 ,1,4,*, 陈选阳,1,2,5,*
,1,4,*, 陈选阳,1,2,5,*Discovery and analysis of NBS-LRR gene family in sweet potato genome
HUANG Xiao-Fang1,2,**, BI Chu-Yun1,2,**, SHI Yuan-Yuan2, HU Yun-Zhuo3, ZHOU Li-Xiang4, LIANG Cai-Xiao4, HUANG Bi-Fang4, XU Ming1,2, LIN Shi-Qiang,1,4,*, CHEN Xuan-Yang,1,2,5,*通讯作者:
收稿日期:2019-11-1接受日期:2020-04-15网络出版日期:2020-08-12
| 基金资助: |
Received:2019-11-1Accepted:2020-04-15Online:2020-08-12
| Fund supported: |
作者简介 About authors
黄小芳,E-mail:1102718600@qq.com,Tel:059183789483。
毕楚韵,E-mail:494028227@qq.com,Tel:059183789483.。

摘要
关键词:
Abstract
Keywords:
PDF (1379KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
黄小芳, 毕楚韵, 石媛媛, 胡韵卓, 周丽香, 梁才晓, 黄碧芳, 许明, 林世强, 陈选阳. 甘薯基因组NBS-LRR类抗病家族基因挖掘与分析[J]. 作物学报, 2020, 46(8): 1195-1207. doi:10.3724/SP.J.1006.2020.94163
HUANG Xiao-Fang, BI Chu-Yun, SHI Yuan-Yuan, HU Yun-Zhuo, ZHOU Li-Xiang, LIANG Cai-Xiao, HUANG Bi-Fang, XU Ming, LIN Shi-Qiang, CHEN Xuan-Yang.
在植物育种过程中, 通常利用抗病R基因(resistance gene)来控制植物病害[1,2,3]。R基因编码的抗性蛋白通过直接或间接的方式识别对应的由病原微生物无毒基因Avr (Avirulence gene)编码的无毒蛋白后, 启动效应因子触发的免疫系统(effector-triggered immunity, ETI), 激活植物体内抗病信号途径, 在侵染部位产生局部的细胞和组织过敏性坏死(hypersensitive response, HR)反应, 最终抵御病原菌的侵染及进一步扩散[4,5]。
目前克隆的抗病R基因大多数都属于NBS-LRR家族[6]。这类基因家族根据其N-末端是否含有TIR结构可分为2个亚类, 一个亚类为TNL型, 含有TIR-NBS-LRR结构, 这个亚类R基因编码的蛋白N-末端有一个白细胞介素受体(toll/interleukin-1 receptor, TIR)的同源结构域; 另一个亚类为非TIR- NBS-LRR (non-TNL)结构, 这类R基因编码的蛋白N-末端不含有TIR结构, 但通常被编码的CC (Coiled-Coil)替代, 称为CC-NBS-LRR(CNL)型[7]。
我国是世界上甘薯种植面积最大的国家, 甘薯总产量稳居世界首位。据联合国粮食及农业组织FAO (
2017年中国科学院上海辰山植物科学研究中心和植物生理生态研究所, 联合德国马克斯普朗克分子遗传研究所和分子植物生理研究所, 利用Illumina测序技术完成对甘薯全基因组序列测序[12], 为甘薯的基础和应用研究建立了扎实的基础。本研究通过生物信息学方法对甘薯全基因组序列进行了CDS (Coding Domain Sequence)区预测, 检索了NBS- LRR家族基因, 并对其进行染色体定位、分类、结构分析和系统进化树研究, 以期为甘薯抗病机制研究及甘薯抗病遗传育种提供科学参考。
1 材料与方法
1.1 甘薯全基因组序列
从NCBI (1.2 snap基因注释
通过snap程序应用拟南芥(At.hmm)、线虫(Ce.hmm)、水稻(Os.hmm)的HMM模型对甘薯染色体序列CDS进行检索[13], 分别得到3个.zff文件, 分别从中随机抽取500组合并, 作为训练甘薯特异HMM模型的序列文件。将甘薯特异的HMM模型文件(Ib.hmm)作为检索工具, 通过snap程序检索预测甘薯染色体CDS, 得到甘薯全基因组蛋白质序列。1.3 含NB-ARC结构域基因预测
从Pfam网站[14,15](1.4 NBS关联的保守结构域分析分类
NBS-LRR家族基因在其N-末端含有CC、TIR[7]、RPW8[27]等结构域, 在C-末端通常含有LRR结构域[28]。从Pfam数据库[14-15] (1.5 甘薯NBS-LRR基因染色体定位
统计甘薯15条染色体上NBS-LRR家族基因的位置信息, 参考Jupe等[31]的方法绘制NBS-LRR家族基因在染色体上的位置。1.6 甘薯CNL、TNL、RPW8亚家族motif分析
将甘薯NBS-LRR家族基因分类整理后, 从中筛选出CC-NBS-LRR、TIR-NBS-LRR、RPW8-NBS三种NBS亚家族类型的90个氨基酸序列[32], 通过MEME程序[33]进行motif分析和识别(设置搜索功能域数为15)。将MEME程序运行后得到的.xml文件利用TBtools软件[34]处理。1.7 甘薯CNL、TNL、RPW8亚家族进化树构建
将筛选得到的CC-NBS-LRR、TIR-NBS-LRR、RPW8-NBS三种NBS-LRR亚家族类型的90个氨基酸序列利用Clustal Omega程序[21]进行多序列比对后, 使用Gblocks[35,36]提取保守序列(允许序列保留50%的缺口数), 进一步利用Jalview[37,38]手动矫正提取到的保守序列, 保存为.fa序列文件。根据比对结果为基础, 应用MEGA X[39]软件, 选择Maximum Likehood法, 设置运行参数模式WAG with Freqs.(+F)model, 校验参数Bootstrap=500, 生成甘薯NBS-LRR基因家族系统进化树。2 结果与分析
2.1 甘薯基因组中含有NB-ARC结构域的基因鉴别及其分类
采用snap程序对甘薯全基因组序列进行CDS区识别预测, 得到甘薯15条染色体CDS序列178,458个。利用hmmsearch程序筛选甘薯基因组中含有NB-ARC结构域的序列, 以E-value≤0.01为标准, 获得相关蛋白序列432个。从中选取高特异蛋白质序列(E-value≤1E-60)构建甘薯特异的NB-ARC HMM模型, 利用甘薯特异的HMM模型再次预测甘薯全基因组序列的NB-ARC保守结构域, 获得甘薯含有NB-ARC结构域的特异蛋白序列735个(E-value≤0.01)。去除氨基酸长度小于200以及结构域缺失严重的序列, 最终得到379个甘薯NBS-LRR家族基因, 占甘薯全基因组基因数目(178,458)的0.212%。根据TIR/RPW8/LRR结构域和CC结构域分为N (NBS)、NL (NBS-LRR)、CNL (CC-NBS- LRR)、TNL (TIL-NBS-LRR)和PN (RPW8-NBS) 4个亚家族类型(表1), 其中N型120个, NL型103个, CNL型133个, TNL型22个, PN型1个。分析发现, NBS/LRR结构域重复数和位置存在差异, 在序列结构域读取文件中含有RX-CC_like结构域, 此结构域为马铃薯抗X病毒和类似蛋白的Coiled-coil结构域(Coiled coil domain of the potato virus X resistance protein and similar proteins)[40], 记为Cx结构域, 以区分Pepcoil程序得到的CC结构域。因此, 根据NBS/LRR结构域重复数和位置差异、CC结构域类型进一步分为34个小类(表1)。Table 1
表1
表1甘薯编码NBS-LRR蛋白的基因数量及其分类
Table 1
| 项目 Item | 预测结构域 Predicted domain | 代码 Code | 甘薯 Ib | 木薯a Me a | 水稻b Os b | 拟南芥c At c | 乌拉尔图小麦d Tu d | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N类型N type | NB | N | 118 | 14 | 45 | 1 | 270 | |||||||
| NCCC | 1 | — | — | — | — | |||||||||
| NB NB | NN | 1 | — | — | — | — | ||||||||
| NL类型NL type | NB LRR | NL | 81 | 52 | 301 | 6 | 31 | |||||||
| NLN | 1 | — | — | — | — | |||||||||
