 ,, 黄杨, 熊洁, 丁戈, 陈伦林,*, 宋来强江西省农业科学院作物研究所, 江西南昌330200
,, 黄杨, 熊洁, 丁戈, 陈伦林,*, 宋来强江西省农业科学院作物研究所, 江西南昌330200QTL mapping and candidate genes screening of earliness traits in Brassica napus L.
LI Shu-Yu,, HUANG Yang, XIONG Jie, DING Ge, CHEN Lun-Lin,*, SONG Lai-QiangInstitute of Crops, Jiangxi Academy of Agricultural Sciences, Nanchang 330200, Jiangxi, China通讯作者:
收稿日期:2020-07-2接受日期:2020-10-14网络出版日期:2021-04-12
| 基金资助: |
Received:2020-07-2Accepted:2020-10-14Online:2021-04-12
| Fund supported: |
作者简介 About authors
E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1047KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李书宇, 黄杨, 熊洁, 丁戈, 陈伦林, 宋来强. 甘蓝型油菜早熟性状QTL定位及候选基因筛选[J]. 作物学报, 2021, 47(4): 626-637. doi:10.3724/SP.J.1006.2021.04145
LI Shu-Yu, HUANG Yang, XIONG Jie, DING Ge, CHEN Lun-Lin, SONG Lai-Qiang.
油菜是我国重要的油料作物, 菜籽油是城乡居民主要的食用植物油[1,2]。在当前我国植物油自给率持续下降的形势下, 稳定甚至增加国内油菜生产对保障我国食用植物油供给安全具有重要的战略性意义[3]。油菜不与粮食争地, 发展南方700万公顷冬闲田种植油菜是解决这一问题的有效方法[4,5]。早熟对于大多数农作物来说是一个重要的农艺性状, 一方面可解决作物复种中的茬口紧张问题, 提高复种指数, 增加全年作物总产; 另一方面, 在高纬度、高海拔、无霜期短的高寒地区不仅可以充分利用高寒地区的光温资源, 还可避免生育前期低温冷害和生育后期的早霜危害, 从而保证作物的产量和品质, 对作物安全生产具有十分重要的意义[6,7]。油菜早熟性状的遗传解析, 对于早熟油菜品种的选育具有重要的理论与现实意义[8]。
对于油菜早熟性状的研究, 前人主要通过QTL连锁分析或GWAS手段解析其遗传基础[9,10,11,12]。Ferreira等[13]首次利用DH群体和含有132个RFLP标记的遗传图谱, 在A9、C2和C6染色体上检测到3个开花期主效QTL, 其中位于A9染色体上的位点表型贡献率达到28%。Long等[14]利用甘蓝型油菜品种TN-DH及其衍生群体, 在11个环境中共检测到42个开花期QTL位点, 其中最大表型贡献率达到52%, 并证实主效区间qFT10-4内BnFLC10基因是油菜开花调控的关键基因。Zhou等利用300份油菜资源和201,817个SNP标记, 对早熟相关性状进行GWAS分析, 检测到131个显著关联的SNP位点, 解释了表型方差的3.27%~13.17%, 此外, 通过单倍型分析和拟南芥同源比对, 筛选得到57个油菜开花期调控基因。Mei等[15]利用早、晚花油菜亲本构建的F2:3家系为材料, 通过2年田间表型考察, 共检测到6个开花期QTL, 解释了表型方差的8.1%~ 30.4%。Xu等[9]利用523份油菜种质资源, 在8个环境下对开花期性状进行全基因组关联分析, 检测到41个显著关联位点, 其表型方差为5.28%~15.75%。截至目前, 在所有19条油菜染色体中均检测到早熟相关性状QTL, 这表明油菜早熟性状具有复杂的遗传基础[16,17]。
目前报道的油菜早熟性状QTL定位主要集中在开花期[18,19,20,21,22], 虽然开花期与生育期呈显著正相关, 但却并不完全一致。开花期可反映开花前生育进程的快慢, 但不能反映开花以后植株生长发育状况。对于油菜开花后一系列生长发育进程相关性状的遗传研究和QTL定位鲜有报道。本研究对影响油菜全生育期的各个发育阶段(开花期、花期持续时间、角果期持续时间)进行研究, 分析各个发育进程的特点及对于油菜早熟形成的影响, 以指导早熟油菜的新品种选育。
1 材料与方法
1.1 试验材料
利用成熟期差异较大的2个油菜品种‘花前早’和‘Global’为亲本, 通过人工杂交获得F1代种子。在初花期对F1植株取样进行小孢子培养, 通过组织培养方法, 小孢子胚进行无菌继代培养, 成苗后移栽至大田。在花期对小孢子苗套袋, 成熟期收获自交种子后即获得该DH群体种子。1.2 田间试验
2016年10月至2017年5月、2018年10月至2019年5月在江西省农业科学院试验基地(2016南昌, 2018南昌), 2016年10月至2017年4月在韶关市农业科学研究所试验基地(2016韶关), 2018年4月至2019年8月在西宁青海大学试验基地(2018西宁)完成了DH群体种植和早熟性状调查工作。该DH群体共包含184个株系及亲本。田间试验采用完全随机区组设计, 2次重复。每小区种3行, 小区厢宽2.0 m, 行距33.3 cm, 小区面积2.0 m2, 单株间平均间距15 cm。田间管理按当地常规标准实施。1.3 性状考察和数据分析
参考《油菜种质资源描述规范和数据标准》调查早熟性状, 其中, 全区25%植株开花为开花期标准, 全区75%以上花序完全谢花(花瓣变色, 开始枯萎)为终花期标准, 全区75%以上角果呈枇杷黄色, 或主轴中段角果内种子开始呈现成熟色为成熟期标准。开花期指从播种到初花期所需的天数。开花期至终花期的时间跨度为花期持续时间, 终花期至成熟期的时间跨度为角果期持续时间。1.4 全基因组重测序
在苗期对DH群体所有株系及亲本取样, 取幼嫩叶片, 采用CTAB法提取DNA。基于Illumina HiSeq平台进行双末端(PE150)测序, 利用cutadapt和trimmomatic软件对测序数据进行质控过滤, 采用BWA软件将测序数据与参考基因组进行比对, 使用Samtools软件和Picard工具进行格式转换和reads排序, 最后使用GATK (v. 3.7)软件进行变异检测以获得SNP变异位点。1.5 遗传图谱构建与QTL定位
参考Xie等[23]提出的方法进行遗传连锁图谱的构建, 首先, 根据子代群体内标记之间的连锁关系推断亲本的基因型, 并根据亲本基因型将子代基因型转换为A和B, 同时也可以与实际获得的亲本基因型进行比较, 以判断亲本材料的真实性; 其次, 基于隐马尔可夫模型(HMM)填补缺失的基因型, 并对部分错误基因型进行修正; 最后, 根据MSTMap软件描述的方法评估标记之间的重组率, 然后使用Kosambi作图函数计算标记之间的遗传图距。使用QTL Cartographer (v. 1.17)软件进行QTL分析, 分析方法选择的是复合区间作图法, LOD阈值用1000次重复排列测验确定(P=0.05)。1.6 候选基因的筛选
利用甘蓝型油菜基因组ZS11-v20200127 (2 结果与分析
2.1 亲本和群体表型
亲本‘花前早’和‘Global’早熟各相关性状差异较大, t测验结果表明, 所有环境中, 双亲在花期、花期持续时间、全生育期的差异均达到极显著水平(表1)。双亲角果期持续时间的差异在不同环境中表现不同(表1)。DH群体各早熟相关性状在不同年份均呈现广泛的变异, 且在各个环境下都呈正态或近似正态分布(图1), 符合数量性状的典型特征, 适宜采用QTL手段解析其早熟性状的遗传基础。Table 1
表1
表1亲本和DH群体在4个环境下的早熟相关性状表型
Table 1
| 环境 Environment | 研究材料 Material | 开花期 Flowering time | 花期持续时间 Flowering period duration | 角果期持续时间 Silique period duration | 全生育期 Full growth period | |
|---|---|---|---|---|---|---|
| 2016南昌 2016 Nanchang | 亲本 Parents | 花前早 Huaqianzao | 76.0 A | 64 A | 32.5 A | 172.5 A |
| Global | 138.5 B | 36.5 B | 30.0 A | 205.0 B | ||
| DH群体 DH population | 最大值 Max. | 141.0 | 77.0 | 36.0 | 205.0 | |
| 最小值 Min. | 76.0 | 26.0 | 22.0 | 172.5 | ||
| 均值 Mean | 118.5 | 43.5 | 29.0 | 191.0 | ||
| 2016韶关 2016 Shaoguan | 亲本 Parents | 花前早 Huaqianzao | 64.5 A | 40.5 A | 27.5 A | 132.5 A |
| Global | 123.5 B | 22.5 B | 37.5 B | 183.5 B | ||
| DH群体 DH population | 最大值 Max. | 131.0 | 58.5 | 44.5 | 185.0 | |
