 ,1,2,**, 郭青青,1,2,*,**, 陈雪1,2, 李加纳1,2, 王瑞,1,2,*
,1,2,**, 郭青青,1,2,*,**, 陈雪1,2, 李加纳1,2, 王瑞,1,2,*Construction of a high-density genetic map using genotyping by sequencing (GBS) for quantitative trait loci (QTL) analysis of pink petal trait in Brassica napus L.
ZHOU Xin-Tong,1,2,**, GUO Qing-Qing,1,2,*,**, CHEN Xue1,2, LI Jia-Na1,2, WANG Rui,1,2,*通讯作者:
收稿日期:2020-05-30接受日期:2020-09-13网络出版日期:2021-04-12
| 基金资助: |
First author contact:
Received:2020-05-30Accepted:2020-09-13Online:2021-04-12
| Fund supported: |
作者简介 About authors
周新桐, E-mail:
郭青青, E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1443KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
周新桐, 郭青青, 陈雪, 李加纳, 王瑞. GBS高密度遗传连锁图谱定位甘蓝型油菜粉色花性状[J]. 作物学报, 2021, 47(4): 587-598. doi:10.3724/SP.J.1006.2021.04115
ZHOU Xin-Tong, GUO Qing-Qing, CHEN Xue, LI Jia-Na, WANG Rui.
油菜属于十字花科(Cruciferace)芸薹属(Brassica)植物, 在中国古代被称为“芸薹”, 是一种栽培历史悠久、适应性强、用途广、经济价值高的油料作物。油菜在世界油料作物生产中产量仅次于大豆, 居第2位, 是中国重要油料作物[1]。近几年随着油菜不同花色品种的示范与推广, 带动了乡村观光旅游, 促进了农民增收。目前已知的油菜花色主要有金黄色、粉色、柠檬黄色、鲜黄色、乳黄色、土黄色、红黄色、桃红色、橘红色、粉白色、粉红色、乳白色、纯白色、微紫丝白花以及黄白嵌合体等。因此, 构建花色分子育种策略和选育不同花色新品种尤为重要, 而对油菜花色性状基因进行定位是最主要的研究内容。
已有一些文献对花色性状基因做了定位研究。早期Chen等[2]利用白花芥蓝与白菜杂交获得甘蓝型油菜白花品系“No 7076”发现, 控制白花性状基因位于C组染色体上。戚存扣等[3]也推测控制白花性状基因位于C组染色体上。分子连锁标记辅助选择花色是选育花色新材料的重要技术手段, 同时也可以用来验证花色性状基因所在区间。董育红等[4]利用集团分离分析法(bulked segregant analysis, BSA)发现与甘蓝型油菜白花基因连锁的随机扩增多态性DNA (random amplified polymorphism DNA, RAPD)标记。通过RAPD标记又发现白花性状基因与高芥酸性状基因具有连锁关系[5]。Huang等[6]证实1个扩增片段长度多态性(amplified fragment length polymorphism, AFLP)标记和2个简单重复序列(simple sequence repeats, SSR)标记与甘蓝型油菜白花基因连锁。利用试验获得的6对AFLP引物扩增151个单株, 确定了离白花基因两侧最近的2个分子标记[7]。张豹[8]结合SSR技术和QTL分析, 将甘蓝型油菜白花基因定位到了C03染色体上。之后又有研究将白花基因定位在了白菜参考基因组A02染色体约162 kb物理区间内[9]。基于二代测序的BSA分析技术为质量性状或主效基因快速准确定位提供了强大工具。淡亚彬[10]推测桔红花基因HS1位于甘蓝型油菜A9连锁群上, 而Yao等[11]用BSA重测序方法将桔红花基因精细定位在甘蓝型油菜C09染色体151 kb区间。利用BSA和全基因组重测序技术, 甘蓝型油菜橙花性状隐性基因BrOF定位到A09染色体41.5 kb区间[12]; 桔黄花性状基因定位在C09染色体区域[13], 并获得了2个连锁的分子标记BnaC09_19和BnaC09_34; 白花性状基因定位在C03染色体52 Mb~55 Mb区间[14], 在此区间760 kb范围内筛选出6个与白花性状基因紧密连锁共分离的SSR标记。尽管甘蓝型油菜花色定位研究较多, 但对于粉色花性状相关基因定位还未见研究报道。
随着高通量测序技术的发展, 测序方法不断更新, 测序成本不断降低。近年来发展的简化GBS测序方法通过与参考组比对获得海量SNP标记, 此类标记易于进行频率估计和基因分型, 已经在柑橘[15]、烟草[16]、玉米[17]、棉花[18]、枣树[19]等植物中应用于聚类分析和基因定位研究。但GBS在甘蓝型油菜图谱构建和基因定位中的应用报道很少。因此, 本研究利用花色分离DH群体, 采用二代简化测序GBS、转录组数据可变剪切、共线性与传统QTL分析方法交叉结合, 期望为甘蓝型油菜粉色花性状相关基因定位与分子标记辅助选择提供新的视角和思路。
1 材料与方法
1.1 试验材料
黄花亲本62和粉花亲本77都是小孢子加倍后的纯系材料。DH群体为(黄花62×粉花77) F1花粉小孢子加倍成功的114个基因型株系。2018年9月至2019年5月将亲本和DH群体种植于重庆市北碚区歇马镇西南大学油菜生物学团队育种基地。每个材料种植3行, 每行8株, 行距40 cm, 株距30 cm, 田间管理按常规方式进行。1.2 DH群体GBS测序和数据处理
2018年12月采集亲本和DH群体植株嫩叶, -20℃保存备用。2019年1月送北京诺禾致源生物信息科技有限公司测序。2个亲本DNA建库类型为DNA-350 bp, 以Illumina HiSeq PE150方法测序, 测序深度为30×。DH子代进行GBS简化基因组测序。利用GBS-SNP-CROP[20]分析流程通过barcode文件一步得到114个DH子代系的双末端测序文件。以法国甘蓝型油菜Darmor-bzh为参考组, BWA-mem软件比对测序reads到参考组, SAMtools 软件参数mpileup 获得114个DH子代系的SNP数据。1.3 花色性状考察和数据分析