| 项目 Item | 预测结构域 Predicted domain | 代码 Code | 甘薯 Ib | 木薯a Me a | 水稻b Os b | 拟南芥c At c | 乌拉尔图小麦d Tu d | |||||||
| NLC | 1 | — | — | — | — | |||||||||
| NB LRR LRR | NLL | 16 | — | — | — | 19 | ||||||||
| NLLC | 1 | — | — | — | — | |||||||||
| NB LRR LRR LRR | NLLL | 1 | — | — | — | 1 | ||||||||
| NB NB LRR | NNL | 2 | — | 3 | — | 1 | ||||||||
| CNL类型CNL type | CC NB | CN | 22 | 11 | 7 | 5 | 55 | |||||||
| CXN | 29 | — | — | — | — | |||||||||
| CC NB LRR | CNL | 15 | 117 | 175 | 51 | 84 | ||||||||
| CXNL | 37 | — | — | — | — | |||||||||
| CNCL | 1 | — | — | — | — | |||||||||
| CNLC | 1 | — | — | — | — | |||||||||
| CXNLC | 1 | — | — | — | — | |||||||||
| LCXNL | 1 | — | — | — | — | |||||||||
| CC NB LRR LRR | CNLL | 4 | — | — | — | 8 | ||||||||
| CXNLL | 5 | — | — | — | — | |||||||||
| CC NB LRR LRR LRR | CXNLLL | 1 | — | — | — | 1 | ||||||||
| CC NB NB LRR LRR | CNNLL | 1 | — | — | — | — | ||||||||
| CC CC NB | CXCN | 4 | — | — | — | — | ||||||||
| CXCXN | 1 | — | — | — | — | |||||||||
| CC CC NB LRR | CCNL | 2 | — | — | — | — | ||||||||
| CXCNL | 6 | — | — | — | — | |||||||||
| CC CC NB LRR LRR | CXCNLL | 1 | — | — | — | — | ||||||||
| CC CC CC CC NB LRR LRR | CCCCNLL | 1 | — | — | — | — | ||||||||
| TNL类型TNL type | TIR NB | TN | 9 | 5 | 3 | 23 | 0 | |||||||
| TIR NB NB | TNN | 1 | — | — | — | — | ||||||||
| TIR NB LRR | TNL | 7 | 29 | — | 88 | 0 | ||||||||
| TIR NB LRR LRR | TNLL | 3 | — | — | — | — | ||||||||
| TIR NB LRR LRR LRR | TNLLL | 2 | — | — | — | — | ||||||||
| PN类型PN type | RPW8 NB | PN | 1 | — | — | — | — | |||||||
| 其他Other | — | 99 | 1 | 33 | 15 | |||||||||
| 总数Total | 379 | 327 | 535 | 207 | 485 | |||||||||
| 基因组中基因总数 Total number of genes in the genome | 178458 | 30666 | 37544 | 25498 | 32265 | |||||||||
| 基因组大小Genome size (Mb) | 633.42 | ~760 | 389 | 125 | 3747.05 | |||||||||
新窗口打开|下载CSV
在NBS-LRR家族基因的N末端区域含有TIR (Toll/inter-leukin-1 receptor)保守结构域的蛋白称为TNL 蛋白, 其他的称为非TNL蛋白。大部分非TNL蛋白的N末端有卷曲螺旋结构(Coiled-coil), 称为CNL蛋白[41]。目前已进行NBS-LRR家族基因信息分析的植物有木薯[42]、水稻[43]、拟南芥[44]、乌拉尔图小麦[45]等, 其中CNL亚家族数目占其NBS家族基因数比例分别为木薯(35.78%)[42]、水稻(32.71%)[43]、拟南芥(24.64%)[44]、乌拉尔图小麦(19.18%)[45]。在甘薯NBS-LRR家族基因序列结构域分类中, 同时含有CC、NBS、LRR结构域的序列有77个, 占NBS-LRR家族基因总数的20.32%; 同时含有TIR、NBS、LRR结构域的序列有10个, 占总基因家族信息数2.64%。
2.2 甘薯NBS-LRR家族基因在染色体上的分布
NBS-LRR家族基因有不同的亚家族, 包括N型(黑色)、NL型(红色)、CNL型(绿色)、TNL型(蓝色)、PN型(青色)(图1)。基因有正链编码(图1中染色体右侧)和负链编码(图1中染色体左侧)。NBS-LRR家族基因数量在甘薯15条染色体上分布并不均匀, 在11号染色体上仅有3个, 而在13号染色体上则有54个。在不同染色体上的基因以单基因或基因簇的形式存在。基因簇是指在200 kb的核苷酸单位中含有的基因群[46,47], 甘薯NBS-LRR家族基因中共有231个基因是以基因簇的形式存在。这231个基因分布在81个基因簇中, 占NBS-LRR家族基因总数的60.9%, 平均每个基因簇含2.9个基因。在这些基因簇中含有2个基因的基因簇数目最多, 有46个, 占总基因簇数的56.8%。其次是3~4个基因的基因簇, 有18个, 12个基因簇含有4个基因。含5个、6个和8个基因的基因簇均只有1个。含有最大基因数的基因簇有2个, 分别分布在9号和13号染色体上, 簇中含有9个基因。图1
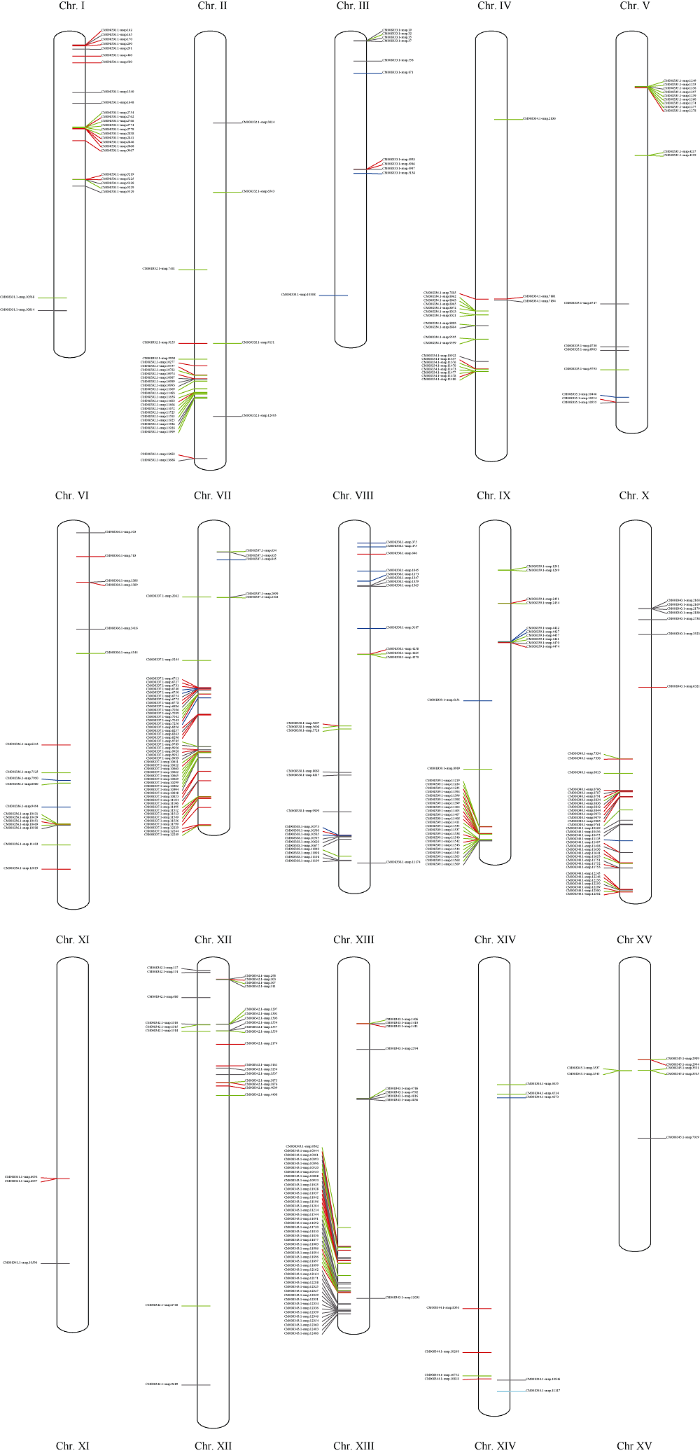 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1甘薯NBS-LRR家族基因在染色体上的分布
Fig. 1Distribution of NBS-LRR family genes on Ipomoea batatas chromosomes
甘薯基因组中, 单独分布在染色体上的NBS- LRR家族基因有148个, 占NBS-LRR基因总数39.1% (图1和表2)。在14号染色体上的9个NBS- LRR家族基因均呈单基因分布, 其中含RPW8结构域的NBS-LRR家族基因也分布于14号染色体。7号染色体基因簇最多达16个, 据此推测7号染色体可能发生NBS-LRR家族抗病基因的大规模复制, 导致该染色体上NBS-LRR家族基因的高密度分布[46]。
Table 2
表2
表2甘薯NBS-LRR基因家族基因簇统计表
Table 2
| 染色体 Chromosome | 基因 Gene | 基因簇/基因数目 Gene cluster/gene number | 最大基因簇 Maximal gene cluster | 百分比 Percentage (%) |
|---|---|---|---|---|
| 1 | 26 | 4/14 | 4 | 53.8 |
| 2 | 28 | 4/13 | 4 | 46.4 |
| 3 | 11 | 2/7 | 4 | 63.6 |
| 4 | 22 | 5/15 | 5 | 68.2 |
| 5 | 18 | 4/13 | 6 | 72.2 |
| 6 | 18 | 2/4 | 2 | 22.2 |
| 7 | 53 | 16/41 | 4 | 77.4 |
| 8 | 29 | 4/9 | 3 | 31.0 |
| 9 | 35 | 8/30 | 9 | 85.7 |
| 10 | 40 | 12/30 | 4 | 75.0 |
| 11 | 3 | 1/2 | 2 | 66.7 |
| 12 | 26 | 5/14 | 4 | 53.8 |
| 13 | 54 | 11/33 | 9 | 61.1 |
| 14 | 9 | 0/0 | 0 | 0 |
| 15 | 7 | 3/6 | 2 | 85.7 |
| 总数Total | 379 | 81/231 | 60.9 |
新窗口打开|下载CSV
2.3 甘薯NBS-LRR家族基因CNL、TNL亚家族motif分析