| 最小值 Min. | 58.0 | 17.0 | 11.0 | 124.0 | ||
| 均值 Mean | 97.5 | 34.2 | 30.2 | 161.8 | ||
| 2018南昌 2018 Nanchang | 亲本 Parents | 花前早 Huaqianzao | 108.0 A | 46.0 A | 29.0 A | 183.0 A |
| Global | 149.0 B | 22 B | 30.0 A | 201.0 B | ||
| DH群体 DH population | 最大值 Max. | 149.0 | 46.0 | 34.0 | 203.0 | |
| 最小值 Min. | 108.0 | 17.0 | 26.0 | 181.0 | ||
| 均值 Mean | 138.1 | 26.3 | 30.1 | 194.5 | ||
| 2018西宁 2018 Xining | 亲本 Parents | 花前早 Huaqianzao | 56.0 A | 31.5 A | 31.5 A | 119.0 A |
| Global | 72.5 B | 41.0 B | 16.0 B | 129.5 B | ||
| DH群体 DH population | 最大值 Max. | 98.0 | 42.0 | 31.5 | 135.5 | |
| 最小值 Min. | 56.0 | 18.0 | 9.5 | 116.5 | ||
| 均值 Mean | 72.5 | 30.1 | 20.5 | 123.9 | ||
新窗口打开|下载CSV
图1
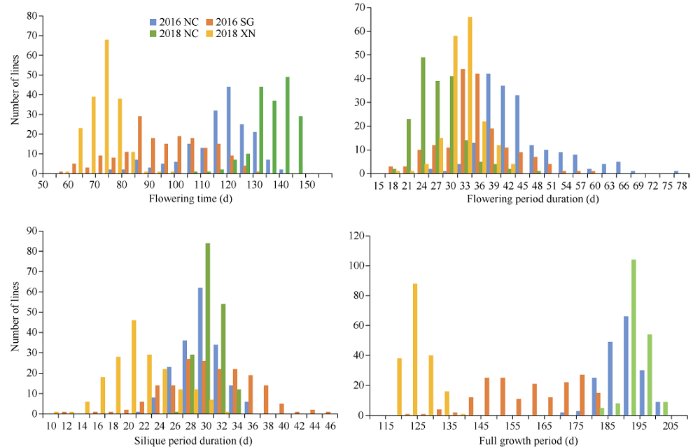 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1DH群体早熟相关性状的频数分布图
2016 NC, 2016 SG, 2018 NC和2018 XN分别指2016年南昌, 2016年韶关, 2018年南昌和2018年西宁。
Fig. 1Phenotype frequency distribution of earliness related traits in DH population
2016 NC, 2016 SG, 2018 NC, and 2018 SG were the code of different environment: 2016 Nanchang, 2016 Shaoguan, 2018 Nanchang and 2018 Xining, respectively.
对上述4个早熟性状进行相关性分析(表2)表明, 开花期、花期持续时间、角果期持续时间等性状与全生育期均显著相关, 其相关系数分别为0.926、-0.701和0.158。
Table 2
表2
表2DH群体4个早熟性状的相关性分析
Table 2
| 开花期 Flowering time | 花期持续时间 Flowering period duration | 角果期持续时间 Silique period duration | 全生育期 Full growth period | |
|---|---|---|---|---|
| 开花期 Flowering time | 1 | -0.880** | 0.041 | 0.926** |
| 花期持续时间 Flowering period duration | 1 | -0.120 | -0.701** | |
| 角果期持续时间 Silique period duration | 1 | 0.158* | ||
| 全生育期 Full growth period | 1 |
新窗口打开|下载CSV
2.2 遗传图谱构建
经过对测序数据进行质控过滤、BWA比对, 变异检测注释等一系列生物信息分析后, 共获得875,740个变异位点。参考Xie等[23]提出的方法, 筛选出874,995个高质量多态性的SNP标记用于构建遗传连锁图谱。各连锁群长度在62.56~ 206.71 cM之间, 平均长度为136.88 cM。对上述SNP位点进行整合得到3905个Bin。各连锁群Bin数目在140~320之间, 平均数目为206个; 各连锁群上相邻bin之间平均距离在0.42~0.98 cM之间。遗传标记在各连锁群上覆盖密度较高, 且分布较为均匀(表3和图2)。Table 3
表3
表3遗传连锁图谱信息统计表
Table 3
| 连锁群 Chr. | 连锁群总长度 Length (cM) | 标记数 No. of markers | 相邻标记间的最大间隔 Max. interval (cM) | Bin数目 No. bins | 相邻bin间平均距离 Bin interval (cM) |
|---|---|---|---|---|---|
| A01 | 110.96 | 48,693 | 4.72 | 183 | 0.61 |
| A02 | 117.38 | 44,545 | 5.95 | 201 | 0.59 |
| A03 | 197.46 | 65,502 | 10.52 | 280 | 0.71 |
| A04 | 119.03 | 43,644 | 6.56 | 190 | 0.63 |
| A05 | 101.61 | 55,238 | 3.39 | 187 | 0.55 |
| A06 | 107.28 | 43,735 | 4.37 | 197 | 0.55 |
| A07 | 118.95 | 40,195 | 5.75 | 212 | 0.56 |
| A08 | 62.56 | 38,113 | 1.50 | 151 | 0.42 |
| A09 | 139.70 | 55,599 | 8.99 | 226 | 0.62 |
| A10 | 95.92 | 44,019 | 3.39 | 185 | 0.52 |
| C01 | 126.88 | 39,781 | 5.48 | 197 | 0.65 |
| C02 | 148.14 | 83,307 | 8.73 | 201 | 0.74 |
| C03 | 206.71 | 85,609 | 7.96 | 320 | 0.65 |
| C04 | 195.49 | 37,202 | 9.81 | 248 | 0.79 |
| C05 | 193.22 | 22,608 | 6.68 | 217 | 0.90 |
| C06 | 161.24 | 27,401 | 17.33 | 165 | 0.98 |
| C07 | 157.87 | 31,921 | 5.80 | 211 | 0.75 |
| C08 | 133.07 | 36,408 | 5.50 | 194 | 0.69 |
| C09 | 107.28 | 31,475 | 6.72 | 140 | 0.77 |
| 平均Average | 136.88 | 46,052 | 6.80 | 206 | 0.67 |
| 合计Total | 2600.73 | 874,995 | — | 3905 | — |
新窗口打开|下载CSV
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2遗传连锁图谱中的标记在各连锁群上的分布情况
Fig. 2Markers distribution of linkage group in genetic linkage map
2.3 多环境下早熟性状QTL定位
利用上述构建好的遗传图谱和DH群体表型数据, 采用QTL Cartographer (v. 1.17)软件对南昌、韶关和西宁2年4个环境下的早熟相关性状分别进行QTL扫描。共检测到30个早熟相关性状QTL位点, 其中开花期、花期持续时间、角果期持续时间和全生育期等分别检测到12、5、4和9个QTL位点, 解释了5.8%~22.4%的表型方差(表4)。其中, qFT.C08-2和qFGP.C08-3位点可在多个环境中重复检测到。发现4、2和1个全生育期QTL置信区间分别与开花期、花期持续时间、角果期持续时间位点置信区间完全或部分重叠(表4), 表明开花期、花期持续时间、角果期持续时间等各个发育阶段均能影响油菜全生育期, 其中开花期影响最大。Table 4
表4
表4不同环境下检测出的早熟相关性状QTL
Table 4
| 位点 Locus | 性状 Trait | 染色体 Chr. | 位置 Pos (cM) | LOD值 LOD value | 贡献率 R2 (%) | 加性效应Additive effect | 置信区间Confidence interval | 环境 Environment |
|---|---|---|---|---|---|---|---|---|
| qFT.A02-1 | FT | A02 | 18.23 | 5.37 | 10.6 | 2.15 | 16.52-18.52 | 2018西宁 2018 Xining |
| qFT.A02-2 | FT | A02 | 60.99 | 3.99 | 9.1 | -1.82 | 58.14-61.84 | 2018西宁 2018 Xining |
| qFT.A06-1 | FT | A06 | 95.90 | 6.73 | 10.9 | 2.39 | 95.61-96.75 | 2018南昌 2018 Nanchang |