油菜盛花期采用分光测色仪(YS3060)测定DH群体花瓣颜色(flower color, FC)。DH群体中每个基因型株系测量5株, 每株测量3次。采用SPSS统计花色性状基本参数。采用R3.6.1制作花色表型数据密度分布直方图。1.4 连锁图谱构建和花色QTL定位
结合甘蓝型油菜62和77两亲本之间变异SNP位点, 对DH群体的SNP位点过滤, 统计DH群体子代SNP多态型并进行基因分型。利用JoinMap 4.0 [21]计算DH群体内SNP位点间遗传距离, perl SVG模块绘制遗传连锁图谱。使用WinQTL Cartographer 2.5 (1.5 共线性分析
使用Python版MCScan (JCVI包)(1.6 转录组测序与可变剪切分析
于初花期采集甘蓝型油菜纯系粉花77和甘蓝型油菜纯系黄花62这2个亲本花瓣样品, 装入2 mL无RNA酶的离心管并迅速置于液氮中, 取样后保存于-80℃备用。提取样品总RNA送北京诺禾致源生物信息科技有限公司, 在Illumina HiSeq 2000平台上完成转录组测序。利用bwa软件将转录组测序数据与法国甘蓝型油菜参考组Darmor-bzh比对, 获得sam文件, 启动Splicing Grapher[22]分析流程, 依据法国甘蓝型油菜基因序列文件和注释文件构建剪切位点模型, 结合R包绘制花色定位区间内花色相关基因的可变剪切视图。2 结果与分析
2.1 遗传图谱构建
过滤筛选后获得3253个作图SNP标记(表1), 利用JoinMap软件构建花色DH群体遗传连锁图谱。19个遗传连锁群总遗传距离为1766.06 cM, 平均每个标记的覆盖范围为0.54 cM, 标记在A和C染色体组上都有分布, 但分布不均匀。lg14 (chrC04)上有最多的410个SNP标记, 平均遗传距离为0.29 cM; lg19 (chrC09)上标记数目最少, 仅含有33个SNP标记, 平均遗传距离为0.60 cM。lg2 (chrA02) 42.469 cM~60.984 cM处出现了18.52 cM最大间距。大区域的标记缺失, 增加了标记的平均遗传距离。SNP标记连锁群分布如图1。Table 1
表1
表1遗传连锁群信息统计
Table 1
| 连锁群 Linkage groups | 标记数量 Number of markers | 遗传距离 Genetic distance (cM) | 平均遗传距离 Average genetic distance (cM) | 标记间最大间隔 Maximum spacing between markers (cM) |
|---|---|---|---|---|
| lg1 | 69 | 93.50 | 1.36 | 15.24 |
| lg2 | 217 | 97.00 | 0.45 | 18.52 |
| lg3 | 146 | 150.84 | 1.03 | 9.17 |
| lg4 | 153 | 99.61 | 0.65 | 6.43 |
| lg5 | 220 | 141.07 | 0.64 | 6.66 |
| lg6 | 256 | 101.53 | 0.40 | 5.51 |
| lg7 | 160 | 131.66 | 0.82 | 18.37 |
| lg8 | 275 | 133.58 | 0.49 | 6.73 |
| lg9 | 222 | 77.51 | 0.35 | 10.69 |
| lg10 | 78 | 84.29 | 1.08 | 13.89 |
| lg11 | 63 | 76.21 | 1.21 | 12.50 |
| lg12 | 349 | 42.19 | 0.12 | 5.53 |
| lg13 | 214 | 88.18 | 0.41 | 4.97 |
| lg14 | 410 | 118.09 | 0.29 | 11.33 |
| lg15 | 72 | 57.28 | 0.80 | 16.00 |
| lg16 | 147 | 97.54 | 0.66 | 9.47 |
| lg17 | 103 | 102.42 | 0.99 | 6.44 |
| lg18 | 66 | 53.73 | 0.81 | 16.87 |
| lg19 总计Total | 33 3253 | 19.83 1766.06 | 0.60 0.54 | 9.65 18.52 |
新窗口打开|下载CSV
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1连锁群标记分布图
X轴表示连锁群; Y轴表示遗传距离(单位: cM); 蓝色为有效SNP标记。
Fig. 1Distribution map of linkage group markers
X-axis represents linkage groups; Y-axis represents genetic distance (cM); blue is a valid SNP marker.
2.2 亲本62、77和DH群体花瓣色泽表型观察
甘蓝型油菜纯系黄花62和甘蓝型油菜纯系粉花77花瓣色泽如图2, 62为鲜黄色, 77粉色花瓣上脉络明显。由于是小孢子加倍材料, 62和77自交仍保持鲜黄色和粉色, 花瓣色泽不会发生分离, 花色相关基因为高度纯合状态。62×77杂交组合F1花瓣颜色为粉白色。粉色花对黄花为部分显性。F1植株花粉小孢子培养秋水仙碱加倍, DH群体花瓣颜色发生了各种分离(图3), 表现为桔红、桔黄、鲜黄、淡黄、暗黄、纯白、淡白、粉白等多种花色表型, 但DH群体中每种基因型都为纯合状态, 自交后花瓣色没有出现分离。与亲本62纯黄色花瓣颜色一样的DH株系比例极少。没有和亲本77粉色花瓣颜色一样的DH株系, 有部分株系表现出粉白色。因而本研究用分光测色仪对DH群体花瓣颜色表型进行数据化采集。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2甘蓝型油菜亲本花瓣表型观察
A1与A2: 母本62; B1与B2: 父本77。
Fig. 2Parental phenotype of pink petal and yellow petal in B. napus
A1 and A2: female parent 62; B1 and B2: male parent 77.
