 ,, 雷建峰, 代培红, 刘超, 李月, 刘晓东,*新疆农业大学农学院 / 棉花教育部工程研究中心, 新疆乌鲁木齐 830052
,, 雷建峰, 代培红, 刘超, 李月, 刘晓东,*新疆农业大学农学院 / 棉花教育部工程研究中心, 新疆乌鲁木齐 830052Efficient screening system of effective sgRNA for cotton CRISPR/Cas9 gene editing
ZHOU Guan-Tong,, LEI Jian-Feng, DAI Pei-Hong, LIU Chao, LI Yue, LIU Xiao-Dong,*College of Agriculture, Xinjiang Agricultural University, Engineering Research Centre of Cotton of Ministry of Education, Urumqi 830052, Xinjiang, China通讯作者:
收稿日期:2020-08-5接受日期:2020-10-14网络出版日期:2021-03-12
| 基金资助: |
Received:2020-08-5Accepted:2020-10-14Online:2021-03-12
| Fund supported: |
作者简介 About authors
E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (4263KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
周冠彤, 雷建峰, 代培红, 刘超, 李月, 刘晓东. 棉花CRISPR/Cas9基因编辑有效sgRNA高效筛选体系的研究[J]. 作物学报, 2021, 47(3): 427-437. doi:10.3724/SP.J.1006.2021.04178
ZHOU Guan-Tong, LEI Jian-Feng, DAI Pei-Hong, LIU Chao, LI Yue, LIU Xiao-Dong.
棉花是我国重要的经济作物和天然纤维来源。而棉花生产面临各种生物、非生物胁迫因子的危害, 同时生产的棉花纤维品质不高, 生产成本日益增加[1,2]。为了从根本上解决上述棉花种植中的问题, 需要棉花功能基因组学来解析各种农艺性状形成的基因调控网络[3,4,5], 并以此为理论基础, 培育出优质高产抗病抗逆的棉花新品种。目前栽培棉花的全基因组测序已经完成, 然而对每个基因的功能解析才刚刚开始。要研究棉花基因的功能, 解析农艺性状形成的基因调控网络, 获得相应基因突变的棉花遗传材料是关键前提。CRISPR/Cas9 基因组编辑技术在棉花上的成功建立为棉花功能基因组学研究提供了强大的技术支持[6]。
基因编辑技术是一种可以针对基因组进行定向靶向修饰的技术[7], 目前主要有3种基因编辑技术, 分别为人工核酸酶介导的锌指核酸酶(zinc-finger nucleases, ZFN)技术、类转录激活效应物核酸酶(transcription activator-like effector nucleases, TALEN)技术和RNA引导的CRISPR/Cas (clustered regularly interspaced short palindromic repeats/CRISPR associated nuclease, Cas)技术[8]。CRISPR/Cas9基因组编辑技术起源于细菌长期进化过程中自身产生的一套免疫机制[9], 包括小片段单链向导RNA (single guide RNA, sgRNA)和具有靶向识别sgRNA的核酸酶Cas9 2个部分。通过带有靶向序列的sgRNA引导Cas9核酸酶在基因组特异性位点(PAM)附近进行切割, 最终由细胞启动基因组同源重组修复(HR)或非同源重组末端连接(NHEJ)导致基因组序列发生可逆或不可逆突变[10,11]。由于靶向序列可以根据基因组不同位点单独设计并且Cas9核酸酶无特异性, 因此与ZFNs和TALENs等基因组编辑技术相比, CRISPR/Cas9基因组编辑技术操作更加简便, 成本更低, 编辑效率更高, 靶向更加精准, 脱靶效应更低[12,13,14], 目前已在多种植物中成功应用[15,16,17,18,19], 在棉花功能基因组学研究中也展现出巨大的应用前景[20,21]。
sgRNA是CRISPR/Cas9基因编辑技术体系的重要元件之一。然而前人研究显示很多sgRNA并不能有效工作[22]。因此在正式运用CRISPR/Cas9基因编辑技术进行稳定转化受体植物前, 一般都需要使用瞬时转化的方法快速验证和筛选出有效的sgRNA, 而原生质体转化法和农杆菌介导注射完整的编辑载体转化植株叶片是2种最常见验证sgRNA活性的方法[23,24]。但由于农杆菌瞬时转化效率不高和Cas9基因太大(大于4000 bp), 导致瞬时转化效率低, 对试验操作者技术要求较高, 基因编辑事件不容易被检测到。
最近的研究表明, 可以利用植物病毒传递sgRNA用于植物基因的靶向编辑, 称为病毒诱导的基因编辑(VIGE)[25,26]。相比传统的转化完整编辑载体的方式, 利用植物病毒导入sgRNA具有如下优势: 首先, sgRNA能够伴随着病毒的侵染和复制快速积累, 在更多细胞里产生更多的sgRNA, 大幅提高基因编辑的检出率; 其次, VIGE可以将多个sgRNAs组装到同一个病毒基因组上实现多基因编辑。一些植物RNA病毒, 如烟草脆裂病毒(TRV)、豌豆早枯病毒(PEBV)、烟草花叶病毒(TMV)和甜菜坏死黄脉病毒(BNYVV)等被相继应用于模式植物拟南芥和烟草的靶向编辑[27,28,29,30]。Hu等[25]利用大麦条纹花叶病毒(BSMV)介导的VIGE系统, 针对六倍体小麦进行基因编辑的应用, 表明了植物RNA病毒介导的VIGE可以有效地用于农作物的基因编辑。随后, 植物DNA病毒介导的VIGE相继被开发利用[26]。然而棉花的VIGE系统还未见报道。
CLCrV是一种双组份DNA病毒由CLCrV-A和CLCrV-B 两个基因组组成。已有研究表明, CLCrV作为病毒诱导的基因沉默(VIGS)载体, 可用于沉默棉花内源基因的表达[31]。为消除Cas9基因过大对基因编辑检出效率的影响, 本研究用Cas9阳性转基因棉花为受体材料, 分别用农杆菌Ti质粒和CLCrV病毒载体投送sgRNA。结果表明, 这2种方法均能够更加高效快捷完成棉花内源基因编辑的有效sgRNA的筛选工作。为后续创制棉花新种质资源和进行棉花功能基因组学研究奠定了重要的技术基础。