分别将甘薯NBS-LRR家族基因中CNL和TNL亚家族序列信息利用MEME程序检测, 筛选得到15个相似度较高的结构域。77个CNL亚家族基因的结构域分布(图2)存在以下规律: motif 10-motif 11- motif 2(P-loop)-motif 4(RNBS-A)-motif 1(Kinase 2)- motif 13(RNBS-B)-motif 5(GLPL)-motif 15-motif 9(RNBS-D)-motif 6(RNBS-D)-motif 12-motif 3(MHDV)-motif 8-motif 14-motif 7。其中motif 15(motif 5和motif 9之间)仅存在于21个序列中, 即位于GLPL结构域和RNBS-D结构域之间。其中RNBS-D保守基序对应于2个结构域, 分别为motif 9和motif 6, 共有70个序列含有motif 6, 其中有17个序列缺失motif 9。TNL亚家族结构域分布(图3)存在以下规律: motif 10(TIR-1)-motif 3-motif 1(TIR-2)-motif 5-(TIR-3)-motif 8(TIR-4)-motif 2(P- loop)-motif 9(Kinase 2)-motif 4(RNBS-B)-motif 13- (RNBS-C)-motif 7(GLPL)-motif 11-motif 15-motif6-(MHDV)-motif 12-motif 14或motif 10(TIR-1)- motif 3-motif 1(TIR-2)-motif 5-(TIR-3)-motif 8(TIR- 4)-motif 2(P-loop)-motif 9(Kinase 2)-motif 4(RNBS- B)-motif 13-(RNBS-C)-motif 7(GLPL)-motif 11-motif 6-(MHDV)-motif 12(LRR)-motif 14(LRR)-motif 15。大部分motif 15存在于motif 11和motif 6之间, 即MHDV结构域之前, 仅有3个序列motif 15位于motif 14之后。图2
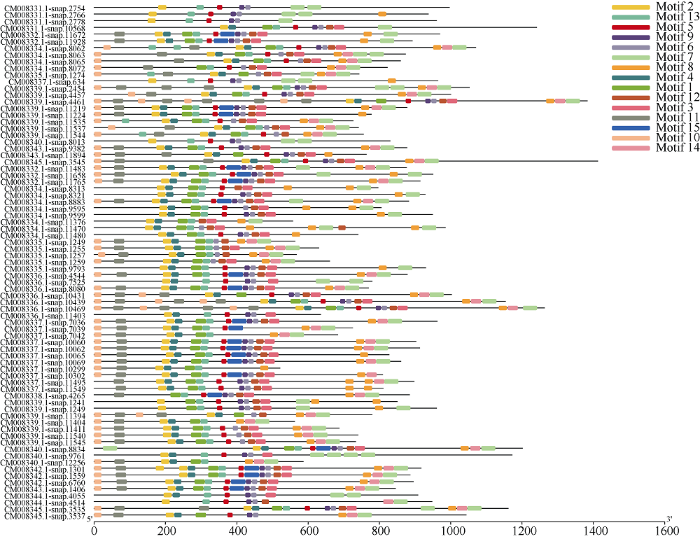 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2甘薯NBS-LRR家族中CNL亚家族保守结构域分布
Fig. 2Distribution of conservative domains of CNL subfamily of Ipomoea batatas NBS-LRR gene family
图3
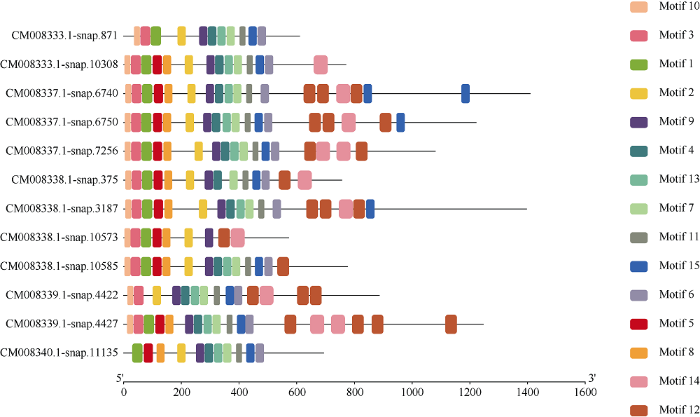 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3甘薯NBS-LRR家族中TNL亚家族保守结构域分布
Fig. 3Distribution of conservative domain of TNL subfamily of Ipomoea batatas NBS-LRR gene family
2.4 CNL、TNL亚家族NB-ARC结构域保守性分析
植物NBS-LRR家族基因中的NB-ARC区域相对保守, 通常有P-loop、RNBS-A、Kinase2、RNBS-B、RNBS-C、GLPL、RNBS-D和MHDV 8个保守基序[48]。对获得的甘薯NBS-LRR家族中CNL和TNL亚家族序列利用Clustal Omega进行多重比对发现, 具有5个保守性较高的序列结构域[44,49-50](图4), 分别为P-loop (Kinase 1)、Kinase 2、RNBS-B、GLPL、MHDV。其中P-loop最可能氨基酸序列为SIVGMGGIGKTTLA (画横线部分为氨基酸保守性较强序列), Kinase 2最可能氨基酸序列为RYLIVLDDIW, RNBS-B最可能的氨基酸序列为GSRI+LTTRLR, GLPL最可能的氨基酸序列为IVSYC+GLPLAIVVVAKRL, MHDV最可能的氨基酸序列为K+TIRMHDLLRDMGR。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4甘薯NB-ARC保守性分析
Fig. 4Conservation analysis of NB-ARC in Ipomoea batatas
根据MEME结构域搜索结果, 对CNL和TNL亚家族基因结构域进行基因定位和识别[51](图5), 发现TNL亚家族和CNL亚家族中分别有10个和7个保守结构域, 其中TNL亚家族NB-ARC结构域中保守基序分别为P-loop、Kinase 2、RNBS-B、RNBS-C、GLPL和MHDV, 缺失RNBS-A以及RNBS-D保守基序; CNL亚家族NB-ARC结构域保守基序分别为P-loop、RNBS-A、Kinase 2、RNBS-B、GLPL、RNBS-D和MHDV, 缺失RNBS-C保守基序。TNL亚家族和CNL亚家族中共有的保守基序分别为P-loop (Kinase 1)、Kinase 2、RNBS-B、GLPL以及MHDV, 与多重比对结果相同。此外, 在TNL亚家族中TIR结构域检测到4个保守基序(TIR1-4)[52]。其中9个NBS-LRR家族基因含有完整的TNL结构域, 占总TNL亚家族基因数的75%, 有2个序列同时缺失TIR 3和TIR 4, 有一个序列缺失TIR 1。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5甘薯NBS-LRR家族基因保守结构域及其氨基酸保守性分析
Fig. 5Conservative domains and amino acids of NBS-LRR family genes in Ipomoea batatas
2.5 甘薯CNL、TNL、RPW8亚家族系统进化树分析
将甘薯NBS-LRR家族基因中CC/TIR/RPW8/ NBS/LRR结构域的序列提取后, 使用MEGA X构建系统进化树。进化树结果中(图6)有3个明显的分支, 分别为CNL型(绿色)、TNL型(蓝色)、PN型(青色)。3种NBS-LRR家族的亚家族存在于不同的进化分支中, 但不存在单独的分支, 说明这3类基因存在相同的进化方式。进化树分支中有些基因分支路径很长, 如CM008340.1-snap12256、CM008331.1- snap10568等, 推测这类基因的“祖先”基因在很早的时候就发生了分化, 相应的基因序列也发生了较大的分化。有些基因序列的分支距离较近, 如CM008334.1-snap8062、CM008342.1-snap6760、CM008334.1-snap11470等, 推测这些基因在短期内发生了复制[46]。在CNL亚家族分支CC结构域中, 马铃薯X病毒类似蛋白[40]的CC结构域(Coiled-coil domain of the potato virus X resistance protein and similar proteins)大致分布于同一小分支, 说明马铃薯X病毒类似蛋白的CC结构域(表1中Cx)与普通CC结构域(表1中C)存在进化关系上的差异。图6
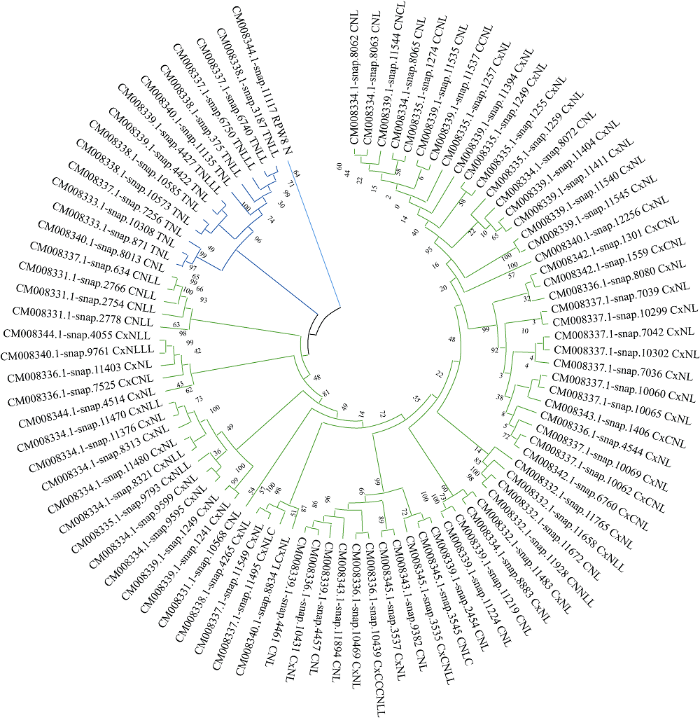 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6甘薯NBS-LRR家族基因系统进化树
Fig. 6Phylogenetic tree of NBS-LRR family genes in Ipomoea batatas
3 讨论
生物信息学方法分析植物抗病基因已被广泛运用于许多作物, 甘薯是重要的粮食作物, 然而, 甘薯基因组尚未注释, 无法直接查找与分析其基因组的R基因。本研究应用snap软件对甘薯全基因组序列进行了外显子预测, 得到甘薯CDS区蛋白质序列, 为甘薯R基因的搜索及其他相关生物信息学分析提供了序列基础。本研究利用HMMER程序对甘薯全基因组序列进行NBS-LRR家族基因检索, 得到735个甘薯NBS-LRR家族基因。经过筛选、验证、剔除等步骤, 最终得到379个序列完整的NBS-LRR家族基因, 占甘薯全基因组数目0.2%, 但与木薯(1.1%)[42]、水稻(1.4%)[43]、拟南芥(0.8%)[44]、乌拉尔图小麦(1.5%)[45]相比数量明显偏少。此外, 甘薯NBS-LRR家族中TNL亚家族基因有22个, 与其他作物有较大差别, 如水稻和乌拉尔图小麦中NBS-LRR家族基因无TNL类型[43,45], 在其他禾本科和草本的单子叶植物中TNL亚家族基因很少[53,54]。本研究分析了甘薯NBS-LRR家族基因的NB-ARC结构域保守性, 分别对CNL和TNL亚家族进行比较, 发现两者在P-loop、Kinase 2、RNBS-B、GLPL、MHDV具有较高的相似性。甘薯P-loop和GLPL基序保守性最高, 其基序分别为SIVGMGGIGKTTLA 和IVSYC+ GLPLAIVVVAKRL, 且GMGGIGKTT区域保守性最高。其次为Kinase 2和RNBS-B, Kinase 2结构域序列为RYLIVLDDIW, 77个CNL类型中, 有70个Kinase 2序列末端氨基酸为W, TNL类型中末端氨基酸主要为E或D, 未发现W。研究表明, Kinase 2序列末端氨基酸W是区分CNL亚家族和TNL亚家族的关键因素[42,55]。同时, TNL类型motif位置的分布比较保守, 为TIR(1-4)-P loop-Kinase 2-RNBS B- RNBS C-GLPL-motif 11。CNL类型motif之间位置的排布规律遵循motif 10-motif 11-P loop-RNBS C-Kinase 2-RNBS B-GLPL的顺序。