| qFT.A09-1 | FT | A09 | 84.60 | 5.22 | 11.8 | 5.56 | 84.03-87.84 | 2016韶关 2016 Shaoguan |
| qFT.A09-2 | FT | A09 | 123.12 | 4.04 | 8.8 | 3.92 | 121.65-126.32 | 2016南昌 2016 Nanchang |
| qFT.C04-1 | FT | C04 | 99.26 | 4.32 | 9.5 | 5.04 | 96.73-99.26 | 2016韶关 2016 Shaoguan |
| qFT.C06-1 | FT | C06 | 145.13 | 5.90 | 9.6 | 2.29 | 144.13-149.13 | 2018南昌 2018 Nanchang |
| qFT.C07-1 | FT | C07 | 81.72 | 3.76 | 5.8 | 1.69 | 78.76-84.10 | 2018南昌 2018 Nanchang |
| qFT.C08-1 | FT | C08 | 15.37 | 7.47 | 12.4 | 2.05 | 10.88-15.37 | 2018西宁 2018 Xining |
| qFT.C08-2 | FT | C08 | 40.61 | 6.60 | 11.1 | 2.01 | 34.77-43.49 | 2018西宁 2018 Xining |
| FT | C08 | 43.49 | 4.65 | 9.0 | 4.00 | 42.63-47.70 | 2016南昌 2016 Nanchang | |
| FT | C08 | 45.80 | 12.87 | 22.4 | 3.71 | 45.80-45.80 | 2018南昌 2018 Nanchang | |
| qFT.C08-3 | FT | C08 | 52.89 | 6.54 | 12.5 | 6.50 | 52.04-53.75 | 2016韶关 2016 Shaoguan |
| qFT.C08-4 | FT | C08 | 104.06 | 4.57 | 7.2 | 2.08 | 103.27-104.94 | 2018南昌 2018 Nanchang |
| qFPD.A02-1 | FPD | A02 | 19.09 | 4.22 | 7.3 | -1.40 | 19.09-21.67 | 2018南昌 2018 Nanchang |
| qFPD.A06-1 | FPD | A06 | 95.61 | 6.14 | 11.0 | -1.54 | 95.33-97.32 | 2018南昌 2018 Nanchang |
| qFPD.A09-1 | FPD | A09 | 84.60 | 4.28 | 8.9 | -2.21 | 84.03-87.25 | 2016韶关 2016 Shaoguan |
| qFPD.C04-1 | FPD | C04 | 123.90 | 3.79 | 8.4 | -2.42 | 118.86-127.70 | 2016南昌 2016 Nanchang |
| qFPD.C08-1 | FPD | C08 | 45.80 | 8.24 | 15.2 | -1.95 | 44.36-46.70 | 2018南昌 2018 Nanchang |
| qSPD.A09-1 | SPD | A09 | 91.99 | 4.59 | 9.1 | -1.19 | 89.73-92.87 | 2018西宁 2018 Xining |
| qSPD.C04-1 | SPD | C04 | 64.02 | 5.07 | 10.3 | -1.30 | 62.14-64.30 | 2018西宁 2018 Xining |
| qSPD.C08-1 | SPD | C08 | 17.20 | 4.77 | 10.2 | 1.71 | 13.88-21.20 | 2016韶关 2016 Shaoguan |
| qSPD.C08-2 | SPD | C08 | 32.35 | 5.86 | 10.1 | -1.27 | 32.35-33.56 | 2018西宁 2018 Xining |
| qFGP.A02-1 | FGP | A02 | 50.67 | 5.50 | 10.7 | -5.35 | 50.10-51.53 | 2016韶关 2016 Shaoguan |
| qFGP.A02-2 | FGP | A02 | 61.56 | 5.72 | 11.9 | -1.53 | 58.71-62.13 | 2018西宁 2018 Xining |
| qFGP.A09-1 | FGP | A09 | 84.60 | 4.48 | 8.8 | 4.21 | 83.74-87.25 | 2016韶关 2016 Shaoguan |
| qFGP.C06-1 | FGP | C06 | 111.50 | 7.34 | 12.0 | 1.34 | 111.16-119.50 | 2018南昌 2018 Nanchang |
| qFGP.C07-1 | FGP | C07 | 79.33 | 6.53 | 10.0 | 1.16 | 78.14-80.33 | 2018南昌 2018 Nanchang |
| qFGP.C08-1 | FGP | C08 | 5.40 | 7.56 | 11.8 | 1.41 | 4.54-9.88 | 2018南昌 2018 Nanchang |
| qFGP.C08-2 | FGP | C08 | 23.27 | 6.16 | 12.5 | 2.15 | 21.20-27.89 | 2016南昌 2016 Nanchang |
| qFGP.C08-3 | FGP | C08 | 42.05 | 3.99 | 7.2 | 1.21 | 40.16-44.65 | 2018西宁 2018 Xining |
| FGP | C08 | 44.07 | 5.42 | 10.7 | 4.84 | 43.49-44.65 | 2016韶关 2016 Shaoguan | |
| qFGP.C08-4 | FGP | C08 | 86.77 | 5.97 | 9.2 | 1.21 | 86.77-87.36 | 2018南昌 2018 Nanchang |
新窗口打开|下载CSV
2.4 候选基因预测
参考甘蓝型油菜基因组ZS11-v20200127, 将30个QTL置信区间内序列和拟南芥的序列进行比对, 分别在A02、A06、A09、C04、C06、C07和C08染色体上的QTL 区间内筛选到29个可能与油菜早熟相关的候选基因(表5)。在开花期QTL区间内, 筛选到12个候选基因, 其中BnaC08G0146200ZS为VRN2基因, 介导春化作用, 参与FLC的转录调控。有2个属于SEP基因家族, 在花分生组织活动中起着核心作用。BnaA02G0048600ZS的拟南芥同源基因AT5G13480影响FCA mRNA加工, 参与开花时间的调控。BnaC08G0123900ZS的拟南芥同源基因AT1G30970为一年生冬性拟南芥延迟开花所必需。在花期持续时间QTL区间内, 筛选到的5个候选基因BnaA02G0058000ZS、BnaA06G0397900ZS、BnaA 09G0527800ZS、BnaC04G0375200ZS和BnaC08G 0148600ZS分别与拟南芥AT5G15160、AT2G03710、AT3G57290、AT5G37020和AT4G17230同源, 其中BnaA02G0058000ZS直接参与开花时间调控, BnaA06G0397900ZS在花分生组织维持中起作用, BnaA09G0527800ZS参与花的发育, 光形态发生, BnaC04G0375200ZS参与花的发育, 对生长素刺激的响应和果实的发育等, BnaC08G0148600ZS为GRAS基因家族的成员, 编码SCL13蛋白质。在角果期持续时间QTL区间内, 筛选到8个候选基因, 有2个候选基因与生长素调控响应相关, 3个候选基因与光合作用及光敏性相关, 2个候选基因编码LEA家族蛋白, 与胚胎晚期发育有关, 1个候选基因编码ENTH/ANTH/VHS超家族蛋白。在全生育期QTL区间内, 筛选到4个候选基因。其中, BnaA02G 0189 000ZS为TAA1基因, 参与调控植物从胚发生到花形成过程, 在乙烯介导的信号传导中起关键作用。Bna C06G0294300ZS编码MADS box家族转录因子, 参与调控根细胞分化和开花时间。BnaC07G02816 00ZS编码MIKC家族转录调控因子, 参与花粉发育。Bna C08G0063300ZS为TLL1基因, 参与昼夜节律。Table 5
表5
表5早熟相关性状候选基因
Table 5
 |
新窗口打开|下载CSV
3 讨论
3.1 油菜遗传图谱构建