1 材料与方法
1.1 供试材料
载体GhU6-5P::GUS-sgRNA[32]和GhU6-5P-2:: GUS-sgRNA由本实验室保存; Cas9基因由中国农业科学院孟志刚惠赠; 用于农杆菌注射的受体材料为上述Cas9转基因阳性棉花植株, 其遗传背景是YZ-1, 带有GFP-Cas9基因和GhALARP-sgRNA, 但该sgRNA与本文中的sgRNA碱基差异很大, 不会与本文试验投送的sgRNA发生竞争, 从而影响实验的正确性, 由石河子大学孙杰课题组惠赠[33]; 载体pClCrVA和pClCrVB由浙江大学周雪平教授惠赠, 限制性内切酶和DNA聚合酶均为NEB和Thermo公司产品, 其余试剂及药品均由国内生物公司代销, 引物合成及测序由上海杰李生物科技有限公司完成。1.2 载体构建
以YZ-1基因组DNA为模板, 克隆GhMAPKKK2 (Gh_D10G0119)、GhAE (Gh_D09: 3656310..3658852)、GhPDS (Gh_D07G1160)、GhCLA1 (Gh_D10G1640)基因部分序列用于寻找合适的靶序列, 靶序列的设计基于以下原则, 一是在四倍体棉花YZ-1中, A组和D组的基因组序列完全一致; 二是GC含量在40%~60%之间; 三是有合适的限制性内切酶位点。将靶序列单链DNA经过DNA复性反应后连接于以Bbs I酶切获得的GhU6- 5P::GUS-sgRNA和GhU6-5P-2::GUS-sgRNA线性化载体。退火反应体系为10 μmol L-1的上下游靶序列各10 μL (表1), 复性程序为: 95℃每5 s降低0.5℃至20℃。经测序验证获得上述GhU6-5P:: MAPKKK2-sgRNA、GhU6-5P::AE-sgRNA质粒后, 再将上述2种质粒以酶切连接的方式分别连接在以Kpn I、Xba I酶切获得的pCAMBIA1300表达载体和和Cas9基因表达载体上, 分别命名为GhU6-5P::MAPKKK2-sgRNA-1300、GhU6-5P::AE- sgRNA-1300和GhU6-5P::MAPKKK2-sgRNA-Cas9、GhU6-5P::AE-sgRNA-Cas9。经测序验证获得上述GhU6-5P-2::PDS-sgRNA、GhU6-5P-2::CLA1-sgRNA、GhU6-5P-2::MAPKKK2-sgRNA和GhU6-5P-2::AE- sgRNA质粒后, 再将其以酶切连接方式连接在以Pac I、Spe I酶切获得pClCrVA病毒载体上, 命名为GhU6-5P-2::PDS-sgRNA-ClCrVA、 GhU6-5P-2::CLA1- sgRNA-ClCrVA、GhU6-5P-2::MAPKKK2-sgRNA- CLCrVA和GhU6-5P-2::AE-sgRNA-CLCrVA。上述重组质粒酶切检测正确后均转化农杆菌GV3101, 命名同表达载体名称。Table 1
表1
表1本研究中使用的引物序列
Table 1
| 引物名称 Primer name | 上游引物序列 Forward sequence (5'-3') | 下游引物序列 Reverse sequence (5'-3') |
|---|---|---|
| GhU6-5P-MAPKKK2-sg | AAGGGTTCCCAGCTGACATA | TATGTCAGCTGGGAACCCTT |
| GhU6-5P-AE-sg | GAGTTTGGAGGGCTTACAAT | ATTGTAAGCCCTCCAAACTC |
| GhU6-5P-2-PDS-sg | GAAGCGAGAGATGTTCTAGG | CCTAGAACATCTCTCGCTTC |
| GhU6-5P-2-CLA1-sg | TATGCTCGCGGAATGATCAG | CTGATCATTCCGCGAGCATA |
| Test MAPKKK2-sg | CCATGTCGTAGCTTATAAAGG | TCATTTACCTTCTCTTCCCAG |
| Test AE-sg | AGACTTGTTTCAATGGACTC | AATAAGCTGACAGCAGTTGG |
| Test PDS-sg | TGCATGATCCATCACTCAAGTTT | GAACGAAAGGCCCTTTCTTTC |
| Test CLA1-sg | GGATCTGAAAGGTGAAAGGAATC | TACCGTGATACTTGTCAGCAGCT |
新窗口打开|下载CSV
1.3 棉花叶片的瞬时转化
将棉花种子在去离子水中浸泡过夜后28℃黑暗发芽24 h。发芽后移入土壤中12 h光照/12 h黑暗条件下培养。当棉花长出子叶或两三片真叶时, 准备进行瞬时转化试验。接种前3 d, 在含有50 μg mL-1卡那霉素和25 μg mL-1利福平的LB琼脂平板上用已转化的农杆菌的甘油储液画线, 28℃孵育24 h。第2天从上述平板中挑取每个载体的单个菌落, 并将其接种到2 mL含有50 μg mL-1卡那霉素和25 μg mL-1利福平的LB培养基中, 28℃, 180转 min-1的速度过夜培养。将上述培养物以1︰100的比例接种至30 mL含有50 μg mL-1卡那霉素、25 μg mL-1利福平、10 mmol L-1 MES和20 μmol L-1乙酰丁香酮的LB培养基的三角瓶中28℃, 180转 min-1过夜培养。将农杆菌细胞4000×g离心10 min后重悬于含有10 mmol L-1 MgCl2, 10 mmol L-1 MES和200 μmol L-1乙酰丁香酮的悬浮液中。将悬浮液的OD600调节至1.5后室温下黑暗放置3 h。用针尖刺破棉花真叶的下表皮, 使用1 mL无针头注射器将GhU6- 5P::MAPKKK2-sgRNA-1300、GhU6-5P::AE-sgRNA- 1300农杆菌注射Cas9转基因阳性植株真叶, GhU6-5P::MAPKKK2-sgRNA-Cas9、GhU6-5P::AE- sgRNA-Cas9农杆菌悬浮液注射野生型对照YZ-1棉花真叶。将pCLCrVB农杆菌分别与等量的GhU6-5P-2::PDS-sgRNA-ClCrVA农杆菌、GhU6-5P- 2::CLA1-sgRNA-ClCrVA农杆菌、GhU6-5P-2:: MAPKKK2-sgRNA-ClCrVA农杆菌和GhU6-5P-2:: AE-sgRNA-ClCrVA农杆菌悬浮液混匀后注射棉花子叶中。棉花转化后黑暗放置24 h, 12 h光照/ 12 h黑暗条件下生长1周。1.4 编辑效应的检测