值得注意的是, 本研究所分析的甘薯基因组是基于二代测序组装的单倍体型基因组[12], 存在数据不完整和一些错误组装的局限性[56], 本研究结果可能难以完整覆盖甘薯栽培种基因组中NBS-LRR家族的所有基因, 在对相关基因进一步深入分析和研究应用时, 还可以结合三浅裂野牵牛(Ipomoea trifida)基因组。但是本研究的分析方法和结果, 可为甘薯NBS-LRR基因发掘、抗性相关机制研究和抗病育种提供参考。
4 结论
预测了甘薯基因组序列的外显子, 得到甘薯染色体组全基因组蛋白序列, 从中筛选了379个NBS-LRR家族基因, 并进行了染色体定位, 发现NBS-LRR家族基因在不同染色体上的分布数量差异很大, 其中有60.9%的NBS-LRR基因序列呈簇状分布。NBS-LRR基因序列有15个保守结构域, 在N端较为保守, 根据对基因的结构分析构建了系统进化树。研究结果可为甘薯进一步开展NBS-LRR家族基因的功能研究和抗性育种提供参考。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1126/science.7732374URLPMID:7732374 [本文引用: 1]

Plant breeders have used disease resistance genes (R genes) to control plant disease since the turn of the century. Molecular cloning of R genes that enable plants to resist a diverse range of pathogens has revealed that the proteins encoded by these genes have several features in common. These findings suggest that plants may have evolved common signal transduction mechanisms for the expression of resistance to a wide range of unrelated pathogens. Characterization of the molecular signals involved in pathogen recognition and of the molecular events that specify the expression of resistance may lead to novel strategies for plant disease control.
[本文引用: 1]
[本文引用: 1]
DOI:10.3389/fpls.2019.00886URLPMID:31354762 [本文引用: 1]
The root-knot nematode (RKN) Meloidogyne incognita severely reduces yields of pepper (Capsicum annuum) worldwide. A single dominant locus, Me7, conferring RKN resistance was previously mapped on the long arm of pepper chromosome P9. In the present study, the Me7 locus was fine mapped using an F2 population of 714 plants derived from a cross between the RKN-susceptible parent C. annuum ECW30R and the RKN-resistant parent C. annuum CM334. CM334 exhibits suppressed RKN juvenile movement, suppressed feeding site enlargement and significant reduction in gall formation compared with ECW30R. RKN resistance screening in the F2 population identified 558 resistant and 156 susceptible plants, which fit a 3:1 ratio confirming that this RKN resistance was controlled by a single dominant gene. Using the C. annuum CM334 reference genome and BAC library sequencing, fine mapping of Me7 markers was performed. The Me7 locus was delimited between two markers G21U3 and G43U3 covering a physical interval of approximately 394.7 kb on the CM334 chromosome P9. Nine markers co-segregated with the Me7 gene. A cluster of 25 putative nucleotide-binding site and leucine-rich repeat (NBS-LRR)-type disease resistance genes were predicted in the delimited Me7 region. We propose that RKN resistance in CM334 is mediated by one or more of these NBS-LRR class R genes. The Me7-linked markers identified here will facilitate marker-assisted selection (MAS) for RKN resistance in pepper breeding programs, as well as functional analysis of Me7 candidate genes in C. annuum.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nature05286URLPMID:17108957 [本文引用: 1]
Many plant-associated microbes are pathogens that impair plant growth and reproduction. Plants respond to infection using a two-branched innate immune system. The first branch recognizes and responds to molecules common to many classes of microbes, including non-pathogens. The second responds to pathogen virulence factors, either directly or through their effects on host targets. These plant immune systems, and the pathogen molecules to which they respond, provide extraordinary insights into molecular recognition, cell biology and evolution across biological kingdoms. A detailed understanding of plant immune function will underpin crop improvement for food, fibre and biofuels production.