分子遗传图谱是进行油菜重要性状QTL定位和基因克隆的基础。早在1991年, Landry等[24]就利用103个RFLP标记, 构建了首张甘蓝型油菜遗传连锁图谱, 此后国内外实验室陆续发布了多张遗传图谱, 但普遍存在密度较低和标记分布不均匀等问题, 这限制了QTL定位的效率和精度。近年来, 分子生物学技术的高速发展极大地推动了油菜高密度遗传图谱的构建。Shi等[25]利用1038个标记和BnaZN F2群体, 构建了一张长度为1763.2 cM的高密度遗传图谱, 其相邻标记距离在1.45~4.01 cM之间, 平均2.19 cM。基于SNP芯片技术, Liu等[26]利用9164个SNP标记和RIL群体, 构建了长度为1832. 9 cM的油菜高密度遗传图谱, 其相邻标记平均距离为0.66 cM, 分辨率大大提高。俎峰等[27]利用甘蓝型油菜60K SNP芯片(Illumina Infinium HD Assay)技术构建的高密度遗传连锁图长度为3838.2 cM, 包含7601个SNP位点。目前报道的高密度遗传连锁图大多采用甘蓝型油菜60K SNP芯片(Illumina Infinium HD Assay)或简化基因组测序技术。本研究利用全基因组重测序筛选出874,995个SNP标记构建高质量遗传连锁图谱, 各连锁群上相邻bin之间平均距离在0.42~0.98 cM之间。遗传标记覆盖密度较高, 且分布较为均匀。基于全基因组重测序技术构建的遗传连锁图谱质量较传统分子标记、芯片或简化基因组测序技术手段获得的遗传图谱更具优势。3.2 不同发育阶段与全生育期的相关性
全生育期是油菜早熟性状鉴定的主要指标, 由于开花期与全生育期高度正相关, 对于油菜早熟性状的研究及品种选育等工作主要围绕开花期性状进行研究[28,29,30,31]。俎峰等[27]认为, 生育期比开花期遗传更复杂, 在其QTL定位研究中需尽可能分解性状, 量化性状判定指标, 从而降低生育期的遗传复杂度, 有利于检测出稳定的遗传位点。本研究对影响油菜全生育期的各个发育阶段(开花期、花期持续时间、角果期持续时间等)进行表型调查和QTL定位分析。各发育阶段的田间表型和QTL定位结果均表明, 它们与全生育期密切相关。其中, 开花期、花期持续时间、角果期持续时间与全生育期的皮尔森相关系数分别为0.926、-0.701和0.158, 相关性均达到显著水平。在检测到9个全生育期QTL位点中, 发现4、2和1个全生育期QTL置信区间分别与开花期、花期持续时间、角果期持续时间位点置信区间完全或部分重叠。以上结果表明, 虽然开花期是早熟性状选择的重要指标, 但开花后的一系列生命活动也参与了油菜的生殖生长发育进程, 与全生育期显著相关。因此, 在早熟性状的研究中, 可以从各个发育阶段入手, 不但有利于使熟期进一步提前, 也可减缓早熟油菜品种过早开花导致的冬前低温寒潮天气的不利影响。3.3 候选基因筛选
对模式植物拟南芥早熟相关基因的研究主要集中在开花期性状[32,33], 其遗传调控网络的研究表明, 主要有以下几种途径参与开花调控, 分别是春化、光周期、赤霉素、自主途径和年龄等, 涉及100多个基因参与[34,35,36,37]。对于油菜早熟相关基因的研究, 也有开花基因被成功克隆的报道[38,39,40,41]。Chen等[42]克隆得到油菜开花基因BnFLC.A2, 并研究其花期调控机制。Hou等[43]克隆出甘蓝型油菜BnFLC.A10基因, 该基因是调控油菜冬春分化的关键因子, 其表达受春化作用的抑制。本研究在油菜早熟性状QTL置信区间内筛选到29个早熟相关基因, 它们主要参与开花调控、花序分生组织活动、春化、光形态建成及生长素响应等生物学过程。其中, 开花期、花期持续时间QTL置信区间内筛选到的候选基因主要影响开花过程, 例如在C08染色体上筛选到的候选基因BnaC08G0146200ZS, 其拟南芥同源基因VRN2介导春化作用, 参与了FLC的转录调控。VRN2编码一种核定位锌指蛋白。在野生型拟南芥中, 春化导致开花抑制因子FLC水平的稳定下降。在vrn2突变体中, 虽然FLC的表达在春化过程中正常下调, 但当植物恢复到正常温度时, FLC mRNA的水平会上升[44]。在A02染色体上筛选到的候选基因BnaA02 G0058000ZS, 其拟南芥同源基因BNQ2与bHLH转录因子结合形成异二聚体并负向调控其表达, 直接受AP3和PI基因的负调控, 为开花期调控所必须[45]。油菜开花受精后, 籽粒发育过程是影响油菜产量、品质、成熟期等性状的关键。对水稻、小麦等作物籽粒灌浆过程的研究表明, 大粒品种和高产品种的籽粒均具有灌浆速率快和灌浆时间长的特点[46]。而在角果持续时间QTL置信区间内筛选到的候选基因主要影响籽粒发育过程。例如, 在A09染色体上的BnaA09G0545800ZS, 其拟南芥同源基因ARGOS为生长素调控基因, 参与籽粒发育。Hu等[47]发现, 受生长素诱导的ARGOS基因在拟南芥器官发育过程中能够通过改变细胞数量, 延长生长发育时间等方式调控籽粒发育。在C08染色体上的BnaC08G0077 800ZS, 其拟南芥同源基因编码LEA家族蛋白, LEA蛋白及其mRNA主要在籽粒发育晚期大量积累, 主要参与应答植物失水胁迫, 与籽粒发育密切相关[48], 其对籽粒发育时间的具体影响还有待进一步研究。4 结论
田间表型和QTL定位结果均表明, 开花期、花期持续时间、角果期持续时间等与全生育期密切相关。筛选到的29个候选基因通过调控花期或籽粒发育等生长发育进程影响油菜早熟。因此, 油菜早熟性状的研究可同时考虑开花和籽粒发育过程。以上工作为油菜早熟性状的遗传改良提供了新的思路。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.7505/j.issn.1007-9084.2014.03.020URL [本文引用: 1]

在当前我国食用植物油对外依存度居高不下的严峻形势下,本文通过对世界及我国油料产需及贸易形 势进行分析,明确了当前我国油料产业发展存在的主要问题,针对存在的问题从政策、技术等方面提出了相应的对策建议。
[本文引用: 1]
URL [本文引用: 1]
]]>
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1073/pnas.1815030116URLPMID:31451662 [本文引用: 1]
The contradiction between
DOI:10.1016/j.tplants.2009.07.005URLPMID:19716745 [本文引用: 1]
Shifting the seasonal timing of reproduction is a major goal of plant breeding efforts to produce novel varieties that are better adapted to local environments and changing climatic conditions. The key regulators of floral transition have been studied extensively in model species, and in recent years a growing number of related genes have been identified in crop species, with some notable exceptions. These sequences and variants thereof, as well as several major genes which were only identified in crop species, can now be used by breeders as molecular markers and for targeted genetic modification of flowering time. This article reviews the major floral regulatory pathways and discusses current and novel strategies for altering bolting and flowering behavior in crop plants.
DOI:10.1093/dnares/dsv035URLPMID:26659471 [本文引用: 2]
Flowering time adaptation is a major breeding goal in the allopolyploid species Brassica napus. To investigate the genetic architecture of flowering time, a genome-wide association study (GWAS) of flowering time was conducted with a diversity panel comprising 523 B. napus cultivars and inbred lines grown in eight different environments. Genotyping was performed with a Brassica 60K Illumina Infinium SNP array. A total of 41 single-nucleotide polymorphisms (SNPs) distributed on 14 chromosomes were found to be associated with flowering time, and 12 SNPs located in the confidence intervals of quantitative trait loci (QTL) identified in previous researches based on linkage analyses. Twenty-five candidate genes were orthologous to