对于pClCrVA病毒载体, 取上部非注射的棉花叶片, 对于其他载体取注射处的棉花叶片, 参照Trans Easypure plant Genomic DNA Kit操作要求提取基因组DNA, 将提取的DNA经过浓度检测后, 取等质量提取的DNA, 采用检测引物进行PCR扩增(表1), PCR反应体系参照NEB公司Phusion超保真DNA聚合酶推荐反应体系。将扩增产物采用靶序列上选择的检测用限制性内切酶(GhMAPKKK2采用Nde I酶切、GhAE采用Mfe I酶切、GhPDS采用Bfa I酶切、GhCLA1采用Bcl I)进行酶切处理, 结果进行琼脂糖凝胶电泳分析。采用EasyPure Quick Gel Extraction Kit (北京全式金生物)回收上述PCR扩增产物, 方法参照试剂盒说明书, 取4 μL产物与 pEASY-Blunt Zero Cloning (北京全式金生物技术有限公司)载体连接, 连接体系为25℃, 5 min, 连接产物转化大肠杆菌感受态Trans T1, 37℃过夜培养, 挑取单克隆进行菌落PCR鉴定, 采用靶序列上选择的检测用限制性内切酶(GhMAPKKK2采用Nde I酶切、GhAE采用Mfe I酶切、GhPDS采用Bfa I酶切、GhCLA1采用Bcl I)对菌落PCR产物酶切处理, 挑取PCR产物未被切开的阳性单克隆接种到含有100 μg mL-1氨苄霉素LB培养基过夜培养后提取质粒用于测序, 测序序列采用DNAstar软件进行比对分析。2 结果与分析
2.1 编辑载体构建
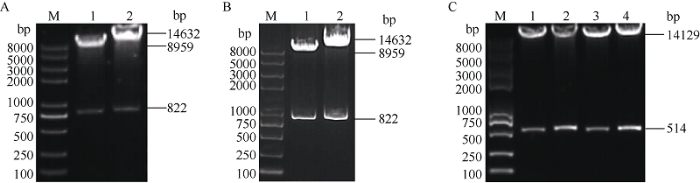
用酶切方式检测上述构建的载体, 酶切结果符合预期设计, 证明GhU6-5P::MAPKKK2-sgRNA- 1300、GhU6-5P::AE-sgRNA-1300、GhU6-5P:: MAPKKK2-sgRNA-Cas9、GhU6-5P::AE-sgRNA-Cas9编辑载体和GhU6-5P-2::PDS-sgRNA-ClCrVA、GhU6-5P-2::CLA1-sgRNA-ClCrVA、GhU6-5P-2:: MAPKKK2-sgRNA-ClCrVA、GhU6-5P-2::AE-sgRNA- ClCrVA病毒载体均构建成功(图1)。14,632 bp条带为Cas9骨架载体大小, 8959 bp条带为p1300骨架载体大小, 14,129 bp为pClCrVA骨架载体大小, 822 bp为GhU6-5P::MAPKKK2-sgRNA和GhU6-5P::AE- sgRNA片段大小, 514 bp为GhU6-5P-2::PDS- sgRNA、GhU6-5P-2::CLA1-sgRNA、GhU6-5P-2:: MAPKKK2-sgRNA和GhU6-5P-2::AE-sgRNA大小。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1基因编辑载体的酶切鉴定
A: 1、2分别为GhU6-5P::MAPKKK2-sgRNA-1300和GhU6-5P::MAPKKK2-sgRNA-Cas9. B: 1、2分别为GhU6-5P::AE-sgRNA-1300和GhU6-5P::AE-sgRNA-Cas9. C: 1~4分别为GhU6-5P-2::PDS-sgRNA-ClCrVA、GhU6-5P-2::CLA1-sgRNA-ClCrVA GhU6-5P-2::MAPKKK2- sgRNA-ClCrVA和GhU6-5P-2::AE-sgRNA-ClCrVA. M: 2K plus II DNA marker。
Fig. 1Identification of gene editing vector by restriction enzyme digestion
A: 1, 2 are GhU6-5P::MAPKKK2-sgRNA-1300 and GhU6-5P::MAPKKK2-sgRNA-Cas9. B: 1, 2 are GhU6-5P::AE-sgRNA-1300 and GhU6-5P:: AE-sgRNA-Cas9. C: 1-4 are GhU6-5P-2::PDS-sgRNA-ClCrVA , GhU6-5P-2::CLA1-sgRNA-ClCrVA, GhU6-5P-2::MAPKKK2-sgRNA- ClCrVA and GhU6-5P-2::AE-sgRNA-ClCrVA. M: 2K plus II DNA marker.
2.2 CRISPR/Cas9基因组编辑效应检测
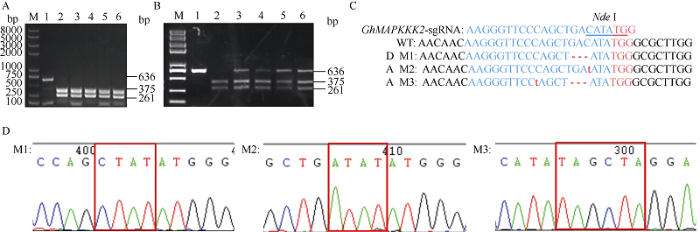
GhU6-5P::MAPKKK2-sgRNA-Cas9编辑载体转化4株YZ-1野生型植株, 转化空载体的YZ-1野生型植株作为对照。GhU6-5P::MAPKKK2-sgRNA-1300编辑载体转化4株YZ-1 Cas9转基因阳性植株, 转化空载体的YZ-1 Cas9转基因阳性植株作为对照。对以上转化获得的棉花叶片基因组DNA进行先PCR扩增后酶切的处理。对于GhMAPKKK2基因, 在转化空载体情况下, 会得到1条完整的条带(636 bp), 用Nde I消化扩增产物产生375 bp和261 bp的2个片段, 而转化GhU6-5P::MAPKKK2-sgRNA-Cas9编辑载体的4株样品中也无完整的未被切开的条带(图2-A), 表明转化GhU6-5P::MAPKKK2-sgRNA-Cas9编辑载体的植株未检测到基因编辑事件, 目标基因靶序列酶切位点处未检测到突变, 琼脂糖凝胶电泳也显示2条条带。然而与此同时转化GhU6-5P:: MAPKKK2-sgRNA-1300载体的样品中却出现完整的未被切开的条带(图2-B), 表明转化GhU6-5P:: MAPKKK2-sgRNA-1300编辑载体的叶片内发生了编辑事件, 目标基因靶序列酶切位点处出现突变, 导致限制性内切酶不能完全切割PCR的产物, 琼脂糖凝胶电泳出现3条带, 636 bp条带明亮, 表明MAPKKK2-sgRNA编辑效率较高。混合回收转化GhU6-5P::MAPKKK2-sgRNA-1300载体的样品残留的636bp条带并克隆测序发现, 在GhMAPKKK靶位点处位于PAM位点上游限制性内切酶位点区域碱基AD组均出现不同类型的突变, 其中包括碱基替换和碱基缺失现象(图2-C和D), 表明该策略可用于验证sgRNA是否有效。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2GhU6-5P::MAPKKK2-sgRNA靶向突变的检测