DOI:10.1016/s0958-1669(03)00035-1URLPMID:12732319 [本文引用: 1]
Activation of local and systemic plant defences in response to pathogen attack involves dramatic cellular reprogramming. Over the past 10 years many novel genes, proteins and molecules have been discovered as a result of investigating plant-pathogen interactions. Most attempts to harness this knowledge to engineer improved disease resistance in crops have failed. Although gene efficacy in transgenic plants has often been good, commercial exploitation has not been possible because of the detrimental effects on plant growth, development and crop yield. Biotechnology approaches have now shifted emphasis towards marker-assisted breeding and the construction of vectors containing highly regulated transgenes that confer resistance in several distinct ways.
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41477-017-0002-zURLPMID:28827752 [本文引用: 2]
Here we present the 15 pseudochromosomes of sweet potato, Ipomoea batatas, the seventh most important crop in the world and the fourth most significant in China. By using a novel haplotyping method based on genome assembly, we have produced a half haplotype-resolved genome from ~296 Gb of paired-end sequence reads amounting to roughly 67-fold coverage. By phylogenetic tree analysis of homologous chromosomes, it was possible to estimate the time of two recent whole-genome duplication events as occurring about 0.8 and 0.5 million years ago. This half haplotype-resolved hexaploid genome represents the first successful attempt to investigate the complexity of chromosome sequence composition directly in a polyploid genome, using sequencing of the polyploid organism itself rather than any of its simplified proxy relatives. Adaptation and application of our approach should provide higher resolution in future genomic structure investigations, especially for similarly complex genomes.
URLPMID:15144565 [本文引用: 1]
[本文引用: 2]
[本文引用: 2]
URLPMID:9918945 [本文引用: 1]
[本文引用: 1]
DOI:10.1093/bioinformatics/btp352URLPMID:19505943 [本文引用: 1]
SUMMARY: The Sequence Alignment/Map (SAM) format is a generic alignment format for storing read alignments against reference sequences, supporting short and long reads (up to 128 Mbp) produced by different sequencing platforms. It is flexible in style, compact in size, efficient in random access and is the format in which alignments from the 1000 Genomes Project are released. SAMtools implements various utilities for post-processing alignments in the SAM format, such as indexing, variant caller and alignment viewer, and thus provides universal tools for processing read alignments. AVAILABILITY: http://samtools.sourceforge.net.
DOI:10.1093/bioinformatics/btr509URLPMID:21903627 [本文引用: 1]
MOTIVATION: Most existing methods for DNA sequence analysis rely on accurate sequences or genotypes. However, in applications of the next-generation sequencing (NGS), accurate genotypes may not be easily obtained (e.g. multi-sample low-coverage sequencing or somatic mutation discovery). These applications press for the development of new methods for analyzing sequence data with uncertainty. RESULTS: We present a statistical framework for calling SNPs, discovering somatic mutations, inferring population genetical parameters and performing association tests directly based on sequencing data without explicit genotyping or linkage-based imputation. On real data, we demonstrate that our method achieves comparable accuracy to alternative methods for estimating site allele count, for inferring allele frequency spectrum and for association mapping. We also highlight the necessity of using symmetric datasets for finding somatic mutations and confirm that for discovering rare events, mismapping is frequently the leading source of errors. AVAILABILITY: http://samtools.sourceforge.net. CONTACT: hengli@broadinstitute.org.
DOI:10.1186/s13007-017-0181-7URLPMID:28465712 [本文引用: 1]
BACKGROUND: PTI and ETI are the two major defence mechanisms in plants. ETI is triggered by the detection of pathogen effectors, or their activity, in the plant cell and most of the time involves internal receptors known as resistance (R) genes. An increasing number of R genes responsible for recognition of specific effectors have been characterised over the years; however, methods to identify R genes are often challenging and cannot always be translated to crop plants. RESULTS: We present a novel method to identify R genes responsible for the recognition of specific effectors that trigger a hypersensitive response (HR) in Nicotiana benthamiana. This method is based on the genome-wide identification of most of the potential R genes of N. benthamiana and a systematic silencing of these potential R genes in a simple transient expression assay. A hairpin-RNAi library was constructed covering 345 R gene candidates of N. benthamiana. This library was then validated using several previously described R genes. Our approach indeed confirmed that Prf, NRC2a/b and NRC3 are required for the HR that is mediated in N. benthamiana by Pto/avrPto (prf, NRC2a/b and NRC3) and by Cf4/avr4 (NRC2a/b and NRC3). We also confirmed that NRG1, in association with N, is required for the Tobacco Mosaic Virus (TMV)-mediated HR in N. benthamiana. CONCLUSION: We present a novel approach combining bioinformatics, multiple-gene silencing and transient expression assay screening to rapidly identify one-to-one relationships between pathogen effectors and host R genes in N. benthamiana. This approach allowed the identification of previously described R genes responsible for detection of avirulence determinants from Pseudomonas, Cladosporium and TMV, demonstrating that the method could be applied to any effectors/proteins originating from a broad range of plant pathogens that trigger an HR in N. benthamiana. Moreover, with the increasing availability of genome sequences from model and crop plants and pathogens, this approach could be implemented in other plants, accelerating the process of identification and characterization of novel resistance genes.
DOI:10.1038/msb.2011.75URLPMID:21988835 [本文引用: 2]
Multiple sequence alignments are fundamental to many sequence analysis methods. Most alignments are computed using the progressive alignment heuristic. These methods are starting to become a bottleneck in some analysis pipelines when faced with data sets of the size of many thousands of sequences. Some methods allow computation of larger data sets while sacrificing quality, and others produce high-quality alignments, but scale badly with the number of sequences. In this paper, we describe a new program called Clustal Omega, which can align virtually any number of protein sequences quickly and that delivers accurate alignments. The accuracy of the package on smaller test cases is similar to that of the high-quality aligners. On larger data sets, Clustal Omega outperforms other packages in terms of execution time and quality. Clustal Omega also has powerful features for adding sequences to and exploiting information in existing alignments, making use of the vast amount of precomputed information in public databases like Pfam.
[本文引用: 1]
DOI:10.1093/nar/gkq769URLPMID:20823090 [本文引用: 1]
Mitochondrial-DNA diseases have no effective treatments. Allotopic expression-synthesis of a wild-type version of the mutated protein in the nuclear-cytosolic compartment and its importation into mitochondria-has been proposed as a gene-therapy approach. Allotopic expression has been successfully demonstrated in yeast, but in mammalian mitochondria results are contradictory. The evidence available is based on partial phenotype rescue, not on the incorporation of a functional protein into mitochondria. Here, we show that reliance on partial rescue alone can lead to a false conclusion of successful allotopic expression. We recoded mitochondrial mt-Nd6 to the universal genetic code, and added the N-terminal mitochondrial-targeting sequence of cytochrome c oxidase VIII (C8) and the HA epitope (C8Nd6HA). The protein apparently co-localized with mitochondria, but a significant part of it seemed to be located outside mitochondria. Complex I activity and assembly was restored, suggesting successful allotopic expression. However, careful examination of transfected cells showed that the allotopically-expressed protein was not internalized in mitochondria and that the selected clones were in fact revertants for the mt-Nd6 mutation. These findings demonstrate the need for extreme caution in the interpretation of functional rescue experiments and for clear-cut controls to demonstrate true rescue of mitochondrial function by allotopic expression.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1371/journal.pgen.1008313URLPMID:31344025 [本文引用: 1]
In many plant species, conflicts between divergent elements of the immune system, especially nucleotide-binding oligomerization domain-like receptors (NLR), can lead to hybrid necrosis. Here, we report deleterious allele-specific interactions between an NLR and a non-NLR gene cluster, resulting in not one, but multiple hybrid necrosis cases in Arabidopsis thaliana. The NLR cluster is RESISTANCE TO PERONOSPORA PARASITICA 7 (RPP7), which can confer strain-specific resistance to oomycetes. The non-NLR cluster is RESISTANCE TO POWDERY MILDEW 8 (RPW8) / HOMOLOG OF RPW8 (HR), which can confer broad-spectrum resistance to both fungi and oomycetes. RPW8/HR proteins contain at the N-terminus a potential transmembrane domain, followed by a specific coiled-coil (CC) domain that is similar to a domain found in pore-forming toxins MLKL and HET-S from mammals and fungi. C-terminal to the CC domain is a variable number of 21- or 14-amino acid repeats, reminiscent of regulatory 21-amino acid repeats in fungal HET-S. The number of repeats in different RPW8/HR proteins along with the sequence of a short C-terminal tail predicts their ability to activate immunity in combination with specific RPP7 partners. Whether a larger or smaller number of repeats is more dangerous depends on the specific RPW8/HR autoimmune risk variant.
