分参与植物体应答逆境和与防御相关的过程[44], 在4种棉属植物中鉴定出1641个RLK基因[45], 在马铃薯、萝卜、苜蓿和二穗短柄草中分别鉴定出479个[46]、292个[47]、329个[48]和268个[49]PLK基因, 本研究在花生全基因组中筛选鉴定得到RLK型抗病基因536个。总共有82个RLP基因在杨树基因组中被鉴别出来[50], 在普通烟草中共鉴定出70个RLP基因[51], 本研究在花生全基因组中筛选鉴定得到RLP型490个, 这些基因可能参与应答青枯菌诱导的防御反应。
抗病基因在染色体上的分布不均匀。如NBS-LRR类抗病基因在甘薯13号染色体有54个而11号染色体仅有3个[52]; 同样的, 番茄中NBS类抗病基因在4号染色体所含数目最多[37]。花生为杂交起源的异源四倍体作物, 包含来自2个祖先物种的全部染色体组, 抗病基因在B组染色体上数目大于A组染色体, 并且花生染色体B02上抗病基因数目最多。
已有研究表明, 花生中AhRRS5[16]和AhRLK1[17]基因在烟草中过表达显著增加了对青枯菌的抗性, 分别下载CDS序列并比对到花生Tifrunner参考基因组[18], 结果显示最高同源序列为染色体B05上Arahy.WIN0ZV基因和A03上Arahy.8BA7G9基因, 将其鉴定为CNL型和RLK型抗病基因。本研究筛选到2类差异表达的抗病候选基因, 第1类是5个在抗病材料接种青枯菌后差异表达高于抗病对照以及感病材料, 推测这些基因是受到青枯菌诱导后上调表达, 初步判定与花生的抗青枯病特性相关; 第2类, 有65个在抗病材料中持续上调表达的差异基因, 这一类基因在接种青枯菌前后, 抗病材料远杂9102中的表达量始终高于感病材料徐州68-4, 其中有27个抗病候选基因在4个时间点以上连续差异上调表达。这2类差异表达基因含11种抗病类别, 基因数目从多到少为NL (14)、N (9)、KIN (8)、RLP (7)、CN (7)、CTNL (6)、RLK (4)、CNL (4)、CK (4)、TNL (2)和TN (2)。本研究还验证了花生青枯病QTL区间的抗病候选基因Arahy.5D95TJ属于第2类持续上调表达抗病基因N型, 在抗病材料远杂9102中4个时间点连续表达。通过Interproscan分析发现, 该基因可能包含与已克隆抗病基因不同的CC和LRR结构域, 推测Arahy.5D95TJ可能与AhRRS5具有相同的靶向位点, 通过相同或相似的信号通路来参与对青枯菌的防御反应。本研究结果可为开展花生青枯病抗病基因的克隆和功能验证, 以及解析花生响应青枯菌胁迫的抗性应答机制提供参考。
4 结论
发掘候选抗病基因4156个, 在染色体上呈不均匀分布; 得到差异表达的5个响应青枯菌诱导型和65个持续上调表达抗病候选基因, 验证了一个持续上调表达抗病候选基因Arahy.5D95TJ。研究结果可为花生抗病基因的克隆、鉴定及其抗病育种奠定基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIPMID [本文引用: 1]

The leucine-rich repeat (LRR) proteins play important roles in the recognition of corresponding ligands and signal transduction networks in plant defence responses. Herein, a novel LRR protein from Capsicum annuum, CaLRR51, was identified and characterized. It was localized to the plasma membrane and transcriptionally up-regulated by Ralstonia solanacearum infection (RSI), as well as the exogenous application of salicylic acid (SA), jasmonic acid (JA) and ethephon (ETH). Virus-induced gene silencing of CaLRR51 significantly increased the susceptibility of pepper to RSI. By contrast, transient overexpression of CaLRR51 in pepper plants activated hypersensitive response (HR)-like cell death, and up-regulated the defence-related marker genes, including PO2, HIR1, PR1, DEF1 and ACO1. Moreover, ectopic overexpression of CaLRR51 in transgenic tobacco plants significantly enhanced the resistance to RSI. Transcriptional expression of the corresponding defence-related marker genes in transgenic tobacco plants was also found to be enhanced by the overexpression of CaLRR51, which was potentiated by RSI. These loss- and gain-of-function assays suggest that CaLRR51 acts as a positive regulator in the response of pepper to RSI. In addition, the putative signal peptide and transmembrane region were found to be required for plasma membrane targeting of CaLRR51, which is indispensable for the role of CaLRR51 in plant immunity.© 2016 BSPP AND JOHN WILEY & SONS LTD.
