 ,1,*, 朱金洁,1,*
,1,*, 朱金洁,1,*Establishment of an efficient genotyping technique based on targeted DNA endonuclease in vitro activity of CRISPR/Cas9 ribonucleoprotein
WANG Nan1, QI Xian-Tao1,2, LIU Chang-Lin1, XIE Chuan-Xiao,1,*, ZHU Jin-Jie,1,*通讯作者:
收稿日期:2019-12-18接受日期:2020-03-24网络出版日期:2020-07-12
| 基金资助: |
Received:2019-12-18Accepted:2020-03-24Online:2020-07-12
| Fund supported: |
作者简介 About authors
E-mail:2720409346@qq.com。

摘要
关键词:
Abstract
Keywords:
PDF (3254KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王南, 祁显涛, 刘昌林, 谢传晓, 朱金洁. 基于CRISPR/Cas9核糖核蛋白体DNA定点内切酶体外活性建立高效基因型分析技术[J]. 作物学报, 2020, 46(7): 978-986. doi:10.3724/SP.J.1006.2020.93064
WANG Nan, QI Xian-Tao, LIU Chang-Lin, XIE Chuan-Xiao, ZHU Jin-Jie.
2012年9月, Jinek等[1]在体外验证了sgRNA/ Cas9核糖核蛋白复合体具有DNA定点剪切的活性。次年, 张峰课题组首次报道了CRISPR/Cas9在人类细胞中定点编辑DNA的活性[2]。自此, 基因编辑技术成为生命科学领域的技术发展前沿, 研究热度呈指数级增长。CRISPR/Cas基因编辑技术不仅可应用于基因敲除、基因插入、定点突变与染色体重组等遗传操作[3,4,5,6], 而且还派生出如碱基编辑、转录激活或抑制、表观修饰以及DNA与RNA原位示踪成像等一系列重要的遗传学工具[7,8,9,10,11]。然而, 对该系统的体外应用却鲜有报道。基于该系统的DNA定点剪切的功能, 该系统可进一步开发为位点限制性内切酶, 拓展普通限制性内切酶无法靶向的位点, 从而满足日益增长的基因型快速鉴定的需求。
目前, 应用最广泛的为组分优化的II型CRISPR/Cas9系统, 由Cas9与sgRNA两个元件组成。在sgRNA的指导下, Cas9蛋白可在PAM基序上游的3 nt处特异性剪切DNA双链[12]。为打破Cas9蛋白对PAM序列的依赖性, 科学家们已开发多种识别不同PAM序列的Cas9蛋白变体[13], 其中, 基于蛋白结构优化获得的Cas9NG的靶向范围最广[14]。目前, 不同版本的CRISPR/Cas9系统已经广泛应用于不同物种的植物基因组编辑[15,16,17]。基于经典的CRISPR/Cas9系统, 我国科学家首次创建了水稻的CRISPR/Cas9突变体库[18], 我们课题组也针对玉米的ZmWx[19,20]、ZmLg1[21]、ZmMTL[22]等多个位点实现DNA的定点编辑, 获得了多个具有重要育种价值的突变体材料。如何对基因编辑突变体进行快速的基因型鉴定成为当前该技术发展亟待解决的重要问题。此外, 除基因编辑突变体具有基因型快速鉴定的需求外, 任何基于DNA序列变异的位点均需要一种快速、经济、可靠、高效与高通量的基因型鉴定技术。例如, 人工诱变及化学诱变群体突变体的快速筛选、基因功能解析中分离群体中个体的基因型快速鉴定、突变体基因型的快速鉴定等, 因此, 建立高效简便的方法对于功能基因组学研究与作物分子育种等均具有重要的应用价值。