[本文引用: 1]
[本文引用: 1]
DOI:10.1126/science.252.5009.1162URLPMID:2031185 [本文引用: 1]
The probability that a residue in a protein is part of a coiled-coil structure was assessed by comparison of its flanking sequences with sequences of known coiled-coil proteins. This method was used to delineate coiled-coil domains in otherwise globular proteins, such as the leucine zipper domains in transcriptional regulators, and to predict regions of discontinuity within coiled-coil structures, such as the hinge region in myosin. More than 200 proteins that probably have coiled-coil domains were identified in GenBank, including alpha- and beta-tubulins, flagellins, G protein beta subunits, some bacterial transfer RNA synthetases, and members of the heat shock protein (Hsp70) family.
URLPMID:16317077 [本文引用: 1]
URLPMID:22336098 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:32637963 [本文引用: 1]
The SARS-CoV-2/COVID-19 pandemic continues to threaten global health and socioeconomic stability. Experiments have revealed snapshots of many of the viral components but remain blind to moving parts of these molecular machines. To capture these essential processes, over a million citizen scientists have banded together through the Folding@home distributed computing project to create the world's first Exascale computer and simulate protein dynamics. An unprecedented 0.1 seconds of simulation of the viral proteome reveal how the spike complex uses conformational masking to evade an immune response, conformational changes implicated in the function of other viral proteins, and 'cryptic' pockets that are absent in experimental snapshots. These structures and mechanistic insights present new targets for the design of therapeutics. This living document will be updated as we perform further analysis and make the data publicly accessible.
DOI:10.1093/oxfordjournals.molbev.a026334URLPMID:10742046 [本文引用: 1]
The use of some multiple-sequence alignments in phylogenetic analysis, particularly those that are not very well conserved, requires the elimination of poorly aligned positions and divergent regions, since they may not be homologous or may have been saturated by multiple substitutions. A computerized method that eliminates such positions and at the same time tries to minimize the loss of informative sites is presented here. The method is based on the selection of blocks of positions that fulfill a simple set of requirements with respect to the number of contiguous conserved positions, lack of gaps, and high conservation of flanking positions, making the final alignment more suitable for phylogenetic analysis. To illustrate the efficiency of this method, alignments of 10 mitochondrial proteins from several completely sequenced mitochondrial genomes belonging to diverse eukaryotes were used as examples. The percentages of removed positions were higher in the most divergent alignments. After removing divergent segments, the amino acid composition of the different sequences was more uniform, and pairwise distances became much smaller. Phylogenetic trees show that topologies can be different after removing conserved blocks, particularly when there are several poorly resolved nodes. Strong support was found for the grouping of animals and fungi but not for the position of more basal eukaryotes. The use of a computerized method such as the one presented here reduces to a certain extent the necessity of manually editing multiple alignments, makes the automation of phylogenetic analysis of large data sets feasible, and facilitates the reproduction of the final alignment by other researchers.
DOI:10.1080/10635150701472164URLPMID:17654362 [本文引用: 1]
Alignment quality may have as much impact on phylogenetic reconstruction as the phylogenetic methods used. Not only the alignment algorithm, but also the method used to deal with the most problematic alignment regions, may have a critical effect on the final tree. Although some authors remove such problematic regions, either manually or using automatic methods, in order to improve phylogenetic performance, others prefer to keep such regions to avoid losing any information. Our aim in the present work was to examine whether phylogenetic reconstruction improves after alignment cleaning or not. Using simulated protein alignments with gaps, we tested the relative performance in diverse phylogenetic analyses of the whole alignments versus the alignments with problematic regions removed with our previously developed Gblocks program. We also tested the performance of more or less stringent conditions in the selection of blocks. Alignments constructed with different alignment methods (ClustalW, Mafft, and Probcons) were used to estimate phylogenetic trees by maximum likelihood, neighbor joining, and parsimony. We show that, in most alignment conditions, and for alignments that are not too short, removal of blocks leads to better trees. That is, despite losing some information, there is an increase in the actual phylogenetic signal. Overall, the best trees are obtained by maximum-likelihood reconstruction of alignments cleaned by Gblocks. In general, a relaxed selection of blocks is better for short alignment, whereas a stringent selection is more adequate for longer ones. Finally, we show that cleaned alignments produce better topologies although, paradoxically, with lower bootstrap. This indicates that divergent and problematic alignment regions may lead, when present, to apparently better supported although, in fact, more biased topologies.
DOI:10.1093/bioinformatics/btp033URLPMID:19151095 [本文引用: 1]
UNLABELLED: Jalview Version 2 is a system for interactive WYSIWYG editing, analysis and annotation of multiple sequence alignments. Core features include keyboard and mouse-based editing, multiple views and alignment overviews, and linked structure display with Jmol. Jalview 2 is available in two forms: a lightweight Java applet for use in web applications, and a powerful desktop application that employs web services for sequence alignment, secondary structure prediction and the retrieval of alignments, sequences, annotation and structures from public databases and any DAS 1.53 compliant sequence or annotation server. AVAILABILITY: The Jalview 2 Desktop application and JalviewLite applet are made freely available under the GPL, and can be downloaded from www.jalview.org.
DOI:10.1093/bioinformatics/btr304URLPMID:21593132 [本文引用: 1]
SUMMARY: JABAWS is a web services framework that simplifies the deployment of web services for bioinformatics. JABAWS:MSA provides services for five multiple sequence alignment (MSA) methods (Probcons, T-coffee, Muscle, Mafft and ClustalW), and is the system employed by the Jalview multiple sequence analysis workbench since version 2.6. A fully functional, easy to set up server is provided as a Virtual Appliance (VA), which can be run on most operating systems that support a virtualization environment such as VMware or Oracle VirtualBox. JABAWS is also distributed as a Web Application aRchive (WAR) and can be configured to run on a single computer and/or a cluster managed by Grid Engine, LSF or other queuing systems that support DRMAA. JABAWS:MSA provides clients full access to each application's parameters, allows administrators to specify named parameter preset combinations and execution limits for each application through simple configuration files. The JABAWS command-line client allows integration of JABAWS services into conventional scripts. AVAILABILITY AND IMPLEMENTATION: JABAWS is made freely available under the Apache 2 license and can be obtained from: http://www.compbio.dundee.ac.uk/jabaws.
DOI:10.1093/molbev/msy096URLPMID:29722887 [本文引用: 1]
The Molecular Evolutionary Genetics Analysis (Mega) software implements many analytical methods and tools for phylogenomics and phylomedicine. Here, we report a transformation of Mega to enable cross-platform use on Microsoft Windows and Linux operating systems. Mega X does not require virtualization or emulation software and provides a uniform user experience across platforms. Mega X has additionally been upgraded to use multiple computing cores for many molecular evolutionary analyses. Mega X is available in two interfaces (graphical and command line) and can be downloaded from www.megasoftware.net free of charge.