DOIURL [本文引用: 2]
DOIURL [本文引用: 2]
DOIPMID [本文引用: 3]
Like many other crops, the cultivated peanut (Arachis hypogaea L.) is of hybrid origin and has a polyploid genome that contains essentially complete sets of chromosomes from two ancestral species. Here we report the genome sequence of peanut and show that after its polyploid origin, the genome has evolved through mobile-element activity, deletions and by the flow of genetic information between corresponding ancestral chromosomes (that is, homeologous recombination). Uniformity of patterns of homeologous recombination at the ends of chromosomes favors a single origin for cultivated peanut and its wild counterpart A. monticola. However, through much of the genome, homeologous recombination has created diversity. Using new polyploid hybrids made from the ancestral species, we show how this can generate phenotypic changes such as spontaneous changes in the color of the flowers. We suggest that diversity generated by these genetic mechanisms helped to favor the domestication of the polyploid A. hypogaea over other diploid Arachis species cultivated by humans.
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIPMID [本文引用: 1]
High-throughput mRNA sequencing (RNA-Seq) promises simultaneous transcript discovery and abundance estimation. However, this would require algorithms that are not restricted by prior gene annotations and that account for alternative transcription and splicing. Here we introduce such algorithms in an open-source software program called Cufflinks. To test Cufflinks, we sequenced and analyzed >430 million paired 75-bp RNA-Seq reads from a mouse myoblast cell line over a differentiation time series. We detected 13,692 known transcripts and 3,724 previously unannotated ones, 62% of which are supported by independent expression data or by homologous genes in other species. Over the time series, 330 genes showed complete switches in the dominant transcription start site (TSS) or splice isoform, and we observed more subtle shifts in 1,304 other genes. These results suggest that Cufflinks can illuminate the substantial regulatory flexibility and complexity in even this well-studied model of muscle development and that it can improve transcriptome-based genome annotation.
DOIURL [本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOI [本文引用: 1]
Pigeonpea is an important legume food crop grown primarily by smallholder farmers in many semi-arid tropical regions of the world. We used the Illumina next-generation sequencing platform to generate 237.2 Gb of sequence, which along with Sanger-based bacterial artificial chromosome end sequences and a genetic map, we assembled into scaffolds representing 72.7% (605.78 Mb) of the 833.07 Mb pigeonpea genome. Genome analysis predicted 48,680 genes for pigeonpea and also showed the potential role that certain gene families, for example, drought tolerance-related genes, have played throughout the domestication of pigeonpea and the evolution of its ancestors. Although we found a few segmental duplication events, we did not observe the recent genome-wide duplication events observed in soybean. This reference genome sequence will facilitate the identification of the genetic basis of agronomically important traits, and accelerate the development of improved pigeonpea varieties that could improve food security in many developing countries.
DOIPMID [本文引用: 1]
Common bean (Phaseolus vulgaris L.) is the most important grain legume for human consumption and has a role in sustainable agriculture owing to its ability to fix atmospheric nitrogen. We assembled 473 Mb of the 587-Mb genome and genetically anchored 98% of this sequence in 11 chromosome-scale pseudomolecules. We compared the genome for the common bean against the soybean genome to find changes in soybean resulting from polyploidy. Using resequencing of 60 wild individuals and 100 landraces from the genetically differentiated Mesoamerican and Andean gene pools, we confirmed 2 independent domestications from genetic pools that diverged before human colonization. Less than 10% of the 74 Mb of sequence putatively involved in domestication was shared by the two domestication events. We identified a set of genes linked with increased leaf and seed size and combined these results with quantitative trait locus data from Mesoamerican cultivars. Genes affected by domestication may be useful for genomics-enabled crop improvement.