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3甘蓝型油菜DH群体花色表型
Fig. 3Phenotype of doubled haploid population for petal color in B. napus
2.3 DH群体花色表型数据分析
对DH群体花色性状进行统计分析(表2), 使用R包制作概率密度曲线频率直方图(图4)。通过Kolmogorov-Smirnov方法检验花色(flower color)表型数据, 花色变异范围为6.21~53.04, 平均数为24.47, 中位数为22.89; 四分位距、偏斜度和峰度分别为12.78、0.85和0.09。显著性Sig值< 0.05, 结果显示花色表型偏正态分布, 表明花色受多个主效基因控制。Table 2
表2
表2甘蓝型油菜花色性状统计参数
Table 2
| 性状 Trait | 平均数 Average | 中位数 Median | 标准偏差 Standard deviation | 最小值 Minimum | 最大值 Maximum | 范围 Range | 四分位距 Interquartile range | 偏斜度 Skewness | 峰度 Kurtosis | 显著性 Significance |
|---|---|---|---|---|---|---|---|---|---|---|
| 花色PC | 24.47 | 22.89 | 11.28 | 6.21 | 53.04 | 46.83 | 12.78 | 0.85 | 0.09 | 0.00 |
新窗口打开|下载CSV
图4
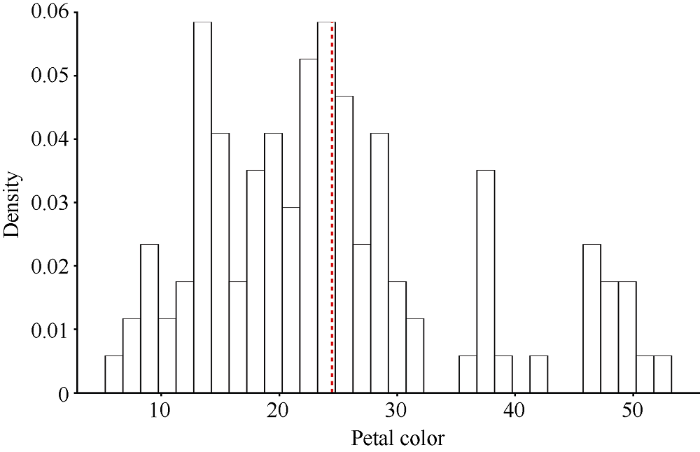 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4花色性状分布密度直方图
X轴代表花色表型组中值, Y轴代表分布密度。
Fig. 4Density distribution histogram of petal color trait
X-axis represents the petal color phenotype; Y-axis represents the distribution density.
2.4 花色性状QTL定位
采用WinQTL Cartographer复合区间作图法对花色性状进行QTL定位分析(图5)。当LOD值>2.5时, 共检测到2个QTL, 分别位于chrA07 (chr7)和chrC03 (chr13) 2条染色体上(表3)。qFC-chr7-1所在定位区间内包含287个基因, LOD值最高为6.62, 位点的加性效应为4.58, 解释表型贡献率为14.29%。而qFC-chr13-1所在定位区间内含有308个基因, LOD峰值最高为14.01, 位点加性效应为6.61, 解释表型变异为34.02%。图5
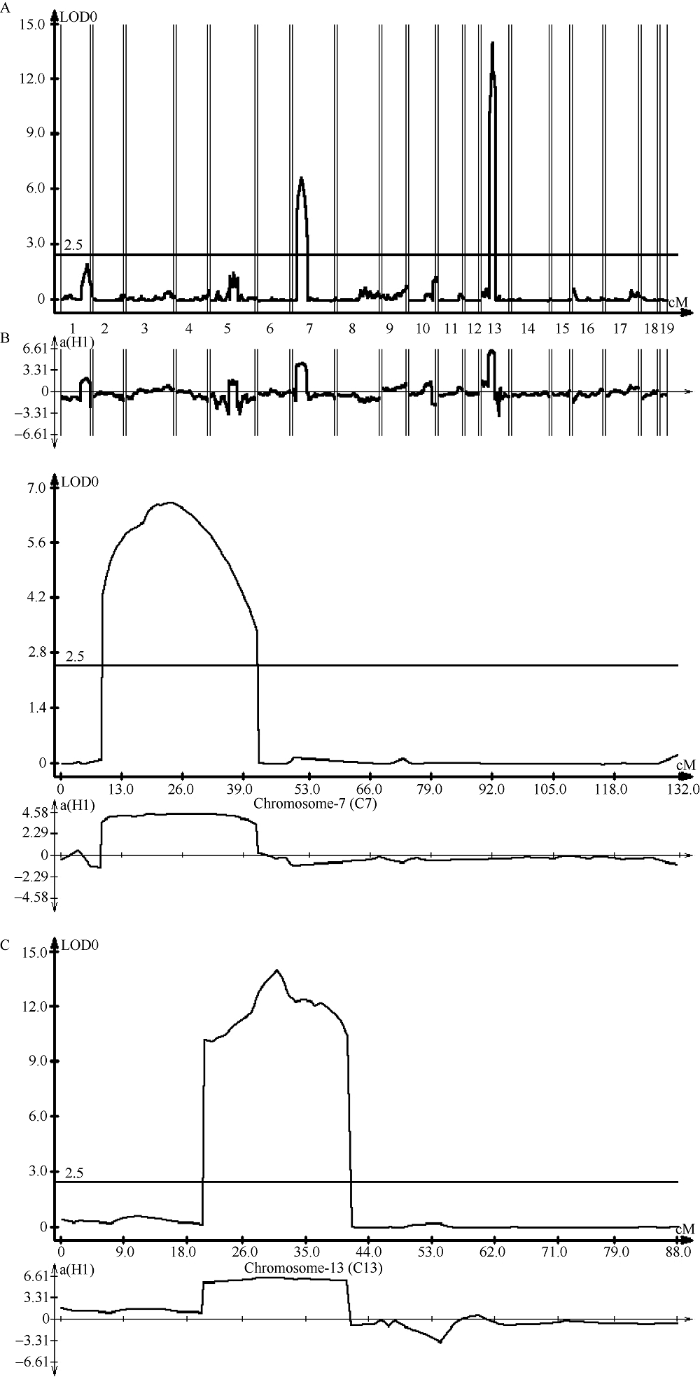 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5WinQTL复合区间方法确定花色QTL连锁群位置
图A中, X轴表示19条染色体, Y轴分别表示LOD值和加性效应a (H1); 横线表示LOD阈值线。图B、图C分别表示在7号染色体和13号染色体检测到的一个花色QTL位点; X轴表示对应染色体上的遗传距离, Y轴分别表示LOD值和加性效应a (H1); 横线表示LOD阈值线。
Fig. 5Chromosomal location of QTL for petal color by WinQTL CIM
X-axis represents 19 chromosomes, and Y-axis represents the LOD value and the additive effect a (H1) in Figure A; the horizontal line represents the LOD threshold line. Figure B, there is a represent QTL of petal color in figure C detected on chromosome 7 and chromosome 13, respectively; X-axis represents the genetic distance on the corresponding chromosome, and is marked with the corresponding linkage marker position, Y-axis represents the LOD value and the additive effect a (H1), respectively; the horizontal line indicates the LOD threshold line.