Arabidopsis thaliana flowering genes. To further our understanding of the genetic factors influencing flowering time in different environments, GWAS was performed on two derived traits, environment sensitivity and temperature sensitivity. The most significant SNPs were found near Bn-scaff_16362_1-p380982, just 13 kb away from BnaC09g41990D, which is orthologous to A. thaliana CONSTANS (CO), an important gene in the photoperiod flowering pathway. These results provide new insights into the genetic control of flowering time in B. napus and indicate that GWAS is an effective method by which to reveal natural variations of complex traits in B. napus.
DOI:10.1093/dnares/dsx052URLPMID:29236947 [本文引用: 1]
0.6) or in 100 kb regions around significant trait-associated SNPs were screened, revealing 57 B. napus genes (33 SNPs) orthologous to 39 Arabidopsis thaliana flowering time genes. These results support the practical and scientific value of novel large-scale SNP data generation in uncovering the genetic control of agronomic traits in B. napus, and also provide a theoretical basis for molecular marker-assisted selection of earliness breeding in rapeseed.]]>
DOI:10.1007/s00122-012-1966-8URL [本文引用: 1]
We identified quantitative trait loci (QTL) underlying variation for flowering time in a doubled haploid (DH) population of vernalisation—responsive canola (Brassica napus L.) cultivars Skipton and Ag-Spectrum and aligned them with physical map positions of predicted flowering genes from the Brassica rapa genome. Significant genetic variation in flowering time and response to vernalisation were observed among the DH lines from Skipton/Ag-Spectrum. A molecular linkage map was generated comprising 674 simple sequence repeat, sequence-related amplified polymorphism, sequence characterised amplified region, Diversity Array Technology, and candidate gene based markers loci. QTL analysis indicated that flowering time is a complex trait and is controlled by at least 20 loci, localised on ten different chromosomes. These loci each accounted for between 2.4 and 28.6?% of the total genotypic variation for first flowering and response to vernalisation. However, identification of consistent QTL was found to be dependant upon growing environments. We compared the locations of QTL with the physical positions of predicted flowering time genes located on the sequenced genome of B. rapa. Some QTL associated with flowering time on A02, A03, A07, and C06 may represent homologues of known flowering time genes in Arabidopsis; VERNALISATION INSENSITIVE 3, APETALA1, CAULIFLOWER, FLOWERING LOCUS C, FLOWERING LOCUS T, CURLY LEAF, SHORT VEGETATIVE PHASE, GA3 OXIDASE, and LEAFY. Identification of the chromosomal location and effect of the genes influencing flowering time may hasten the development of canola varieties having an optimal time for flowering in target environments such as for low rainfall areas, via marker-assisted selection.]]>
DOI:10.1007/s11032-014-0139-7URL [本文引用: 1]
DOI:10.1007/BF00222140URLPMID:24174034 [本文引用: 1]
Rapeseed cultivars (Brassica napus L.) can be classified into annual and biennial groups according to their requirement for vernalization in order to induce flowering. The genetic control of these phenotypic differences is not well understood, but this information could be valuable for the design of breeding approaches to accelerate rapeseed improvement. In order to map loci controlling this variation, a doubled haploid population, derived from a cross between annual and biennial cultivars, was evaluated for vernalization requirement and days-to-flowering in a replicated field experiment using three treatments: no vernalization, 4 weeks of vernalization and 8 weeks of vernalization. A linkage map of 132 RFLP loci was used to locate loci controlling these traits. Marker segregation in one region of linkage group 9 was strongly associated with the annual/biennial growth habit in the unvernalized treatment and with days-to-flowering in all three treatments. Two other regions with smaller effects on days-to-flowering were also identified.