A: M: 2K plus II DNA marker; 1、2分别为对照载体基因组PCR扩增产物酶切前后结果; 3~6为转化GhU6-5P::MAPKKK2-sgRNA-Cas9单株样品基因组PCR扩增产物酶切后结果。B: M: 2K plus II DNA marker; 1、2分别为对照载体基因组PCR扩增产物酶切前后结果; 3~6为转化GhU6-5P::MAPKKK2-sgRNA-1300单株样品基因组PCR扩增产物酶切后结果。C: GhMAPKKK2-sgRNA靶序列及突变PCR产物的测序。M1发生在D亚组的GhMAPKKK2, M2、M3发生在A亚组GhMAPKKK2。D: 突变PCR产物克隆的测序峰图, 红色方框指示碱基相比对照发生突变区域。PCR/RE表示PCR扩增产物酶切分析。
Fig. 2GhU6-5P::MAPKKK2-sgRNA targeted mutations
A: M: 2K plus II DNA marker; 1, 2: PCR/RE assay of negative control genome before and after the enzyme digestion; 3-6: PCR/RE assay of GhU6-5P::MAPKKK2-sgRNA-Cas9 genome of single plant sample after enzyme digestion. B: M: 2K plus II DNA marker; 1, 2: PCR/RE assay of negative control genome before and of single plant sample after the enzyme digestion; 3-6: PCR/RE assay of GhU6-5P::MAPKKK2-sgRNA-1300 genome after enzyme digestion. C: sequencing of GhMAPKKK2-sgRNA target sequence and mutant PCR products. M1 occurred at GhMAPKKK2 from D subgenome, M2, M3 occurred at GhMAPKKK2 from A subgenome. D: sequencing peak diagram of the mutant PCR product clone, the red box indicates that the base has a mutation region compared to the control. PCR/RE indicates PCR/restriction enzyme assays.
同样采用上述策略验证AE-sgRNA的有效性。GhU6-5P::AE-sgRNA-Cas9编辑载体转化10株YZ-1野生型植株, 转化空载体的YZ-1野生型植株作为对照。GhU6-5P::AE-sgRNA-1300编辑载体转化10株YZ-1 Cas9转基因阳性植株, 转化空载体的YZ-1 Cas9转基因阳性植株作为对照。对以上转化获得的棉花叶片基因组DNA进行先PCR扩增后酶切的处理, 转化空载体的试验, 得到了1条完整的条带(659 bp), 用Mfe I消化扩增产物产生436 bp和223 bp的2个片段(图3-A), 而转化GhU6-5P::AE-sgRNA-1300和GhU6-5P::AE-sgRNA-Cas9编辑载体的样品中均无完整的未被切开的条带(图3)。结果显示, 转化GhU6-5P::AE-sgRNA-Cas9、和GhU6-5P::AE-sgRNA- 1300编辑载体的植株均未检测到基因编辑事件, 表明AE-sgRNA可能无效或非常低效。那么AE-sgRNA是否确实不能用于下一步实验?
图3
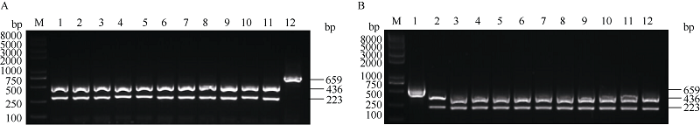 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3GhU6-5P::AE-sgRNA靶向突变的检测
A: M: 2K plus II DNA marker; 12、11分别为对照载体基因组PCR扩增产物酶切前后结果; 3~12为转化GhU6-5P::AE-sgRNA-Cas9单株样品基因组PCR扩增产物酶切后结果。B: M: 2K plus II DNA marker; 1、2分别为对照载体基因组PCR扩增产物酶切前后结果; 3~12为转化GhU6-5P::AE-sgRNA-1300单株样品基因组PCR扩增产物酶切后结果。PCR/RE表示PCR扩增产物酶切分析。
Fig. 3GhU6-5P::AE-sgRNA targeted mutations
A: M: 2K plus II DNA marker; 12, 11: PCR/RE assay of negative control genome before and after the enzyme digestion; 3-12: PCR/RE assay of GhU6-5P::AE-sgRNA-Cas9 of single plant sample after enzyme digestion. B: M: 2K plus II DNA marker; 1, 2: PCR/RE assay of negative control genome before and after the enzyme digestion; 3-12: PCR/RE assay of GhU6-5P::AE-sgRNA-1300 of single plant sample genome after enzyme digestion. PCR/RE indicates PCR/restriction enzyme assays.