Table 3
表3
表3WinQTL复合区间作图花色QTL结果
Table 3
| QTL | LOD峰值 LOD peak | 位置 Position (cM) | 左端标记 Left marker | 右端标记 Right marker | 99%置信 区间范围 99% CI (cM) | 加性效应 Additive effect | 贡献率 R2 (%) | 区间基因数量 Number of genes |
|---|---|---|---|---|---|---|---|---|
| qFC-chr7-1 | 6.62 | 23.8 | mk2056 | mk2036 | 8.383-42.019 | 4.58 | 14.29 | 287 |
| qFC-chr13-1 | 14.01 | 30.8 | mk4000 | mk3898 | 20.305-41.353 | 6.61 | 34.02 | 308 |
新窗口打开|下载CSV
2.5 甘蓝型油菜粉色花定位区间基因与甘蓝、白菜同源基因共线性分析
WinQTL Cartographer复合区间作图法确定的QTL区间为遗传图谱区间。GBS测序数据与参考组比对获得的SNP标记具有准确的染色体物理位置, 花色QTL区间两端的SNP标记比对到法国甘蓝型油菜参考组和注释文件上, SNP标记区间内就能得到参考组对应的注释基因。根据基因序列同源性, Python (https://gsthub.com/tanghaibao/jcvi/Mcscan)流程对花色定位区间内基因与甘蓝、白菜同源基因进行共线性分析。1个花色QTL区间比对到甘蓝型油菜A07染色体13.27 Mb~14.95 Mb区间共有287个注释基因, 244个同源基因共线性比对到白菜A02染色体17.39 Mb~22.05 Mb区间, 183个同源基因共线性比对到甘蓝C06染色体0.65 Mb~12.85 Mb区间。另一个花色QTL区间比对到甘蓝型油菜C03染色体51.00 Mb~54.25 Mb区间, 共308个注释基因, 113个同源基因共线性比对到白菜A08染色体55.10 Mb~57.89 Mb区间, 230个同源基因共线性比对到甘蓝C03染色体10.58 Mb~14.99 Mb区间(图6)。图6
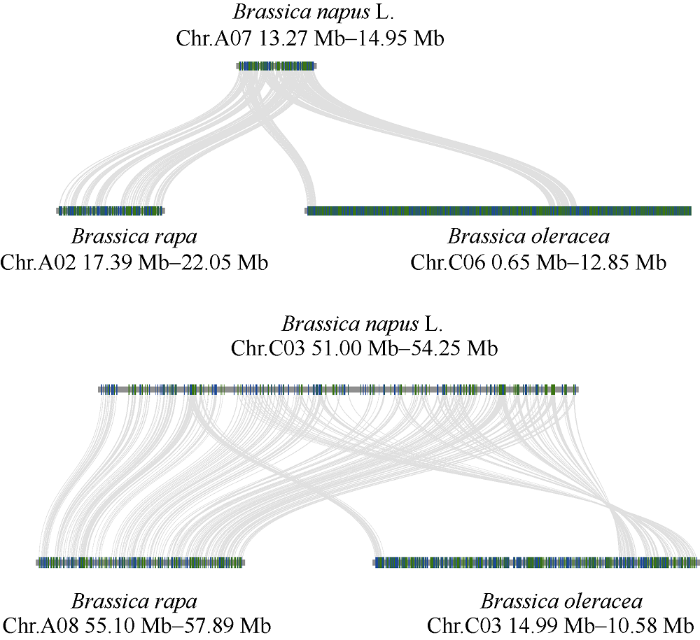 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6甘蓝型油菜WinQTL复合区间花色QTLs物理区间与甘蓝、白菜同源基因共线性分析
Fig. 6Synteny analysis of candidate interval for petal color between B. napus and B. oleracea or B. rapa by WinQTL CIM
白菜和甘蓝进行种间杂交可以人工合成甘蓝型油菜, 3个物种基因共线性分析反映了花色定位区间内基因由二倍体物种到四倍体物种的同源进化关系。
2.6 亲本花色定位区间内基因可变剪切比较
利用hisat2软件处理亲本77粉色花瓣和亲本62黄色花瓣转录组测序数据, 整理成sam文件, 使用SpliceGrapher分析流程, 统计两亲本花色定位区间内基因转录本的剪切情况(表4)。在A07染色体花色定位区间内共有287个基因, 亲本77和62发生可变剪切的基因数都为26个, 亲本77比亲本62内含子保留、外显子跳跃、3°端可变剪接都多1个, 5°端可变剪接少2个; 在C03染色体花色定位区间内共有308个基因, 亲本77有14个基因发生可变剪切, 而亲本62有11个基因可变剪切。亲本77内含子保留出现10次, 3°端可变剪接出现5次, 5°端可变剪接仅出现1次。亲本62共有11个基因发生可变剪切, 其中内含子保留出现8次, 3°端可变剪接出现5次, 5°端可变剪接出现2次; 这些结果显示花色定位区间内的基因转录本在2个亲本花瓣中多数为正常剪切, 基因转录本可变剪切所占的比例约为10%~20%, 多种可变剪切类型并存。Table 4
表4
表4甘蓝型油菜亲本花色定位区间内基因可变剪切类型
Table 4
| 方法 Method | 亲本Parent | 染色体 Chromosome | 定位区间内基因总数 Total number of genes in location interval | 发生可变剪切基因总数 Number of genes with alternative splicing | 可变剪切出现次数 Number of the alternative splicings | ||||
|---|---|---|---|---|---|---|---|---|---|