DOI:10.1534/genetics.107.080705URLPMID:18073439 [本文引用: 1]
Most agronomical traits exhibit quantitative variation, which is controlled by multiple genes and are environmentally dependent. To study the genetic variation of flowering time in Brassica napus, a DH population and its derived reconstructed F(2) population were planted in 11 field environments. The flowering time varied greatly with environments; 60% of the phenotypic variation was attributed to genetic effects. Five to 18 QTL at a statistically significant level (SL-QTL) were detected in each environment and, on average, two new SL-QTL were discovered with each added environment. Another type of QTL, micro-real QTL (MR-QTL), was detected repeatedly from at least 2 of the 11 environments; resulting in a total of 36 SL-QTL and 6 MR-QTL. Sixty-three interacting pairs of loci were found; 50% of them were involved in QTL. Hundreds of floral transition genes in Arabidopsis were aligned with the linkage map of B. napus by in silico mapping; 28% of them aligned with QTL regions and 9% were consistent with interacting loci. One locus, BnFLC10, in N10 and a QTL cluster in N16 were specific to spring- and winter-cropped environments respectively. The number of QTL, interacting loci, and aligned functional genes revealed a complex genetic network controlling flowering time in B. napus.
DOI:10.1111/pbr.2009.128.issue-5URL [本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
]]>
[本文引用: 1]
DOI:10.1007/s11032-015-0425-zURL [本文引用: 1]
DOI:10.1371/journal.pone.0102611URLPMID:25061822 [本文引用: 1]
Time of flowering is a key adaptive trait in plants and is conditioned by the interaction of genes and environmental cues including length of photoperiod, ambient temperature and vernalisation. Here we investigated the photoperiod responsiveness of summer annual-types of Brassica napus (rapeseed, canola). A population of 131 doubled haploid lines derived from a cross between European and Australian parents was evaluated for days to flowering, thermal time to flowering (measured in degree-days) and the number of leaf nodes at flowering in a compact and efficient glasshouse-based experiment with replicated short and long day treatments. All three traits were under strong genetic control with heritability estimates ranging from 0.85-0.93. There was a very strong photoperiod effect with flowering in the population accelerated by 765 degree-days in the long day versus short day treatments. However, there was a strong genetic correlation of line effects (0.91) between the long and short day treatments and relatively low genotype x treatment interaction indicating that photoperiod had a similar effect across the population. Bivariate analysis of thermal time to flowering in short and long days revealed three main effect quantitative trait loci (QTLs) that accounted for 57.7% of the variation in the population and no significant interaction QTLs. These results provided insight into the contrasting adaptations of Australian and European varieties. Both parents responded to photoperiod and their alleles shifted the population to earlier flowering under long days. In addition, segregation of QTLs in the population caused wide transgressive segregation in thermal time to flowering. Potential candidate flowering time homologues located near QTLs were identified with the aid of the Brassica rapa reference genome sequence. We discuss how these results will help to guide the breeding of summer annual types of B. napus adapted to new and changing environments.
URLPMID:26419915 [本文引用: 1]
DOI:10.1007/s00122-006-0324-0URL [本文引用: 1]
Unadapted germplasm may contain alleles that could improve hybrid cultivars of spring oilseed Brassica napus. Quantitative trait loci (QTL) mapping was used to identify potentially useful alleles from two unadapted germplasm sources, a Chinese winter cultivar and a re-synthesized B. napus, that increase seed yield when introgressed into a B. napus spring hybrid combination. Two populations of 160 doubled haploid (DH) lines were created from crosses between the unadapted germplasm source and a genetically engineered male-fertility restorer line (P1804). A genetically engineered male-sterile tester line was used to create hybrids with each DH line (testcrosses). The two DH line populations were evaluated in two environments and the two testcross populations were evaluated in three or four environments for seed yield and other agronomic traits. Several genomic regions were found in the two testcross populations which contained QTL for seed yield. The map positions of QTL for days to flowering and resistance to a bacterial leaf blight disease coincided with QTL for seed yield and other agronomic traits, suggesting the occurrence of pleiotropic or linked effects. For two hybrid seed yield QTL, the favorable alleles increasing seed yield originated from the unadapted parents, and one of these QTL was detected in multiple environments and in both populations. In this QTL region, a chromosome rearrangement was identified in P1804, which may have affected seed yield.]]>
DOI:10.1186/1471-2148-9-271URLPMID:19939256 [本文引用: 1]
BACKGROUND: The gene FLOWERING LOCUS T (FT) and its orthologues play a central role in the integration of flowering signals within Arabidopsis and other diverse species. Multiple copies of FT, with different cis-intronic sequence, exist and appear to operate harmoniously within polyploid crop species such as Brassica napus (AACC), a member of the same plant family as Arabidopsis. RESULTS: We have identified six BnFT paralogues from the genome of B. napus and mapped them to six distinct regions, each of which is homologous to a common ancestral block (E) of Arabidopsis chromosome 1. Four of the six regions were present within inverted duplicated regions of chromosomes A7 and C6. The coding sequences of BnFT paralogues showed 92-99% identities to each other and 85-87% identity with that of Arabidopsis. However, two of the paralogues on chromosomes A2 and C2, BnA2.FT and BnC2.FT, were found to lack the distinctive CArG box that is located within intron 1 that has been shown in Arabidopsis to be the binding site for theFLC protein. Three BnFT paralogues (BnA2.FT, BnC6.FT.a and BnC6.FT.b) were associated with two major QTL clusters for flowering time. One of the QTLs encompassing two BnFT paralogues (BnC6.FT.a and BnC6.FT.b) on chromosome C6 was resolved further using near isogenic lines, specific alleles of which were both shown to promote flowering. Association analysis of the three BnFT paralogues across 55 cultivars of B. napus showed that the alleles detected in the original parents of the mapping population used to detect QTL (NY7 and Tapidor) were ubiquitous amongst spring and winter type cultivars of rapeseed. It was inferred that the ancestral FT homologues in Brassica evolved from two distinct copies, one of which was duplicated along with inversion of the associated chromosomal segment prior to the divergence of B. rapa (AA) and B. oleracea (CC). At least ten such inverted duplicated blocks (IDBs) were identified covering a quarter of the whole B. napus genome. CONCLUSION: Six orthologues of Arabidopsis FT were identified and mapped in the genome of B. napus which sheds new light on the evolution of paralogues in polyploidy species. The allelic variation of BnFT paralogues results in functional differences affecting flowering time between winter and spring type cultivars of oil seed Brassica. The prevalent inverted duplicated blocks, two of which were located by four of the six BnFT paralogues, contributed to gene duplications and might represent predominant pathway of evolution in Brassica.