近几年利用植物病毒投送sgRNA用于植物基因靶向编辑的VIGE策略, 已展示出更高效的基因编辑效率。为此我们在棉花上采用该策略测试了上述AE-sgRNA是否确实非常低效或无效。构建的GhU6-5P-2::MAPKKK2-sgRNA-ClCrVA病毒载体转化4株YZ-1 Cas9转基因阳性植株的子叶, 构建的GhU6-5P-2::AE-sgRNA-ClCrVA病毒载体转化5株YZ-1 Cas9转基因阳性植株的子叶取新生长的真叶叶片作检测, 转化空载体的YZ-1 Cas9转基因阳性植株作为对照。对以上转化获得的棉花叶片基因组DNA进行先PCR扩增后酶切的处理, 对于GhMAPKKK2基因, 转化GhU6-5P::MAPKKK2- sgRNA-CLCrV载体的样品中有残留的未被切开的条带(636 bp)(图4-B), 将残留条带混合回收后克隆, 挑取单克隆以Test MAPKKK2-sgF和Test MAPKKK2-sgR为引物进行菌液PCR后用Nde I酶切其产物, 菌液PCR产物被切开说明该单克隆为野生型, 未被切开说明该单克隆为样品基因组靶序列处发生了基因编辑事件导致基因突变(图4-C)。对于GhAE基因, 转化GhU6-5P::AE-sgRNA-CLCrV载体的样品也有残留的未被切开的条带(659 bp)(图5-B), 但残留条带相对较弱。将残留条带混合回收后克隆, 挑取单克隆以M13F和M13R为引物进行菌液PCR后用Mfe I酶切其产物, 菌液PCR产物被切开说明该单克隆为野生型, 未被切开说明该单克隆为样品基因组靶序列处发生了基因编辑事件导致基因突变(图5-C)。结果显示利用病毒载体投送sgRNA策略, 转化GhU6-5P-2::MAPKKK2-sgRNA-ClCrVA和GhU6- 5P-2::AE-sgRNA-ClCrVA病毒载体的样品均发生了基因编辑事件, 证明GhU6-5P::MAPKKK2-sgRNA和GhU6-5P::AE-sgRNA均是有效的。与投送GhU6-5P::AE-sgRNA-1300载体但并未检测到突变的结果相比, GhU6-5P-2::AE-sgRNA-ClCrVA能够明显检测到基因编辑的事件, 只是效率并不高。说明病毒载体投送sgRNA的验证策略更高效、更可靠。
图4
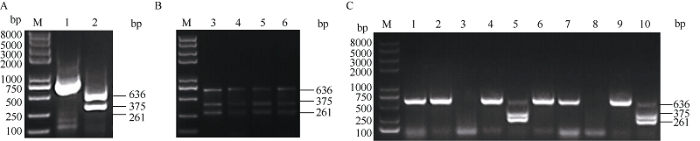 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4GhU6-5P-2::MAPKKK2-sgRNA-CLCrVA部分编辑效应测序检测结果
A: M: 2K plus II DNA marker; 1、2分别为对照载体基因组PCR扩增产物酶切前后结果。B: M: 2K plus II DNA marker; 3~6为转化GhU6-5P-2::MAPKKK2-sgRNA-CLCrVA单株样品基因组PCR扩增产物酶切后结果。C: M: 2K plus II DNA marker; 1~10为单克隆菌液PCR产物酶切后结果。PCR/RE表示PCR扩增产物酶切分析。
Fig. 4Results of GhU6-5P-2::MAPKKK2-sgRNA-CLCrVA partial editing effect sequencing
A: M: 2K plus II DNA marker; 1, 2: PCR/RE assay of negative control genome before and after the enzyme digestion. B: M: 2K plus II DNA marker; 3-6: PCR/RE assay of GhU6-5P::MAPKKK2-sgRNA-CLCrVA genome of single plant sample after enzyme digestion. C: M: 2K plus II DNA marker, 1-10: clony PCR product after enzyme digestion. PCR/RE indicates PCR/restriction enzyme assays.
图5
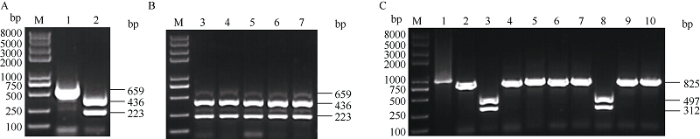 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5GhU6-5P-2::AE-sgRNA-CLCrVA部分编辑效应测序检测结果
A: M: 2K plus II DNA marker; 1、2分别为对照载体基因组PCR扩增产物酶切前后结果。B: M: 2K plus II DNA marker; 3~7为转化GhU6-5P-2::AE-sgRNA-CLCrVA单株样品基因组PCR扩增产物酶切后结果。C: M: 2K plus II DNA marker; 1~10为单克隆菌液PCR产物酶切后结果。PCR/RE表示PCR扩增产物酶切分析。
Fig. 5GhU6-5P-2::AE-sgRNA-CLCrVA partial editing effect sequencing results
A: M: 2K plus II DNA marker; 1, 2: PCR/RE assay of negative control genome before and after the enzyme digestion. B: M: 2K plus II DNA marker, 3-7: PCR/RE assay of GhU6-5P::AE-sgRNA-CLCrVA genome of single plant sample after enzyme digestion. C: M: 2K plus II DNA marker; 1-10: clony PCR product after enzyme digestion. PCR/RE indicates PCR/restriction enzyme assays.
为进一步验证基于棉花叶皱缩病毒(CLCrV)的VIGE系统验证sgRNA的可靠性, 本研究选择了前人已报道过的棉花PDS和CLA1这两个基因的有效sgRNA, 来验证该系统的可靠性[33]。GhU6-5P-2:: PDS-sgRNA-ClCrVA病毒载体转化8株YZ-1 Cas9转基因阳性植株的子叶, GhU6-5P-2::PDS-sgRNA- ClCrVA和GhU6-5P-2::CLA1-sgRNA-ClCrVA病毒载体转化3株YZ-1 Cas9转基因阳性植株的子叶, 取新生长的真叶叶片作检测, 转化空载体的YZ-1Cas9转基因阳性植株作为对照。对以上转化获得的棉花叶片基因组DNA进行先PCR扩增后酶切的处理, 对于GhPDS基因, 转化空载体, 得到了1条完整的条带(486 bp), 用Bfa I消化扩增产物产生了340 bp和146 bp的2个片段(图6-A), 转化GhU6-5P-2::PDS- sgRNA-ClCrVA病毒载体的样品中有完整的未被切开的条带(图6-B)。对于GhCLA1基因, 转化空载体得到了1条完整的条带(621 bp), 用Bcl I消化扩增产物产生417 bp和204 bp的2个片段(图7-A), 转化GhU6-5P-2::CLA1-sgRNA-ClCrVA病毒载体的样品中有完整的未被切开的条带(图7-B)。表明转化GhU6-5P-2::PDS-sgRNA-ClCrVA和GhU6-5P-2::CLA1- sgRNA-ClCrVA病毒载体的叶片内发生了基因编辑事件, 目标基因靶序列酶切位点处出现突变, 导致限制性内切酶不能切割PCR的产物, 琼脂糖凝胶电泳显示3条带。转化GhU6-5P-2::PDS-sgRNA-ClCrVA载体的486 bp条带较亮(图6-B), 表明GhU6-5P-2:: PDS-sgRNA编辑效率较高, 而转化GhU6-5P-2:: CLA1-sgRNA-ClCrVA载体的621 bp条带较弱(图7-B), 表明CLA1-sgRNA在基因编辑事件中可以工作但效率可能较低。混合回收转化GhU6-5P-2::PDS- sgRNA-ClCrVA和GhU6-5P-2:CLA1-sgRNA-ClCrVA载体的样品残留的486 bp和621 bp条带并克隆测序发现, 在GhPDS和GhCLA1靶位点处位于PAM位点上游限制性内切酶位点区域的碱基组成AD组均出现不同类型的突变, 其中包括碱基替换、碱基插入和碱基缺失现象(图6-C, D; 图7-C, D), 结果表明该策略有效并且可以在非注射部位检测到突变。采用带有sgRNA的Cas9载体和p1300载体的GhAE-sgRNA在Cas9载体和p1300载体中未能检测到突变, 而采用带有sgRNA的pCLCrV载体的策略在非注射的叶片中也能高效检测到该sgRNA发生了基因编辑的事件, 说明该策略更为高效和准确。表明用病毒载体投送sgRNA可以更加稳定、可靠地鉴定sgRNA的有效性。