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIPMID [本文引用: 1]
Nucleotide-binding site (NBS) disease resistance genes play an integral role in defending plants from a range of pathogens and insect pests. Consequently, a number of recent studies have focused on NBS-encoding genes in molecular disease resistance breeding programmes for several important plant species. Little information, however, has been reported with an emphasis on systematic analysis and a comparison of NBS-encoding genes in maize. In the present study, 109 NBS-encoding genes were identified based on the complete genome sequence of maize (Zea mays cv. B73), classified as four different subgroups, and then characterized according to chromosomal locations, gene duplications, structural diversity and conserved protein motifs. Subsequent phylogenetic comparisons indicated that several maize NBS-encoding genes possessed high similarity to function-known NBS-encoding genes, and revealed the evolutionary relationships of NBS-encoding genes in maize comparede to those in other model plants. Analyses of the physical locations and duplications of NBS-encoding genes showed that gene duplication events of disease resistance genes were lower in maize than in other model plants, which may have led to an increase in the functional diversity of the maize NBS-encoding genes. Various expression patterns of maize NBS-encoding genes in different tissues were observed using an expressed-sequence tags database and, alternatively, after southern leaf blight infection or the application of exogenous salicylic acid. The results reported in the present study contribute to an improved understanding of the NBS-encoding gene family in maize.© 2012 The Authors Journal compilation © 2012 FEBS.
PMID [本文引用: 1]
The availability of the rice genome sequence enabled the global characterization of nucleotide-binding site (NBS)-leucine-rich repeat (LRR) genes, the largest class of plant disease resistance genes. The rice genome carries approximately 500 NBS-LRR genes that are very similar to the non-Toll/interleukin-1 receptor homology region (TIR) class (class 2) genes of Arabidopsis but none that are homologous to the TIR class genes. Over 100 of these genes were predicted to be pseudogenes in the rice cultivar Nipponbare, but some of these are functional in other rice lines. Over 80 other NBS-encoding genes were identified that belonged to four different classes, only two of which are present in dicotyledonous plant sequences present in databases. Map positions of the identified genes show that these genes occur in clusters, many of which included members from distantly related groups. Members of phylogenetic subgroups of the class 2 NBS-LRR genes mapped to as many as ten different chromosomes. The patterns of duplication of the NBS-LRR genes indicate that they were duplicated by many independent genetic events that have occurred continuously through the expansion of the NBS-LRR superfamily and the evolution of the modern rice genome. Genetic events, such as inversions, that inhibit the ability of recently duplicated genes to recombine promote the divergence of their sequences by inhibiting concerted evolution.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
PMID [本文引用: 1]
The Arabidopsis genome contains approximately 200 genes that encode proteins with similarity to the nucleotide binding site and other domains characteristic of plant resistance proteins. Through a reiterative process of sequence analysis and reannotation, we identified 149 NBS-LRR-encoding genes in the Arabidopsis (ecotype Columbia) genomic sequence. Fifty-six of these genes were corrected from earlier annotations. At least 12 are predicted to be pseudogenes. As described previously, two distinct groups of sequences were identified: those that encoded an N-terminal domain with Toll/Interleukin-1 Receptor homology (TIR-NBS-LRR, or TNL), and those that encoded an N-terminal coiled-coil motif (CC-NBS-LRR, or CNL). The encoded proteins are distinct from the 58 predicted adapter proteins in the previously described TIR-X, TIR-NBS, and CC-NBS groups. Classification based on protein domains, intron positions, sequence conservation, and genome distribution defined four subgroups of CNL proteins, eight subgroups of TNL proteins, and a pair of divergent NL proteins that lack a defined N-terminal motif. CNL proteins generally were encoded in single exons, although two subclasses were identified that contained introns in unique positions. TNL proteins were encoded in modular exons, with conserved intron positions separating distinct protein domains. Conserved motifs were identified in the LRRs of both CNL and TNL proteins. In contrast to CNL proteins, TNL proteins contained large and variable C-terminal domains. The extant distribution and diversity of the NBS-LRR sequences has been generated by extensive duplication and ectopic rearrangements that involved segmental duplications as well as microscale events. The observed diversity of these NBS-LRR proteins indicates the variety of recognition molecules available in an individual genotype to detect diverse biotic challenges.
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}