DOI:10.1074/jbc.M113.517417URLPMID:24194517 [本文引用: 2]
The potato (Solanum tuberosum) disease resistance protein Rx has a modular arrangement that contains coiled-coil (CC), nucleotide-binding (NB), and leucine-rich repeat (LRR) domains and mediates resistance to potato virus X. The Rx N-terminal CC domain undergoes an intramolecular interaction with the Rx NB-LRR region and an intermolecular interaction with the Rx cofactor RanGAP2 (Ran GTPase-activating protein 2). Here, we report the crystal structure of the Rx CC domain in complex with the Trp-Pro-Pro (WPP) domain of RanGAP2. The structure reveals that the Rx CC domain forms a heterodimer with RanGAP2, in striking contrast to the homodimeric structure of the CC domain of the barley disease resistance protein MLA10. Structure-based mutagenesis identified residues from both the Rx CC domain and the RanGAP2 WPP domain that are crucial for their interaction and function in vitro and in vivo. Our results reveal the molecular mechanism underlying the interaction of Rx with RanGAP2 and identify the distinct surfaces of the Rx CC domain that are involved in intramolecular and intermolecular interactions.
DOI:10.1186/1756-0500-2-197URLPMID:19785756 [本文引用: 1]
BACKGROUND: Plant resistance (R) gene products recognize pathogen effector molecules. Many R genes code for proteins containing nucleotide binding site (NBS) and C-terminal leucine-rich repeat (LRR) domains. NBS-LRR proteins can be divided into two groups, TIR-NBS-LRR and non-TIR-NBS-LRR, based on the structure of the N-terminal domain. Although both classes are clearly present in gymnosperms and eudicots, only non-TIR sequences have been found consistently in monocots. Since most studies in monocots have been limited to agriculturally important grasses, it is difficult to draw conclusions. The purpose of our study was to look for evidence of these sequences in additional monocot orders. FINDINGS: Using degenerate PCR, we amplified NBS sequences from four monocot species (C. blanda, D. marginata, S. trifasciata, and Spathiphyllum sp.), a gymnosperm (C. revoluta) and a eudicot (C. canephora). We successfully amplified TIR-NBS-LRR sequences from dicot and gymnosperm DNA, but not from monocot DNA. Using databases, we obtained NBS sequences from additional monocots, magnoliids and basal angiosperms. TIR-type sequences were not present in monocot or magnoliid sequences, but were present in the basal angiosperms. Phylogenetic analysis supported a single TIR clade and multiple non-TIR clades. CONCLUSION: We were unable to find monocot TIR-NBS-LRR sequences by PCR amplification or database searches. In contrast to previous studies, our results represent five monocot orders (Poales, Zingiberales, Arecales, Asparagales, and Alismatales). Our results establish the presence of TIR-NBS-LRR sequences in basal angiosperms and suggest that although these sequences were present in early land plants, they have been reduced significantly in monocots and magnoliids.
DOI:10.1186/s12864-015-1554-9URLPMID:25948536 [本文引用: 4]
BACKGROUND: Plant resistance genes (R genes) exist in large families and usually contain both a nucleotide-binding site domain and a leucine-rich repeat domain, denoted NBS-LRR. The genome sequence of cassava (Manihot esculenta) is a valuable resource for analysing the genomic organization of resistance genes in this crop. RESULTS: With searches for Pfam domains and manual curation of the cassava gene annotations, we identified 228 NBS-LRR type genes and 99 partial NBS genes. These represent almost 1% of the total predicted genes and show high sequence similarity to proteins from other plant species. Furthermore, 34 contained an N-terminal toll/interleukin (TIR)-like domain, and 128 contained an N-terminal coiled-coil (CC) domain. 63% of the 327 R genes occurred in 39 clusters on the chromosomes. These clusters are mostly homogeneous, containing NBS-LRRs derived from a recent common ancestor. CONCLUSIONS: This study provides insight into the evolution of NBS-LRR genes in the cassava genome; the phylogenetic and mapping information may aid efforts to further characterize the function of these predicted R genes.
DOI:10.1007/s00438-004-0990-zURLPMID:15014983 [本文引用: 4]
A complete set of candidate disease resistance ( R) genes encoding nucleotide-binding sites (NBSs) was identified in the genome sequence of japonica rice ( Oryza sativaL. var. Nipponbare). These putative R genes were characterized with respect to structural diversity, phylogenetic relationships and chromosomal distribution, and compared with those in Arabidopsis thaliana. We found 535 NBS-coding sequences, including 480 non-TIR (Toll/IL-1 receptor) NBS-LRR (Leucine Rich Repeat) genes. TIR NBS-LRR genes, which are common in A. thaliana, have not been identified in the rice genome. The number of non-TIR NBS-LRR genes in rice is 8.7 times higher than that in A. thaliana, and they account for about 1% of all of predicted ORFs in the rice genome. Some 76% of the NBS genes were located in 44 gene clusters or in 57 tandem arrays, and 16 apparent gene duplications were detected in these regions. Phylogenetic analyses based both NBS and N-terminal regions classified the genes into about 200 groups, but no deep clades were detected, in contrast to the two distinct clusters found in A. thaliana. The structural and genetic diversity that exists among NBS-LRR proteins in rice is remarkable, and suggests that diversifying selection has played an important role in the evolution of R genes in this agronomically important species. (Supplemental material is available online at http://gattaca.nju.edu.cn.)
DOI:10.1105/tpc.009308URLPMID:12671079 [本文引用: 4]
The Arabidopsis genome contains approximately 200 genes that encode proteins with similarity to the nucleotide binding site and other domains characteristic of plant resistance proteins. Through a reiterative process of sequence analysis and reannotation, we identified 149 NBS-LRR-encoding genes in the Arabidopsis (ecotype Columbia) genomic sequence. Fifty-six of these genes were corrected from earlier annotations. At least 12 are predicted to be pseudogenes. As described previously, two distinct groups of sequences were identified: those that encoded an N-terminal domain with Toll/Interleukin-1 Receptor homology (TIR-NBS-LRR, or TNL), and those that encoded an N-terminal coiled-coil motif (CC-NBS-LRR, or CNL). The encoded proteins are distinct from the 58 predicted adapter proteins in the previously described TIR-X, TIR-NBS, and CC-NBS groups. Classification based on protein domains, intron positions, sequence conservation, and genome distribution defined four subgroups of CNL proteins, eight subgroups of TNL proteins, and a pair of divergent NL proteins that lack a defined N-terminal motif. CNL proteins generally were encoded in single exons, although two subclasses were identified that contained introns in unique positions. TNL proteins were encoded in modular exons, with conserved intron positions separating distinct protein domains. Conserved motifs were identified in the LRRs of both CNL and TNL proteins. In contrast to CNL proteins, TNL proteins contained large and variable C-terminal domains. The extant distribution and diversity of the NBS-LRR sequences has been generated by extensive duplication and ectopic rearrangements that involved segmental duplications as well as microscale events. The observed diversity of these NBS-LRR proteins indicates the variety of recognition molecules available in an individual genotype to detect diverse biotic challenges.
[本文引用: 4]
[本文引用: 4]
[本文引用: 3]
[本文引用: 3]
DOI:10.1038/35080508URLPMID:11433358 [本文引用: 1]
Plant pathology was born after the nineteenth-century potato famine, and since then insightful genetic experiments have contributed to the great progress in our understanding of disease control. Our current view of plant resistance focuses on numerous polymorphic resistance loci, which contain genes known as R genes. The complete sequence of the Arabidopsis thaliana genome provides a framework for exploring the 'big bang' of R genes that occurred and how R genes evolved in plants from their associations with microorganisms, and for improving strategies for more sustainable deployment of disease resistance in crops.
DOI:10.1007/s13205-019-1714-8URLPMID:31065502 [本文引用: 1]
As one of the most important resistance (R) gene families in plants, the NBS-LRR genes, encoding proteins with nucleotide-binding site (NBS) and leucine-rich repeat (LRR) domains, play significant roles in resisting pathogens. The published genomic data for cabbage (Brassica oleracea L.) provide valuable data to identify and characterize the genomic organization of cabbage NBS-LRR genes. Ultimately, we identified 105 TIR (N-terminal Toll/interleukin-1 receptor)-NBS-LRR (TNL) genes and 33 CC (coiled-coil)-NBS-LRR (CNL) genes. Further research indicated that 50.7% of the 138 NBS-LRR genes exist in 27 clusters and there are large differences among the gene structures and protein characteristics. Conserved motif and phylogenetic analysis showed that the structures of TNLs and CNLs were similar, with some differences. These NBS-LRRs are evolved under negative selection and mostly arose from whole-genome duplication events during evolution. Tissue-expression profiling of NBS-LRR genes revealed that 37.1% of the TNL genes are highly or specifically expressed in roots, especially the genes on chromosome 7 (76.5%). Digital gene expression and reverse transcription PCR analyses revealed the expression patterns of the NBS-LRR genes upon challenge by Fusarium oxysporum f.sp. conglutinans: nine genes were upregulated, and five were downregulated. The major resistance gene Foc1 probably works together with the other four genes in the same cluster to resist F. oxysporum infection.