| IR | ES | A3SS | A5SS | 总计 Total | |||||
| WinQTL Cart | 77 | A07 | 287 | 26 | 26 | 2 | 5 | 0 | 33 |
| 62 | A07 | 287 | 26 | 25 | 1 | 4 | 2 | 32 | |
| 77 | C03 | 308 | 14 | 10 | 0 | 5 | 1 | 16 | |
| 62 | C03 | 308 | 11 | 8 | 0 | 5 | 2 | 15 | |
新窗口打开|下载CSV
已有研究表明, 花色素苷的合成代谢受到转录因子的调控[9], 目前已鉴定了MYB家族成员、bHLH蛋白和WD40因子3类转录因子参与花色素苷代谢合成。花色定位区间内基因BnaC03g62830D、BnaC03g64160D、BnaA07g16010D与转录因子MYB有关; BnaC03g62910D、BnaC03g62920D和BnaC03g62930D为3个WD40重复蛋白基因; 基因BnaA07g17500D参与转录因子bHLH调控; 基因BnaA07g15980D 参与类胡萝卜素合成途径。花色定位区间内的8个花色相关基因在2个亲本花瓣中有6个基因转录本都是正常剪切。而BnaA07g15980D基因在亲本62黄色花瓣中正常剪切(图7-A)、在亲本77粉色花瓣中为1个内含子保留可变剪切(图7-B); BnaA07g17500D基因在亲本62黄色花瓣中正常剪切(图7-C)、在亲本77粉色花瓣中为2个内含子保留可变剪切(图7-D)。因此, 考察花色定位区间内花色相关基因的转录状态和剪切类型可为分析推定花色候选基因提供更多线索和信息。
图7
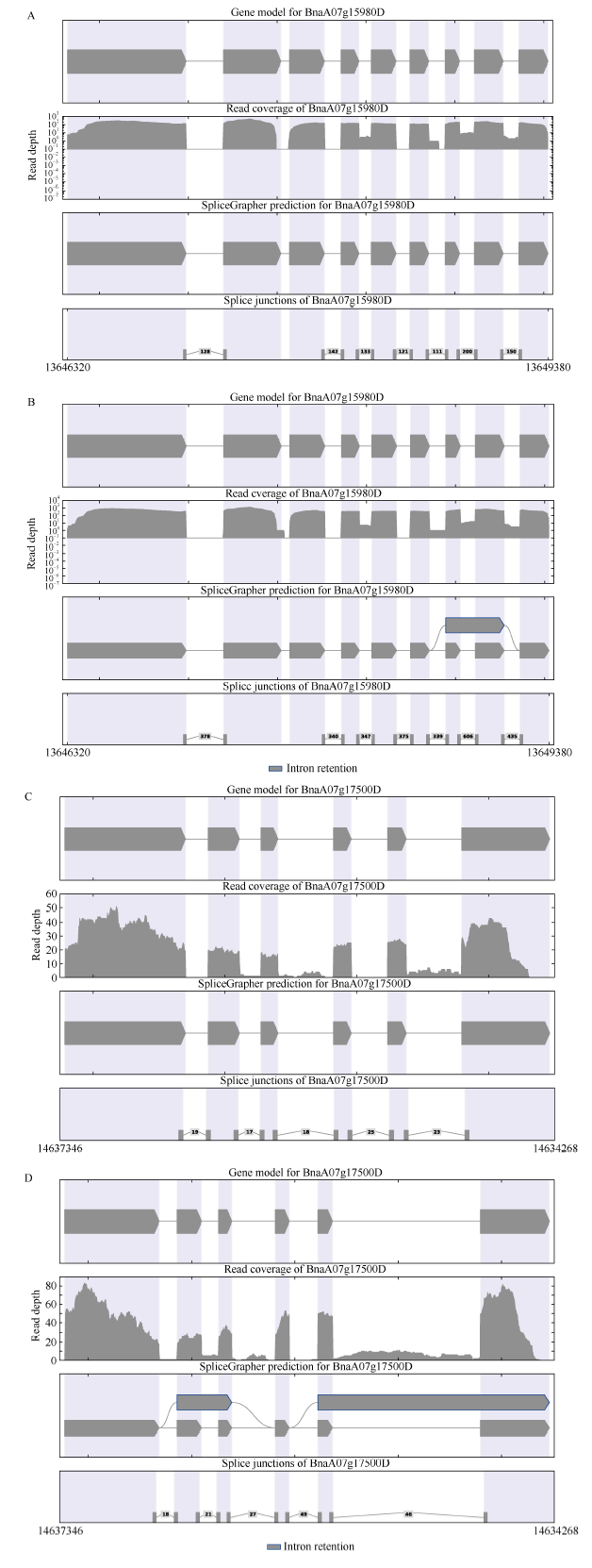 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7两亲本花瓣中花色相关基因可变剪切视图
A: BnaA07g15980D在黄花亲本62花瓣中转录本剪切视图; B: BnaA07g15980D在粉花亲本77花瓣中转录本剪切视图; C: BnaA07g17500D在黄花亲本62花瓣花瓣中转录本剪切视图; D: BnaA07g17500D在粉花亲本77花瓣中转录本剪切视图。灰色五边形为外显子, 白色区域为内含子, 连线表示不同剪接方式。图片包含4个区域, 第1部分为注释文件基因模型, 第2部分是测序结果, 第3部分为测序结果与注释文件比较基因剪切模型(带有代表性的isoform同源蛋白异构体), 第4部分为测序文件中支持外显子reads数目。左下角、右下角数字为该基因的起始位点和终止位点。
Fig. 7Splice graphs for genes related to petal color between 77 and 62
A: splice graph for BnaA07g15980D in 62 petals; B: splice graph for BnaA07g15980D in 77 petals; C: splice graph for BnaA07g17500D in 62 petals; D: splice graph for BnaA07g17500D in 77 petals. The gray pentagons represent exons, the white region represent introns, the lines between them represent different ways of splicing. The picture is split into four parts, the first part is the gene model from the annotation file, the second part is the sequencing result, the third part is the gene splicing model between the sequencing result and the annotation file (with a representative protein isoform), the fourth part is the number of reads supporting each exon in the sequencing file. The numbers in the lower left and right corners represent the start and stop sites of the gene, respectively.