图6
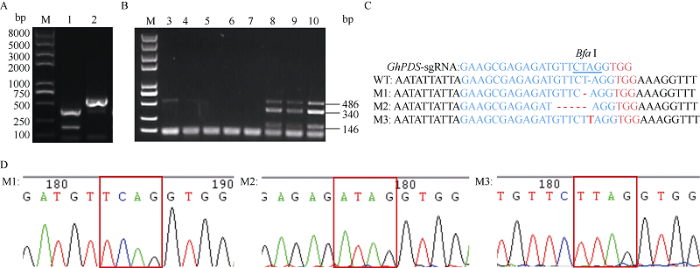 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6GhU6-5P-2::PDS-sgRNA-CLCrVA部分编辑效应测序检测结果
A: M: 2K plus II DNA marker; 1、2分别为对照载体基因组PCR扩增产物酶切后和酶切前结果。B: M: 2K plus II DNA marker; 3~5为转化GhU6-5P-2::PDS-sgRNA-CLCrVA单株样品基因组PCR扩增产物酶切后结果。C: GhPDS-sgRNA靶序列及突变PCR产物的测序。M1、M2发生在A亚组的GhPDS, M3发生在D亚组的GhPDS。D: 突变PCR产物的测序峰图, 红色方框指示碱基相比对照发生突变区域。PCR/RE表示PCR扩增产物酶切分析。
Fig. 6Results of GhU6-5P-2::PDS-sgRNA-CLCrVA partial editing effect sequencing
A: M: 2K plus II DNA marker; 1, 2: PCR/RE assay of negative control genome after and before the enzyme digestion. B: M: 2K plus II DNA marker; 3-5: PCR/RE assay of GhU6-5P-2::PDS-sgRNA-CLCrVA of single plant sample genome after enzyme digestion. C: sequencing of GhPDS-sgRNA target sequence and mutant PCR products. M1, M2 occurred at GhPDS from A subgenome; M3 occurred at GhPDS from D subgenome. D: sequencing peak diagram of the mutant PCR product; the red box indicates that the base has a mutation region compared to the control. PCR/RE indicates PCR/restriction enzyme assays.
图7
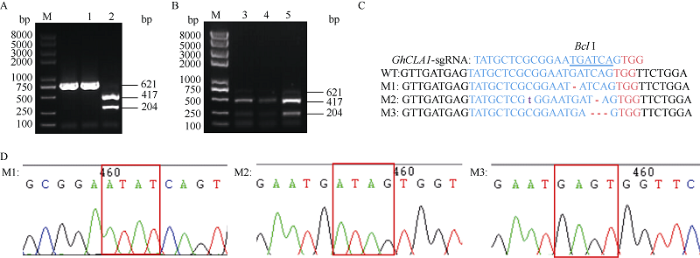 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7GhU6-5P-2::CLA1-sgRNA-CLCrVA部分编辑效应测序检测结果
A: M: 2K plus II DNA marker; 1、2分别为对照载体基因组PCR扩增产物酶切前后结果。B: M: 2K plus II DNA marker; 3~5为转化GhU6-5P-2::CLA1-sgRNA-CLCrVA单株样品基因组PCR扩增产物酶切后结果。C: GhCLA1-sgRNA靶序列及突变PCR产物的测序。M1, M3发生在D亚组的GhCLA1, M2发生在A亚组的GhCLA1。D: 突变PCR产物的测序峰图, 红色方框指示碱基相比对照发生突变区域。PCR/RE表示PCR扩增产物酶切分析。
Fig. 7Results of GhU6-5P-2::CLA1-sgRNA-CLCrVA partial editing effect sequencing
A: M: 2K plus II DNA marker; 1, 2: PCR/RE assay of negative control genome before and after the enzyme digestion. B: M: 2K plus II DNA marker, 3-5: PCR/RE assay of GhU6-5P::CLA1-sgRNA-CLCrVA genome of single plant sample after enzyme digestion. C: sequencing of GhCLA1-sgRNA target sequence and mutant PCR products. M1, M3 occurred at GhCLA1 from D subgenome; M2 occurred at GhCLA1 from A subgenome. D: sequencing peak diagram of the mutant PCR product; the red box indicates that the base has a mutation region compared to the control. PCR/RE indicates PCR/restriction enzyme assays.
在设计靶序列前为了避免植株个体差异和排除PCR扩增导致上述的碱基突变, 我们在YZ-1野生型植株对GhMAPKKK2、GhCBP、GhAE、GhPDS、GhCLA1基因进行同样的PCR扩增, 并对其扩增产物进行克隆测序。在20个阳性克隆测序结果中, 靶序列区域碱基组成无任何变化(结果略)。表明靶序列区碱基的突变确实是CRISPR/Cas9系统基因编辑的结果。
3 讨论
3.1 棉花sgRNA有效性的验证体系的研究现状
CRISPR/Cas9介导的基因组编辑技术在植物中研究已逐渐趋于成熟, 目前棉花的CRISPR/Cas9基因组编辑载体在陆地棉中的稳定转化过程存在体细胞胚胎发生困难、效率低、转化周期长等问题。由于sgRNA时常存在无效或者低效的现象因此在正式进行CRISPR/Cas9基因组编辑载体转化棉花前, 采用瞬时转化法来快速验证设计的sgRNA的有效性就显得尤为重要。早期对sgRNA有效性的验证采用的是把完整的CRISPR/Cas9基因组编辑载体瞬时转化原生质体或者叶片中[23,24]。原生质体转化法, 转化效率较高, 但实验过程费时费力费钱, 对试验操作者的试验技能要求较高, 尤其是对于原生质体制备效率比较低的棉花等材料来说更是如此[34,35]; 农杆菌介导的注射叶片法, 操作简单, 省时、省力和省钱, 但转化效率低, 很难准确验证sgRNA的有效性。本研究在前期实验的基础上, 先进行了原生质体转化, 尝试以陆地棉多种组织作为供试材料, 虽获得符合要求的原生质体, 但所有sgRNA并未检测到突变。后尝试农杆菌注射法, 将完整的带sgRNA和Cas9的载体转化农杆菌注射棉花叶片, 依然是大多数sgRNA没有检测到基因编辑事件。3.2 有效sgRNA高效验证体系的优化
采用传统方法验证sgRNA的有效性成功率较低, 不能有效检测到基因编辑事件, 但并不能由此判定sgRNA是无效的或者是低效的, 未检测到突变可能的另外一个原因是完整编辑载体的瞬时转化效率低。而这个原因与Cas9基因太大(大于4000 bp)密切相关。Cas9基因作为CRISPR/Cas9基因编辑体系的重要组成部分, 由于其基因片段很大, 会导致DNA转化效率低, 同时即使转入棉花细胞, 可能也会因为基因巨大, 进而导致瞬时表达量过低, 也会导致编辑效率低下。因此Cas9基因是解决上述问题的核心关键。基于CRISPR/Cas9基因组编辑系统的原理: 无论靶向编辑哪个基因, Cas9基因都是不用更换的。因此我们选择采用先得到Cas9转基因阳性植株,然后以此为受体材料再转化编码sgRNA载体的策略。该策略的优势在于测试的受体材料已经稳定表达了Cas9蛋白, 所需转化的载体无需Cas9基因, 只需构建编码sgRNA的投送载体即可。不仅载体小, 而且构建载体更快捷, 更省时更省事。本研究采用了2种投送sgRNA的策略发现, 对于GhMAPKKK2, 用带有sgRNA的p1300载体转化Cas9转基因阳性植株可以高效检测到突变, 而对应完整的CRISPR/Cas9基因编辑载体转化的野生型植株没有检测到; 对于GhAE基因的sgRNA, 即使单独转化sgRNA的策略也未能检测到编辑事件。但采用病毒载体投送GhAE基因的sgRNA, 可以检测到基因突变, 只是基因编辑的效率较低, 是一个不建议优先使用的sgRNA。试验结果显示与转化完整基因编辑载体的对照相比, 采用的2种投送sgRNA的策略都能有效提高基因编辑的检出率。这2个策略都能有效消除DNA转化效率低的原因, 能更准确地完成对sgRNA有效性的验证。相比较第1种策略, 本研究的第2种策略是用病毒载体去投送sgRNA。pCLCrV载体是病毒的侵染性克隆载体[31], 转化农杆菌, 注射棉花叶片后, 编码sgRNA的基因可以伴随病毒在棉花叶片中大量扩散, 大量累积, 且可以随着棉花生长扩散到新的组织中, 因此该策略可以在非注射的叶片中也能高效检测到基因编辑的事件。本研究结果显示, GhU6-5P-2:: AE-sgRNA-ClCrVA病毒载体的样品能检测到基因编辑事件, 而转化GhU6-5P-2::AE-sgRNA-1300编辑载体的样品中未检测到基因突变, 表明病毒载体投送sgRNA的策略可以更准确的验证sgRNA的有效性。