目前, 用于植物突变体基因型鉴定的技术主要包括PCR限制性内切酶酶切(PCR/RE)、错配特异性核酸酶酶切(T7E I/Surveyor assay)、Sanger测序、二代测序(Next-generation sequencing, NGS)及高分辨率溶解曲线分析(High-resolution melting analysis, HRMA)等, 这些技术都存在自身的优点, 但同时也有局限性。PCR/RE技术严重依赖于突变碱基附近的限制性内切酶识别位点, 限制了靶点设计的自由度从而限制了其范围[23]。基于DNA双链错配原理的T7E I或CEL I酶切检测, 商业酶价格高昂, 技术上也存在不能有效区分纯合突变与野生型以及杂合突变与双等位基因突变的限制[24]。Sanger测序虽可直观地呈现靶点突变信息, 但其价格昂贵, 不适用于大规模突变群体检测。NGS及衍生的Hi-TOM技术主要适用于高通量筛选鉴定, 无法满足对小量与中量样本突变体材料早期快速鉴定的需要[25]。2018年高彩霞团队首先尝试开发了基于CRISPR-Cas9系统的PCR/RNP技术[26], 该技术不受限制性酶切位点选择的局限, 同时可精准区分野生型、纯合突变及杂合突变等基因型, 成本低廉、快速高效, 可实现突变体高通量筛选, 但检测体系仍需要优化, 进一步提高检测效率、特异性与灵敏度, 才能进一步推广应用。
本研究的目标是建立与优化基于CRISPR/Cas9与CRISPR/Cas9NG核糖核蛋白体定点DNA内切酶体外活性的基因型高效分析技术, 为该技术的高效应用奠定完善的技术方案。本研究利用原核表达并纯化的Cas9蛋白或Cas9NG蛋白为DNA内切酶, 以体外转录的靶向ZmWx基因靶点的sgRNA或esgRNA为向导RNA分子, 通过体外组装为sgRNA/Cas9-RNP复合体, 对实验室通过基因编辑手段获得的玉米ZmWx突变体靶点PCR产物进行酶切, 从而实现对突变体的基因型分析。本研究将为突变体的高效基因型分析提供技术参考。
1 材料与方法
1.1 材料
基于ZC01玉米自交系背景创制的靶向ZmWx基因CRISPR-Cas9基因编辑等待基因型分析的玉米材料[19], BL21(DE3)大肠杆菌表达感受态(北京全式金生物技术有限公司, 货号3CD801), PET30a-Cas9载体(构建), PET 30a-Cas9NG载体(构建)。1.2 方法
1.2.1 Cas9及Cas9NG蛋白的原核表达载体构建从本实验室保存载体上设计引物(表1), 扩增获得Cas9片段, 使用限制性内切酶Nco I和Nhe I双酶切PET 30a载体, 琼脂糖凝胶电泳分离后, 进行胶回收纯化。回收纯化的Cas9片段与PET 30a线性载体, 用NEB组装酶(北京NEB公司) 50℃, 1 h进行组装, 转化至BL21(DE3)大肠杆菌表达感受态中, 均匀涂布在卡那抗性的LB固体培养基上, 37℃过夜培养, 挑取单克隆酶切鉴定, 将获得的阳性克隆质粒送上海英潍捷基公司测序。测序结果与Snapgene载体图谱比对一致, 即获得PET 30a-Cas9载体。用上述方法, 将限制性内切酶Nhe I替换为Hind III, 构建得到PET 30a-Cas9NG载体。
Table 1
表1
表1本研究所用引物
Table 1
| 引物名称 Primer name | 序列 Sequence (5′-3′) |
|---|---|
| Cas9 F | GACGACGACGACAAGGCCATGATGGACAAGAAGTACTCCA |
| Cas9 R | TCGACGGAGCTCGCTAGCGTCGCCGCCGAGCTGGGAGAGG |
| Cas9NG F | ACGACGACGACAAGGCCATGGACAAGAAGTACTCCATCGGC |
| Cas9NG R | CTCGAGTGCGGCCGCAAGCTTGTCGCCGCCGAGCTGGCTCAG |
| sgRNA scaffold F | GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAG |
| sgRNA scaffold R | AAAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAA |
| esgRNA scaffold F | GTTTCAGAGCTATGCTGGAAACAGCATAGCAAGTTGAAATAAGGCTAGTCCGTTATC |
| esgRNA scaffold R | AAAAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTC |
| ZmWx F | CATACTTCTCCGGACCATACGGTAA |
| ZmWx R | TCCCTGCTGGGGTCCCACTC |