3 讨论
本研究采用GBS技术对DH花色分离群体进行简化测序, 与全基因组重测序比较, GBS测序操作简单、成本低廉、省工省时。通过barcode[20]接头文件从总测序数据中一次分出114个DH子代株系测序数据, 将这些数据与法国甘蓝型油菜参考组序列比对, 结合亲本重测序之间SNP变异位点, 快速获得具有染色体物理位置的SNP基因型分型数据。GBS与SNP芯片相比较, 芯片设计开发成本相对较高并且检测位点固定, 部分SNP无法被检测导致检测位点过少。但是GBS测序也存在一些局限性, 如酶切反应有一定偏好性, 一种限制性内切酶进行酶切时只能识别特定核苷酸序列, 测序覆盖范围受到一定程度影响。GBS测序过程聚合酶链式反应(polymerase chain reaction, PCR)可能产生非特异性扩增使测序错误增多; 接头二聚体过滤不完全也会在测序中造成一定损失[23]。然而在遗传图谱构建和QTL定位分析研究中, 通过GBS测序获得的SNP标记完全能够满足要求。Landry等[24]用103个限制性内切酶片段长度多态性(restriction fragment length polymorphism, RFLP)标记构建甘蓝型油菜遗传连锁图谱。Mahmood等[25]用108个插入缺失标记(Insertion-Deletion, InDel)和89个SSR标记, 刘雪平[26]用255个SSR、相关序列扩增多态性(sequence-related amplified polymorphism, SRAP)和RAPD标记, Wang等[27]用11,458个SNP和57个SSR标记分别构建了甘蓝型油菜标记连锁图谱。而本研究应用GBS简化测序技术避免了以往人工全染色体设计引物跑标记计算标记遗传距离构建连锁图谱的繁琐弊端, 且GBS测序数据与法国甘蓝型油菜参考组比对获得的SNP标记有准确的染色体对应物理位置, 定位QTL更为准确直观, 定位区间可以直接利用法国甘蓝型油菜参考组注释文件基因染色体物理位置信息。
WinQTL Cartographer是美国北卡罗来纳州立大学开发的一款软件, 在DH、F2群体基因定位中广泛应用。结合连锁图谱SNP标记染色体物理位置, 此方法通过复合区间作图定位1个花色QTL区间在甘蓝型油菜A07染色体13.27 Mb~14.95 Mb, 另一个花色QTL区间在甘蓝型油菜C03染色体51.00 Mb~ 54.25 Mb。很多研究者已将甘蓝型油菜白花基因定位于C03染色体上。刘雪平[26]将甘蓝型油菜白花基因定位于C03染色体; Zhang等[28]通过图位克隆在甘蓝型油菜C03染色体上鉴定到一个白花性状QTL; 陈雪等[14]应用二代测序技术把白花基因定位在甘蓝型油菜C03染色体52 Mb~55 Mb区间内。本研究利用WinQTLCart复合区间作图法定位1个花色主效QTL在C03染色体的结果与陈雪等[14]一致, 且区间部分重合。
根据基因序列同源性, 对花色定位区间内基因进行甘蓝型油菜、白菜、甘蓝3个物种之间共线性分析。白菜A02染色体17.39 Mb~22.05 Mb区间与甘蓝型油菜A07染色体花色QTL区间具有基因同源共线性, 与杨晓刚[29]定位的白菜白花基因位于A02染色体20.99 Mb~22.97 Mb区间存在重合, 与赵君伟[9]定位的白菜白花基因位于A02染色体21.69 Mb~21.86 Mb区间也部分重合。甘蓝C03染色体10.58 Mb~14.99 Mb区间与甘蓝型油菜C03染色体花色QTL区间具有基因同源共线性。而杨茹涵等[30]将白花基因定位于甘蓝C03染色体48.08 Mb~ 48.92 Mb区间。本课题组利用重测序BSA方法将甘蓝型油菜桔红花基因定位于A07染色体19 Mb~20 Mb区间(结果待发表), 与WinQTL Cartographer定位另一个花色QTL区间在甘蓝型油菜A07染色体13.27 Mb~14.95 Mb结果十分相近。因此, 粉色花性状主效基因QTL染色体定位区间与已报道的文献基本一致, 定位区间部分重叠以及芸薹属共线性区间展示了花色定位区间内基因由二倍体物种到四倍体物种的基因同源关系。尤其是C03染色体上的粉色花主效基因QTL与前人定位的甘蓝型油菜白花基因C03染色体区间基本一致, 因此猜测C03染色体粉色花主效基因和前人定位的白花基因是同一个基因。
田间花色育种实践中, 甘蓝型油菜粉色花纯系自交不分离。但粉色花与甘蓝型黄花杂交构建F2群体或DH群体时, 花瓣色会有纯白和桔红2种主要花色的分离, 还有大量的淡红、淡黄、粉白等中间色, 花色表型数据呈多峰特点, 表现出主效-微效基因控制的数量性状遗传特点。而通过WinQTL Cartographer定位2个粉色花QTL分别分布在C03和A07染色体上, 推测甘蓝型油菜粉色花性状至少有2个主效基因, 粉色花与黄花杂交分离世代中, 2个主效基因分离, 多个微效基因作用, 从而导致F2和DH群体呈现多种花瓣颜色的分离。
对甘蓝型油菜亲本77粉色花瓣和亲本62黄色花瓣转录组数据进行可变剪切事件分析发现, 花色定位区间6个花色相关基因BnaC03g62830D、BnaC03g64160D、BnaC03g62910D、BnaC03g62 920D、BnaC03g62930D、BnaA07g16010D的转录本在亲本62黄色花瓣和亲本77粉色花瓣中都为正常剪切。而花色相关基因BnaA07g15980D和BnaA07 g17500D在亲本粉色花瓣中都发生内含子保留可变剪切。BnaA07g15980D 直接参与类胡萝卜素合成, BnaA07g17500D参与花色苷代谢。转录本的可变剪切增加了基因表达的复杂程度与蛋白质功能的多样性, 可能是影响甘蓝型油菜发育过程中花瓣颜色发生改变的因素之一。
传统QTL区间定位方法和二代简化测序、共线性比对、转录组测序和可变剪切等分析技术相结合, 有助于进一步缩小和锁定甘蓝型油菜粉色花相关基因的范围, 为粉色花候选基因精细定位和开发甘蓝型油菜花色基因分子连锁标记提供更多信息。
4 结论
GBS测序技术分析DH群体筛选出3253个SNP标记, 构建了全长1766.06 cM甘蓝型油菜连锁图谱, 标记间平均遗传距离为0.54 cM; 粉色花性状2个QTL分别定位到A07和C03染色体上; 定位区间内基因与甘蓝和白菜线性比对, 找到一些存在于3个物种的同源基因; 定位区间花色相关基因可变剪切分析发现, 2个花色相关基因在粉花亲本花瓣中发生内含子保留可变剪切。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.7505/j.issn.1007-9084.2014.03.020URL [本文引用: 1]

在当前我国食用植物油对外依存度居高不下的严峻形势下,本文通过对世界及我国油料产需及贸易形 势进行分析,明确了当前我国油料产业发展存在的主要问题,针对存在的问题从政策、技术等方面提出了相应的对策建议。
[本文引用: 1]
DOI:10.1111/pbr.1988.100.issue-2URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
一个由甘蓝型油菜品种Quantum (黄花、低芥酸)和人工合成的甘蓝型油菜品系No.2127-17(白花、高芥酸)为亲本材料建立的DH群体中芥酸呈现单基因的遗传模式。为了发展与芥酸紧密连锁的分子标记对其实行有效的控制,随机选择121个结实正常的DH系为作图群体,利用SSR和RAPD标记构建了一张甘蓝型油菜的遗传连锁图谱。在亲本间检测