URLPMID:20498060 [本文引用: 2]
DOI:10.1139/g91-084URL [本文引用: 1]
DOI:10.1038/srep14481URLPMID:26434411 [本文引用: 1]
To facilitate the pseudochromosomes assembly and gene cloning in rapeseed, we developed a reference genetic population/map (named BnaZNF2) from two sequenced cultivars, Zhongshuang11 and No.73290, those exhibit significant differences in many traits, particularly yield components. The BnaZNF2 genetic map exhibited perfect collinearity with the physical map of B. napus, indicating its high quality. Comparative mapping revealed several genomic rearrangements between B. napus and B. rapa or B. oleracea. A total of eight and 16 QTLs were identified for pod number and seed number per pod, respectively, and of which three and five QTLs are identical to previously identified ones, whereas the other five and 11 are novel. Two new major QTL respectively for pod number and seed number per pod, qPN.A06-1 and qSN.A06-1 (R(2 )= 22.8% and 32.1%), were colocalised with opposite effects, and only qPN.A06-1 was confirmed and narrowed by regional association analysis to 180 kb including only 33 annotated genes. Conditional QTL analysis and subsequent NILs test indicated that tight linkage, rather than pleiotropy, was the genetic causation of their colocalisation. Our study demonstrates potential of this reference genetic population/map for precise QTL mapping and as a base for positional gene cloning in rapeseed.
DOI:10.1371/journal.pone.0083052URLPMID:24386142 [本文引用: 1]
A high density genetic linkage map for the complex allotetraploid crop species Brassica napus (oilseed rape) was constructed in a late-generation recombinant inbred line (RIL) population, using genome-wide single nucleotide polymorphism (SNP) markers assayed by the Brassica 60 K Infinium BeadChip Array. The linkage map contains 9164 SNP markers covering 1832.9 cM. 1232 bins account for 7648 of the markers. A subset of 2795 SNP markers, with an average distance of 0.66 cM between adjacent markers, was applied for QTL mapping of seed colour and the cell wall fiber components acid detergent lignin (ADL), cellulose and hemicellulose. After phenotypic analyses across four different environments a total of 11 QTL were detected for seed colour and fiber traits. The high-density map considerably improved QTL resolution compared to the previous low-density maps. A previously identified major QTL with very high effects on seed colour and ADL was pinpointed to a narrow genome interval on chromosome A09, while a minor QTL explaining 8.1% to 14.1% of variation for ADL was detected on chromosome C05. Five and three QTL accounting for 4.7% to 21.9% and 7.3% to 16.9% of the phenotypic variation for cellulose and hemicellulose, respectively, were also detected. To our knowledge this is the first description of QTL for seed cellulose and hemicellulose in B. napus, representing interesting new targets for improving oil content. The high density SNP genetic map enables navigation from interesting B. napus QTL to Brassica genome sequences, giving useful new information for understanding the genetics of key seed quality traits in rapeseed.
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00018-011-0673-yURL [本文引用: 1]
Plants undergo a major physiological change as they transition from vegetative growth to reproductive development. This transition is a result of responses to various endogenous and exogenous signals that later integrate to result in flowering. Five genetically defined pathways have been identified that control flowering. The vernalization pathway refers to the acceleration of flowering on exposure to a long period of cold. The photoperiod pathway refers to regulation of flowering in response to day length and quality of light perceived. The gibberellin pathway refers to the requirement of gibberellic acid for normal flowering patterns. The autonomous pathway refers to endogenous regulators that are independent of the photoperiod and gibberellin pathways. Most recently, an endogenous pathway that adds plant age to the control of flowering time has been described. The molecular mechanisms of these pathways have been studied extensively in Arabidopsis thaliana and several other flowering plants.
URLPMID:22992955 [本文引用: 1]
DOI:10.1016/j.cell.2010.04.024URLPMID:20434991 [本文引用: 1]
URLPMID:17908925 [本文引用: 1]
DOI:10.1126/science.1072147URLPMID:12114624 [本文引用: 1]
Arabidopsis VRN genes mediate vernalization, the process by which a long period of cold induces a mitotically stable state that leads to accelerated flowering during later development. VRN1 encodes a protein that binds DNA in vitro in a non-sequence-specific manner and functions in stable repression of the major target of the vernalization pathway, the floral repressor FLC. Overexpression of VRN1 reveals a vernalization-independent function for VRN1, mediated predominantly through the floral pathway integrator FT, and demonstrates that VRN1 requires vernalization-specific factors to target FLC.
DOI:10.1186/1471-2229-12-238URLPMID:23241244 [本文引用: 1]
BACKGROUND: Rapeseed (Brassica napus L.) has spring and winter genotypes adapted to different growing seasons. Winter genotypes do not flower before the onset of winter, thus leading to a longer vegetative growth period that promotes the accumulation and allocation of more resources to seed production. The development of winter genotypes enabled the rapeseed to spread rapidly from southern to northern Europe and other temperate regions of the world. The molecular basis underlying the evolutionary transition from spring- to winter- type rapeseed is not known, however, and needs to be elucidated. RESULTS: We fine-mapped the spring environment specific quantitative trait locus (QTL) for flowering time, qFT10-4,in a doubled haploid (DH) mapping population of rapeseed derived from a cross between Tapidor (winter-type) and Ningyou7 (semi-winter) and delimited the qFT10-4 to an 80-kb region on chromosome A10 of B. napus. The BnFLC.A10 gene, an ortholog of FLOWERING LOCUS C (FLC) in Arabidopsis, was cloned from the QTL. We identified 12 polymorphic sites between BnFLC.A10 parental alleles of the TN-DH population in the upstream region and in intron 1. Expression of both BnFLC.A10 alleles decreased during vernalization, but decreased more slowly in the winter parent Tapidor. Haplotyping and association analysis showed that one of the polymorphic sites upstream of BnFLC.A10 is strongly associated with the vernalization requirement of rapeseed (r2 = 0.93, chi2 = 0.50). This polymorphic site is derived from a Tourist-like miniature inverted-repeat transposable element (MITE) insertion/deletion in the upstream region of BnFLC.A10. The MITE sequence was not present in the BnFLC.A10 gene in spring-type rapeseed, nor in ancestral 'A' genome species B. rapa genotypes. Our results suggest that the insertion may have occurred in winter rapeseed after B. napus speciation. CONCLUSIONS: Our findings strongly suggest that (i) BnFLC.A10 is the gene underlying qFT10-4, the QTL for phenotypic diversity of flowering time in the TN-DH population, (ii) the allelic diversity caused by MITE insertion/deletion upstream of BnFLC.A10 is one of the major causes of differentiation of winter and spring genotypes in rapeseed and (iii) winter rapeseed has evolved from spring genotypes through selection pressure at the BnFLC.A10 locus, enabling expanded cultivation of rapeseed along the route of Brassica domestication.