DOI:10.1007/s11103-008-9293-9URLPMID:18247136 [本文引用: 1]
As the largest class of disease resistance R genes, the genes encoding nucleotide binding site and leucine-rich repeat proteins (
DOI:10.1371/journal.pone.0034775URLPMID:22493716 [本文引用: 1]
The majority of disease resistance (R) genes identified to date in plants encode a nucleotide-binding site (NBS) and leucine-rich repeat (LRR) domain containing protein. Additional domains such as coiled-coil (CC) and TOLL/interleukin-1 receptor (TIR) domains can also be present. In the recently sequenced Solanum tuberosum group phureja genome we used HMM models and manual curation to annotate 435 NBS-encoding R gene homologs and 142 NBS-derived genes that lack the NBS domain. Highly similar homologs for most previously documented Solanaceae R genes were identified. A surprising approximately 41% (179) of the 435 NBS-encoding genes are pseudogenes primarily caused by premature stop codons or frameshift mutations. Alignment of 81.80% of the 577 homologs to S. tuberosum group phureja pseudomolecules revealed non-random distribution of the R-genes; 362 of 470 genes were found in high density clusters on 11 chromosomes.
DOI:10.1111/mpp.12797URLPMID:30957942 [本文引用: 1]
Improving genetic resistance is a preferred method to manage Verticillium wilt of cotton and other hosts. Identifying host resistance is difficult because of the dearth of resistance genes against this pathogen. Previously, a novel candidate gene involved in Verticillium wilt resistance was identified by a genome-wide association study using a panel of Gossypium hirsutum accessions. In this study, we cloned the candidate resistance gene from cotton that encodes a protein sharing homology with the TIR-NBS-LRR receptor-like defence protein DSC1 in Arabidopsis thaliana (hereafter named GhDSC1). GhDSC1 expressed at higher levels in response to Verticillium wilt and jasmonic acid (JA) treatment in resistant cotton cultivars as compared to susceptible cultivars and its product was localized to nucleus. The transfer of GhDSC1 to Arabidopsis conferred Verticillium resistance in an A. thaliana dsc1 mutant. This resistance response was associated with reactive oxygen species (ROS) accumulation and increased expression of JA-signalling-related genes. Furthermore, the expression of GhDSC1 in response to Verticillium wilt and JA signalling in A. thaliana displayed expression patterns similar to GhCAMTA3 in cotton under identical conditions, suggesting a coordinated DSC1 and CAMTA3 response in A. thaliana to Verticillium wilt. Analyses of GhDSC1 sequence polymorphism revealed a single nucleotide polymorphism (SNP) difference between resistant and susceptible cotton accessions, within the P-loop motif encoded by GhDSC1. This SNP difference causes ineffective activation of defence response in susceptible cultivars. These results demonstrated that GhDSC1 confers Verticillium resistance in the model plant system of A. thaliana, and therefore represents a suitable candidate for the genetic engineering of Verticillium wilt resistance in cotton.
DOI:10.1046/j.1365-313x.2002.01404.xURLPMID:12366802 [本文引用: 1]
The Toll/interleukin-1 receptor (TIR) domain is found in one of the two large families of homologues of plant disease resistance proteins (R proteins) in Arabidopsis and other dicotyledonous plants. In addition to these TIR-NBS-LRR (TNL) R proteins, we identified two families of TIR-containing proteins encoded in the Arabidopsis Col-0 genome. The TIR-X (TX) family of proteins lacks both the nucleotide-binding site (NBS) and the leucine rich repeats (LRRs) that are characteristic of the R proteins, while the TIR-NBS (TN) proteins contain much of the NBS, but lack the LRR. In Col-0, the TX family is encoded by 27 genes and three pseudogenes; the TN family is encoded by 20 genes and one pseudogene. Using massively parallel signature sequencing (MPSS), expression was detected at low levels for approximately 85% of the TN-encoding genes. Expression was detected for only approximately 40% of the TX-encoding genes, again at low levels. Physical map data and phylogenetic analysis indicated that multiple genomic duplication events have increased the numbers of TX and TN genes in Arabidopsis. Genes encoding TX, TN and TNL proteins were demonstrated in conifers; TX and TN genes are present in very low numbers in grass genomes. The expression, prevalence, and diversity of TX and TN genes suggests that these genes encode functional proteins rather than resulting from degradation or deletions of TNL genes. These TX and TN proteins could be plant analogues of small TIR-adapter proteins that function in mammalian innate immune responses such as MyD88 and Mal.
DOI:10.1038/nature02253URLPMID:14685245 [本文引用: 1]
Mutations in BLM, which encodes a RecQ helicase, give rise to Bloom's syndrome, a disorder associated with cancer predisposition and genomic instability. A defining feature of Bloom's syndrome is an elevated frequency of sister chromatid exchanges. These arise from crossing over of chromatid arms during homologous recombination, a ubiquitous process that exists to repair DNA double-stranded breaks and damaged replication forks. Whereas crossing over is required in meiosis, in mitotic cells it can be associated with detrimental loss of heterozygosity. BLM forms an evolutionarily conserved complex with human topoisomerase IIIalpha (hTOPO IIIalpha), which can break and rejoin DNA to alter its topology. Inactivation of homologues of either protein leads to hyper-recombination in unicellular organisms. Here, we show that BLM and hTOPO IIIalpha together effect the resolution of a recombination intermediate containing a double Holliday junction. The mechanism, which we term double-junction dissolution, is distinct from classical Holliday junction resolution and prevents exchange of flanking sequences. Loss of such an activity explains many of the cellular phenotypes of Bloom's syndrome. These results have wider implications for our understanding of the process of homologous recombination and the mechanisms that exist to prevent tumorigenesis.
DOI:10.1038/sj.emboj.7601164URLPMID:16724105 [本文引用: 1]
RNA silencing is an evolutionarily conserved system that functions as an antiviral mechanism in higher plants and insects. To counteract RNA silencing, viruses express silencing suppressors that interfere with both siRNA- and microRNA-guided silencing pathways. We used comparative in vitro and in vivo approaches to analyse the molecular mechanism of suppression by three well-studied silencing suppressors. We found that silencing suppressors p19, p21 and HC-Pro each inhibit the intermediate step of RNA silencing via binding to siRNAs, although the molecular features required for duplex siRNA binding differ among the three proteins. None of the suppressors affected the activity of preassembled RISC complexes. In contrast, each suppressor uniformly inhibited the siRNA-initiated RISC assembly pathway by preventing RNA silencing initiator complex formation.
DOI:10.1007/s00438-003-0868-5URLPMID:12827500 [本文引用: 1]
Plant disease resistance genes (R genes) show significant similarity amongst themselves in terms of both their DNA sequences and structural motifs present in their protein products. Oligonucleotide primers designed from NBS (Nucleotide Binding Site) domains encoded by several R-genes have been used to amplify NBS sequences from the genomic DNA of various plant species, which have been called Resistance Gene Analogues (RGAs) or Resistance Gene Candidates (RGCs). Using specific primers from the NBS and TIR (Toll/Interleukin-1 Receptor) regions, we identified twelve classes of RGCs in cassava (Manihot esculenta Crantz). Two classes were obtained from the PCR-amplification of the TIR domain. The other 10 classes correspond to the NBS sequences and were grouped into two subfamilies. Classes RCa1 to RCa5 are part of the first subfamily and were linked to a TIR domain in the N terminus. Classes RCa6 to RCa10 corresponded to non-TIR NBS-LRR encoding sequences. BAC library screening with the 12 RGC classes as probes allowed the identification of 42 BAC clones that were assembled into 10 contigs and 19 singletons. Members of the two TIR and non-TIR NBS-LRR subfamilies occurred together within individual BAC clones. The BAC screening and Southern hybridization analyses showed that all RGCs were single copy sequences except RCa6 that represented a large and diverse gene family. One BAC contained five NBS sequences and sequence analysis allowed the identification of two complete RGCs encoding two highly similar proteins. This BAC was located on linkage group J with three other RGC-containing BACs. At least one of these genes, RGC2, is expressed constitutively in cassava tissues.
URLPMID:30389915 [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}