[本文引用: 1]
DOI:10.1080/01140671.2013.863211URL [本文引用: 1]
DOI:10.3969/j.issn.1000-7091.2012.01.018URL [本文引用: 1]
甘蓝型油菜的白花性状是一种有用的指示性状,本研究以甘蓝型白花油菜及其分离群体为材料,对甘蓝型油菜的白花性状的遗传规律进行了研究。结果表明,甘蓝型油菜的白花性状受1对不完全显性基因控制。利用集团分离法(BSA) 对白花基因进行AFLP分析,在256对AFLP引物中共获得6个与白花基因紧密连锁的分子标记。EA11MC04与EA12MC01为基因两侧最近的分子标记,其遗传距离分别为0.8,1.0 cM。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s10681-017-1959-4URL [本文引用: 1]
DOI:10.1007/s11032-018-0907-xURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3864/j.issn.0578-1752.2020.06.003URL [本文引用: 3]
2群体为研究对象,通过二代测序技术,对白花性状基因候选区间定位,开发与白花性状连锁的分子标记,为定位白花候选基因和选育白花新材料提供新思路。【方法】以甘蓝型油菜DH纯系黄花Y05和甘蓝型油菜纯系白花W01杂交,观察F1和F2群体的花色分离,分析白花性状遗传模式。在F2群体中选取30株纯白花和30株纯黄花构建DNA叶片子代池和RNA花瓣子代池,对亲本和DNA叶片子代池进行30×重测序,对RNA花瓣子代池进行5×测序。以法国甘蓝型油菜Darmor-bzh、中双11、Darmor、Tapidor为参考序列,重测序QTL-seq分析流程计算2个DNA子代池的SNP-index和delta(SNP-index)。利用R包画出SNP-index和delta(SNP-index)滑窗分析图,鉴定候选区间。转录组MMAPPR分析流程以法国甘蓝型油菜Darmor-bzh为参考序列,计算SNP频率,ED 4(Loess fit)检测峰值和鉴定候选区间。利用MISA进行重复序列鉴定,使用Prime3在候选区间进行SSR引物设计,在F2群体中采用聚丙烯酰胺凝胶电泳方法对SSR引物进行筛选。【结果】甘蓝型油菜黄花与白花杂交F2群体中,白花和黄花性状分离比符合3﹕1,暗示白花性状受1对显性主效基因控制。全基因组重测序区间定位结果显示,白花性状基因候选区间在Darmor-bzh C03染色体52—55 Mb。同时以甘蓝型油菜中双11、Darmor、Tapidor分别为参考序列,均鉴定出白花基因候选区间在C03染色体上的一致性和稳定性。转录组测序定位白花性状基因位于Darmor-bzh C03染色体54—55 Mb。转录组测序和重测序定位染色体结果高度一致。在此区间内MISA和Primer3结合设计SSR引物,聚丙烯酰胺凝胶电泳筛选到6个与白花性状紧密连锁共分离的SSR标记。6个SSR标记区间范围在760 kb(52.81—53.57 Mb)。此候选区间与甘蓝、白菜共线性分析,对应白菜A02染色体56.76—57.40 Mb区间,对应甘蓝C03染色体10.99—11.28 Mb区间。【结论】甘蓝型油菜白花性状由1对显性主效基因控制。白花性状基因候选区间在法国甘蓝型油菜Darmor-bzh C03染色体52—55 Mb区间内。此区间760 kb范围内筛选出6个与白花性状基因紧密连锁共分离的SSR标记。]]>
[本文引用: 3]
DOI:10.3864/j.issn.0578-1752.2017.09.012URL [本文引用: 1]
GBS(genotyping-by-sequencing)是一种高效而经济的SNP(单核苷酸多态性)发掘和基因分型技术。采用GBS技术对240份宽皮柑橘进行基因分型,以阐明一些野生宽皮柑橘和地方品种的遗传背景,为其起源和演化研究提供更可靠的证据。【方法】选用国家柑橘种质重庆资源圃保存的具有广泛遗传多样性和地理起源的240份宽皮柑橘作为材料,利用EcoR I限制性内切酶消化基因组DNA后构建GBS文库;然后进行Illumina HiSeq PE150二代测序获得短读序列,通过BWA软件将序列映射到克里曼丁参考基因组上,再利用SAMTOOLS软件鉴定SNP位点。依据SNP的基因分型结果,采用邻近法构建系统演化树,并进行主成分分析。【结果】利用GBS简化基因组测序技术对240份宽皮柑橘进行测序,共获得96.3 Gb的测序数据,平均每个样本测序数据为401.26 Mb,经过测序深度为4X、Miss0.2、次要等位基因频率(MAF)>0.01的筛选条件过滤,最后共获得了114 200个高质量的SNP位点。主成分分析结果显示240份宽皮柑橘被分为4大类,其中温州蜜柑亚群、野生宽皮柑橘亚群可明显区分于其他宽皮柑橘。利用系统演化树可将240份宽皮柑橘划分到11个类群中。系统演化树和主成分分析都揭示了不同地理来源和特定形态的宽皮柑橘在遗传水平上存在明显的差异,比如来源于日本的温州蜜柑、欧美的克里曼丁橘及其杂种后代,以及中国南方的野生宽皮柑橘由于地理分布不同而形成了较为独特的类型,彼此间能够相互区分开。进化树结果表明中国南、北不同地域的宽皮柑橘可能存在不同的演化路径,南岭山脉及南方地区的野生宽皮柑橘、酸橘和目前南方地区栽培的砂糖橘存在较近的起源演化联系,而北方宽皮柑橘的演化却与宽皮柑橘中的古老地方品种存在紧密联系。人工杂交育种、长期的人工选择和驯化形成了不同类型的宽皮柑橘,同时也导致宽皮柑橘遗传多样性的增加。另外,一些宽皮柑橘资源中的可疑亲本也通过GBS技术得以准确鉴定。本研究表明沃柑与金诺橘有较近的亲缘关系。【结论】GBS技术用于柑橘种质资源的基因分型高效可靠,建立的系统演化树可以对240份宽皮柑橘进行准确划分,与用植物形态学划分的结果高度吻合。另外,GBS技术用于资源材料的准确鉴定和亲缘关系的研究,可为柑橘植物新品种权的保护提供可靠的技术支撑。]]>
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s10681-016-1788-xURL [本文引用: 1]
DOI:10.1007/s11295-016-1032-9URL [本文引用: 1]
DOI:10.1186/s12859-016-0879-yURL [本文引用: 2]
DOI:10.1111/j.1365-313X.1993.00739.xURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1139/g91-084URL [本文引用: 1]
.
DOI:10.1007/s11032-016-0501-zURL [本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.3389/fpls.2015.01164URLPMID:26779193 [本文引用: 1]