4 结论
采用带有sgRNA的pCLCrV载体和p1300载体转化Cas9转基因阳性棉花的CRISPR/Cas9基因组编辑技术体系的策略, 可以有效消除转化效率低的因素, 从而高效检测到内源基因的突变, 带有sgRNA的pCLCrV载体的策略在非注射的叶片中也能高效检测到基因编辑的事件, 且更为高效和准确。证明了该策略在棉花中用于筛选有效sgRNA的快速和高效性, 为棉花功能基因组学研究提供了重要的技术基础。证明了该策略在棉花中用于筛选有效sgRNA的快速和高效性, 为棉花功能基因组学研究提供了重要的技术基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3389/fpls.2016.01928URLPMID:28066481 [本文引用: 1]

Genome editing technologies enable precise modifications of DNA sequences in vivo and offer a great promise for harnessing plant genes in crop improvement. The precise manipulation of plant genomes relies on the induction of DNA double-strand breaks by sequence-specific nucleases (SSNs) to initiate DNA repair reactions that are based on either non-homologous end joining (NHEJ) or homology-directed repair (HDR). While complete knock-outs and loss-of-function mutations generated by NHEJ are very valuable in defining gene functions, their applications in crop improvement are somewhat limited because many agriculturally important traits are conferred by random point mutations or indels at specific loci in either the genes' encoding or promoter regions. Therefore, genome modification through SSNs-mediated HDR for gene targeting (GT) that enables either gene replacement or knock-in will provide an unprecedented ability to facilitate plant breeding by allowing introduction of precise point mutations and new gene functions, or integration of foreign genes at specific and desired
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:24906146 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/pbi.12468URLPMID:26360626 [本文引用: 1]
The Streptococcus-derived CRISPR/Cas9 system is being widely used to perform targeted gene modifications in plants. This customized endonuclease system has two components, the single-guide RNA (sgRNA) for target DNA recognition and the CRISPR-associated protein 9 (Cas9) for DNA cleavage. Ubiquitously expressed CRISPR/Cas9 systems (UC) generate targeted gene modifications with high efficiency but only those produced in reproductive cells are transmitted to the next generation. We report the design and characterization of a germ-line-specific Cas9 system (GSC) for Arabidopsis gene modification in male gametocytes, constructed using a SPOROCYTELESS (SPL) genomic expression cassette. Four loci in two endogenous genes were targeted by both systems for comparative analysis. Mutations generated by the GSC system were rare in T1 plants but were abundant (30%) in the T2 generation. The vast majority (70%) of the T2 mutant population generated using the UC system were chimeras while the newly developed GSC system produced only 29% chimeras, with 70% of the T2 mutants being heterozygous. Analysis of two loci in the T2 population showed that the abundance of heritable gene mutations was 37% higher in the GSC system compared to the UC system and the level of polymorphism of the mutations was also dramatically increased with the GSC system. Two additional systems based on germ-line-specific promoters (pDD45-GT and pLAT52-GT) were also tested, and one of them was capable of generating heritable homozygous T1 mutant plants. Our results suggest that future application of the described GSC system will facilitate the screening for targeted gene modifications, especially lethal mutations in the T2 population.