DOI:10.1093/jxb/8.1.1URL [本文引用: 1]
[本文引用: 1]
DOI:10.1023/a:1006064514311URLPMID:9678571 [本文引用: 1]
The Arabidopsis thaliana CONSTANS (CO) gene which promotes flowering in long days was recently isolated by chromosome walking. The mapping of QTLs controlling flowering time in Brassica species has identified genomic regions that contain homologues of the CO gene. Four genes homologous to the Arabidopsis CO gene were isolated from a pair of homoeologous loci in each of two doubled-haploid Brassica napus lines displaying different flowering times, N-o-1 and N-o-9. The four genes, BnCOa1, BnCOa9, BnCOb1 and BnCOb9, are located on linkage groups N10 and N19, and are highly similar to each other and to the Arabidopsis CO gene. Two regions of the proteins are particularly well conserved, a N-terminal region with two putative zinc fingers and a C-terminal region which may contain a nuclear localization signal. All four genes appear to be expressed in B. napus. The BnCOa1 allele was shown to complement the co-2 mutation in Arabidopsis in a dosage-dependent manner causing earlier flowering than in wild type under both long- and short-day conditions.
DOI:10.1016/j.molp.2017.09.020URLPMID:29024744 [本文引用: 1]
DOI:10.1186/1471-2229-12-238URLPMID:23241244 [本文引用: 1]
BACKGROUND: Rapeseed (Brassica napus L.) has spring and winter genotypes adapted to different growing seasons. Winter genotypes do not flower before the onset of winter, thus leading to a longer vegetative growth period that promotes the accumulation and allocation of more resources to seed production. The development of winter genotypes enabled the rapeseed to spread rapidly from southern to northern Europe and other temperate regions of the world. The molecular basis underlying the evolutionary transition from spring- to winter- type rapeseed is not known, however, and needs to be elucidated. RESULTS: We fine-mapped the spring environment specific quantitative trait locus (QTL) for flowering time, qFT10-4,in a doubled haploid (DH) mapping population of rapeseed derived from a cross between Tapidor (winter-type) and Ningyou7 (semi-winter) and delimited the qFT10-4 to an 80-kb region on chromosome A10 of B. napus. The BnFLC.A10 gene, an ortholog of FLOWERING LOCUS C (FLC) in Arabidopsis, was cloned from the QTL. We identified 12 polymorphic sites between BnFLC.A10 parental alleles of the TN-DH population in the upstream region and in intron 1. Expression of both BnFLC.A10 alleles decreased during vernalization, but decreased more slowly in the winter parent Tapidor. Haplotyping and association analysis showed that one of the polymorphic sites upstream of BnFLC.A10 is strongly associated with the vernalization requirement of rapeseed (r2 = 0.93, chi2 = 0.50). This polymorphic site is derived from a Tourist-like miniature inverted-repeat transposable element (MITE) insertion/deletion in the upstream region of BnFLC.A10. The MITE sequence was not present in the BnFLC.A10 gene in spring-type rapeseed, nor in ancestral 'A' genome species B. rapa genotypes. Our results suggest that the insertion may have occurred in winter rapeseed after B. napus speciation. CONCLUSIONS: Our findings strongly suggest that (i) BnFLC.A10 is the gene underlying qFT10-4, the QTL for phenotypic diversity of flowering time in the TN-DH population, (ii) the allelic diversity caused by MITE insertion/deletion upstream of BnFLC.A10 is one of the major causes of differentiation of winter and spring genotypes in rapeseed and (iii) winter rapeseed has evolved from spring genotypes through selection pressure at the BnFLC.A10 locus, enabling expanded cultivation of rapeseed along the route of Brassica domestication.
DOI:10.1016/s0092-8674(01)00573-6URLPMID:11719192 [本文引用: 1]
The acceleration of flowering by a long period of low temperature, vernalization, is an adaptation that ensures plants overwinter before flowering. Vernalization induces a developmental state that is mitotically stable, suggesting that it may have an epigenetic basis. The VERNALIZATION2 (VRN2) gene mediates vernalization and encodes a nuclear-localized zinc finger protein with similarity to Polycomb group (PcG) proteins of plants and animals. In wild-type Arabidopsis, vernalization results in the stable reduction of the levels of the floral repressor FLC. In vrn2 mutants, FLC expression is downregulated normally in response to vernalization, but instead of remaining low, FLC mRNA levels increase when plants are returned to normal temperatures. VRN2 function therefore stably maintains FLC repression after a cold treatment, serving as a mechanism for the cellular memory of vernalization.
DOI:10.1105/tpc.109.065946URLPMID:20305124 [本文引用: 1]
The Arabidopsis thaliana MADS box transcription factors APETALA3 (AP3) and PISTILLATA (PI) heterodimerize and are required to specify petal identity, yet many details of how this regulatory process is effected are unclear. We have identified three related genes, BHLH136/BANQUO1 (BNQ1), BHLH134/BANQUO2 (BNQ2), and BHLH161/BANQUO3 (BNQ3), as being directly and negatively regulated by AP3 and PI in petals. BNQ1, BNQ2, and BNQ3 encode products belonging to a family of atypical non-DNA binding basic helix-loop-helix (bHLH) proteins that heterodimerize with and negatively regulate bHLH transcription factors. We show that bnq3 mutants have pale-green sepals and carpels and decreased chlorophyll levels, suggesting that BNQ3 has a role in regulating light responses. The ap3 bnq3 double mutant displays pale second-whorl organs, supporting the hypothesis that BNQ3 is downstream of AP3. Consistent with a role in light response, we show that the BNQ gene products regulate the function of HFR1 (for LONG HYPOCOTYL IN FAR-RED1), which encodes a bHLH protein that regulates photomorphogenesis through modulating phytochrome and cryptochrome signaling. The BNQ genes also are required for appropriate regulation of flowering time. Our results suggest that petal identity is specified in part through downregulation of BNQ-dependent photomorphogenic and developmental signaling pathways.
DOI:10.7505/j.issn.1007-9084.2013.06.007URL [本文引用: 1]
为进一步了解特大粒甘蓝型油菜种质DL01的大粒机理,以普通籽粒H8为对照,田间调查并比较分析它们的生育期、主花序角果和籽粒在形态、重量、灌浆速率和相对含水量等特征及变化。结果表明,特大籽粒DL01的平均全生育期为207d,比H8长17d,尤其是灌浆期长12d以上;在开花当天,DL01的花梗直径、子房的长度和宽度及重量分别是H8的1.30、1.21、1.39、1.46倍;其最大角果皮光合面积和角果鲜重分别是H8的2.2和3.0倍;单荚胚珠数二者相当,但DL01的着粒数比H8少,最大差异为64%;DL01最大籽粒鲜重(百粒重1.20g)、干重、体积和灌浆速率均是H8的2倍;在含水量方面,在荚角成熟前DL01的果皮含水量比H8略高(3%~5%),但H8荚果高含水量(>75%)期只能维持68d,而DL01荚果高含水量(>80%)期可维持80d;H8籽粒鲜重在47d时达到最高,其含水量为52%,DL01籽粒鲜重在62d时达到最高,其含水量为50%。因此结论是,DL01大粒源自大子房、粗花梗、大的角果皮表面积、低着粒数、高的果皮与籽粒含水量并且灌浆持续时间较长。
[本文引用: 1]
URLPMID:12953103 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}