The apetalous genotype is a morphological ideotype for increasing seed yield and should be of considerable agricultural use; however, only a few studies have focused on the genetic control of this trait in Brassica napus. In the present study, a recombinant inbred line, the AH population, containing 189 individuals was derived from a cross between an apetalous line 'APL01' and a normally petalled variety 'Holly'. The Brassica 60 K Infinium BeadChip Array harboring 52,157 single nucleotide polymorphism (SNP) markers was used to genotype the AH individuals. A high-density genetic linkage map was constructed based on 2,755 bins involving 11,458 SNPs and 57 simple sequence repeats, and was used to identify loci associated with petalous degree (PDgr). The linkage map covered 2,027.53 cM, with an average marker interval of 0.72 cM. The AH map had good collinearity with the B. napus reference genome, indicating its high quality and accuracy. After phenotypic analyses across five different experiments, a total of 19 identified quantitative trait loci (QTLs) distributed across chromosomes A3, A5, A6, A9 and C8 were obtained, and these QTLs were further integrated into nine consensus QTLs by a meta-analysis. Interestingly, the major QTL qPD.C8-2 was consistently detected in all five experiments, and qPD.A9-2 and qPD.C8-3 were stably expressed in four experiments. Comparative mapping between the AH map and the B. napus reference genome suggested that there were 328 genes underlying the confidence intervals of the three steady QTLs. Based on the Gene Ontology assignments of 52 genes to the regulation of floral development in published studies, 146 genes were considered as potential candidate genes for PDgr. The current study carried out a QTL analysis for PDgr using a high-density SNP map in B. napus, providing novel targets for improving seed yield. These results advanced our understanding of the genetic control of PDgr regulation in B. napus.
DOI:10.1111/nph.13335URLPMID:25690717 [本文引用: 1]
In Brassica napus, yellow petals had a much higher content of carotenoids than white petals present in a small number of lines, with violaxanthin identified as the major carotenoid compound in yellow petals of rapeseed lines. Using positional cloning we identified a carotenoid cleavage dioxygenase 4 gene, BnaC3.CCD4, responsible for the formation of flower colour, with preferential expression in petals of white-flowered B. napus lines. Insertion of a CACTA-like transposable element 1 (TE1) into the coding region of BnaC3.CCD4 had disrupted its expression in yellow-flowered rapeseed lines. alpha-Ionone was identified as the major volatile apocarotenoid released from white petals but not from yellow petals. We speculate that BnaC3.CCD4 may use delta- and/or alpha-carotene as substrates. Four variations, including two CACTA-like TEs (alleles M1 and M4) and two insertion/deletions (INDELs, alleles M2 and M3), were identified in yellow-flowered Brassica oleracea lines. The two CACTA-like TEs were also identified in the coding region of BcaC3.CCD4 in Brassica carinata. However, the two INDELs were not detected in B. napus and B. carinata. We demonstrate that the insertions of TEs in BolC3.CCD4 predated the formation of the two allotetraploids.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}