DOI:10.1111/jipb.12474URL [本文引用: 1]
Streptococcus pyogenes (SpCas9) cleaves double-stranded DNA targeted by a chimeric single-guide RNA (sgRNA). For plant genome editing, Agrobacterium-mediated T-DNA transformation has been broadly used to express Cas9 proteins and sgRNAs under the control of CaMV 35S and U6/U3 promoter, respectively. We here developed a simple and high-throughput binary vector system to clone a 19−20 bp of sgRNA, which binds to the reverse complement of a target locus, in a large T-DNA binary vector containing an SpCas9 expressing cassette. Two-step cloning procedures: (1) annealing two target-specific oligonucleotides with overhangs specific to the AarI restriction enzyme site of the binary vector; and (2) ligating the annealed oligonucleotides into the two AarI sites of the vector, facilitate the high-throughput production of the positive clones. In addition, Cas9-coding sequence and U6/U3 promoter can be easily exchanged via the GatewayTM system and unique EcoRI/XhoI sites on the vector, respectively. We examined the mutation ratio and patterns when we transformed these constructs into Arabidopsis thaliana and a wild tobacco, Nicotiana attenuata. Our vector system will be useful to generate targeted large-scale knock-out lines of model as well as non-model plant.]]>
URLPMID:27349768 [本文引用: 1]
DOI:10.1073/pnas.0914991107URLPMID:20508152 [本文引用: 1]
We report here an efficient method for targeted mutagenesis of Arabidopsis genes through regulated expression of zinc finger nucleases (ZFNs)-enzymes engineered to create DNA double-strand breaks at specific target loci. ZFNs recognizing the Arabidopsis ADH1 and TT4 genes were made by Oligomerized Pool ENgineering (OPEN)-a publicly available, selection-based platform that yields high quality zinc finger arrays. The ADH1 and TT4 ZFNs were placed under control of an estrogen-inducible promoter and introduced into Arabidopsis plants by floral-dip transformation. Primary transgenic Arabidopsis seedlings induced to express the ADH1 or TT4 ZFNs exhibited somatic mutation frequencies of 7% or 16%, respectively. The induced mutations were typically insertions or deletions (1-142 bp) that were localized at the ZFN cleavage site and likely derived from imprecise repair of chromosome breaks by nonhomologous end-joining. Mutations were transmitted to the next generation for 69% of primary transgenics expressing the ADH1 ZFNs and 33% of transgenics expressing the TT4 ZFNs. Furthermore, approximately 20% of the mutant-producing plants were homozygous for mutations at ADH1 or TT4, indicating that both alleles were disrupted. ADH1 and TT4 were chosen as targets for this study because of their selectable or screenable phenotypes (adh1, allyl alcohol resistance; tt4, lack of anthocyanins in the seed coat). However, the high frequency of observed ZFN-induced mutagenesis suggests that targeted mutations can readily be recovered by simply screening progeny of primary transgenic plants by PCR and DNA sequencing. Taken together, our results suggest that it should now be possible to obtain mutations in any Arabidopsis target gene regardless of its mutant phenotype.
DOI:10.1038/nature07992URLPMID:19404259 [本文引用: 1]
Agricultural biotechnology is limited by the inefficiencies of conventional random mutagenesis and transgenesis. Because targeted genome modification in plants has been intractable, plant trait engineering remains a laborious, time-consuming and unpredictable undertaking. Here we report a broadly applicable, versatile solution to this problem: the use of designed zinc-finger nucleases (ZFNs) that induce a double-stranded break at their target locus. We describe the use of ZFNs to modify endogenous loci in plants of the crop species Zea mays. We show that simultaneous expression of ZFNs and delivery of a simple heterologous donor molecule leads to precise targeted addition of an herbicide-tolerance gene at the intended locus in a significant number of isolated events. ZFN-modified maize plants faithfully transmit these genetic changes to the next generation. Insertional disruption of one target locus, IPK1, results in both herbicide tolerance and the expected alteration of the inositol phosphate profile in developing seeds. ZFNs can be used in any plant species amenable to DNA delivery; our results therefore establish a new strategy for plant genetic manipulation in basic science and agricultural applications.
URLPMID:19404258 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1534/genetics.115.175166URLPMID:25695951 [本文引用: 1]
Success with genome editing by the RNA-programmed nuclease Cas9 has been limited by the inability to predict effective guide RNAs and DNA target sites. Not all guide RNAs have been successful, and even those that were, varied widely in their efficacy. Here we describe and validate a strategy for Caenorhabditis elegans that reliably achieved a high frequency of genome editing for all targets tested in vivo. The key innovation was to design guide RNAs with a GG motif at the 3' end of their target-specific sequences. All guides designed using this simple principle induced a high frequency of targeted mutagenesis via nonhomologous end joining (NHEJ) and a high frequency of precise DNA integration from exogenous DNA templates via homology-directed repair (HDR). Related guide RNAs having the GG motif shifted by only three nucleotides showed severely reduced or no genome editing. We also combined the 3' GG guide improvement with a co-CRISPR/co-conversion approach. For this co-conversion scheme, animals were only screened for genome editing at designated targets if they exhibited a dominant phenotype caused by Cas9-dependent editing of an unrelated target. Combining the two strategies further enhanced the ease of mutant recovery, thereby providing a powerful means to obtain desired genetic changes in an otherwise unaltered genome.
URLPMID:28287154 [本文引用: 2]
[本文引用: 2]
URLPMID:31273916 [本文引用: 2]
URLPMID:26450012 [本文引用: 2]
DOI:10.1016/j.molp.2015.02.011URLPMID:25749112 [本文引用: 1]
URLPMID:29051052 [本文引用: 1]
DOI:10.1104/pp.17.00411URLPMID:28663331 [本文引用: 1]
Development of CRISPR/Cas9 transient gene editing screening tools in plant biology has been hindered by difficulty of delivering high quantities of biologically active single guide RNAs (sgRNAs). Furthermore, it has been largely accepted that in vivo generated sgRNAs need to be devoid of extraneous nucleotides, which has limited sgRNA expression by delivery vectors. Here, we increased cellular concentrations of sgRNA by transiently delivering sgRNAs using a Tobacco mosaic virus-derived vector (TRBO) designed with 5' and 3' sgRNA proximal nucleotide-processing capabilities. To demonstrate proof-of-principle, we used the TRBO-sgRNA delivery platform to target GFP in Nicotiana benthamiana (16c) plants, and gene editing was accompanied by loss of GFP expression. Surprisingly, indel (insertions and deletions) percentages averaged nearly 70% within 7 d postinoculation using the TRBO-sgRNA constructs, which retained 5' nucleotide overhangs. In contrast, and in accordance with current models, in vitro Cas9 cleavage assays only edited DNA when 5' sgRNA nucleotide overhangs were removed, suggesting a novel processing mechanism is occurring in planta. Since the Cas9/TRBO-sgRNA platform demonstrated sgRNA flexibility, we targeted the N. benthamiana NbAGO1 paralogs with one sgRNA and also multiplexed two sgRNAs using a single TRBO construct, resulting in indels in three genes. TRBO-mediated expression of an RNA transcript consisting of an sgRNA adjoining a GFP protein coding region produced indels and viral-based GFP overexpression. In conclusion, multiplexed delivery of sgRNAs using the TRBO system offers flexibility for gene expression and editing and uncovered novel aspects of CRISPR/Cas9 biology.
URLPMID:30565826 [本文引用: 1]
URLPMID:24521483 [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}