| T7 F | AAGCTAATACGACTCACTATAGGAGGTTCAGCTCCGGGTAGTGTTTTAGAGCTAGAAA |
| eT7 F | AAGCTAATACGACTCACTATAGGAGGTTCAGCTCCGGGTAGTGTTTCAGAGCTATGCT |
| Target1 T7 F | AAGCTAATACGACTCACTATAGGTTCAGCTCCGGGTAGTCGGGTTTCAGAGCTATGCT |
| Target2 T7 F | AAGCTAATACGACTCACTATAGCTCCGGGTAGTCGGAGAAGGGTTTCAGAGCTATGCT |
| Target3 T7 F | AAGCTAATACGACTCACTATAGCTTCTCCGACTACCCGGAGCGTTTCAGAGCTATGCT |
| Target4 T7 F | AAGCTAATACGACTCACTATAGTTCGCCTTCTCCGACTACCCGTTTCAGAGCTATGCT |
| Target5 T7 F | AAGCTAATACGACTCACTATAGGCCTTCTCCGACTACCCGGAGTTTCAGAGCTATGCT |
| Target6 T7 F | AAGCTAATACGACTCACTATAGCAGCTCCGGGTAGTCGGAGAGTTTCAGAGCTATGCT |
| T7 R | AAAAAGCACCGACTCGGTGCCACTTTTT |
新窗口打开|下载CSV
1.2.2 Cas9及Cas9NG蛋白的原核表达与纯化
将构建好的融合His标签的Cas9和Cas9NG蛋白原核表达载体转化到原核表达菌株BL21(DE3)中。挑取单克隆, 2次扩大培养至OD600在0.7~0.8之间。冰浴后加入0.5 mol L-1 IPTG溶液诱导蛋白表达。以下步骤均在4℃进行: 超声破碎裂解后, 取上清, 进行Ni-NTA琼脂糖介质亲和层析, 然后进行阳离子交换柱层析, 收集各组分进行10% SDS-PAGE鉴定, 根据凝胶电泳结果收集含有洗脱蛋白的各组分, 最后, 用100,000-MWCO蛋白超滤管将含有洗脱蛋白的组分缓冲液替换为蛋白存储液(50 mmol L-1 Hepes, 150 mmol L-1 KCl, 1 mmol L-1 TCEP, 20% Glycerol, pH 7.5)。分装后液氮速冻, 于-80°C冰箱保存备用。
1.2.3 Cas9及Cas9NG蛋白的定量 对待测样品进行BSA (bovine serum albumin)粗定量后, 使用BCA蛋白快速测定试剂盒进行蛋白精确定量(上海英潍捷基公司), 首先制备蛋白标准品梯度稀释液和BCA工作液, 然后分别取标准品和待测样品20 μL加入到微孔板中, 之后加入20 μL工作试剂, 充分混匀, 震荡30 s, 室温静置5 min, 用分光光度计测量蛋白标准品和待测样品在480 nm处的吸光度。最后作出蛋白标准品吸光度相对于浓度的标准曲线, 根据待测样品在480 nm处的吸光度, 在标准曲线上查出待测样品相当于标准蛋白的量, 从而计算得出待测样品的浓度。
1.2.4 RNA体外转录 利用引物重叠PCR原理, 设计引物sgRNA scaffold F、sgRNA scaffold R (表1)扩增获得sgRNA转录模板, 用2%琼脂糖凝胶电泳分离后, 回收纯化。使用T7转录试剂盒(北京NEB公司)进行体外转录, 体外转录体系为2 μL模板, 15 μL NTP Mix, 3 μL T7转录酶, RNase-free H2O补至40 μL。37℃孵育3.5 h。结束后, 加入2 μL DNase I、8 μL RNase-free H2O消化模板DNA。然后用RNA纯化浓缩试剂盒(ZYM公司)进行纯化浓缩, 分光光度计测得浓度后, 通过10% Urea-PAGE鉴定sgRNA的质量。液氮冷冻, -80℃保存备用。采用相同方法获得高纯度esgRNA。
1.2.5 ZmWx突变体DNA提取 切取玉米ZmWx突变体种子部分, 充分研磨, 置于1.5 mL EP管中, 加入1 mL裂解缓冲液(10 mmol L-1 Tris-HCl, 1 mmol L-1 EDTA, pH 8.0, 0.1% SDS, pH 8.0), 混匀后于65℃水浴1 h, 10,000×g离心2 min, 小心吸取上清液于一支新的1.5 mL EP管中, 加入等体积的酚/氯仿/异戊醇(25:24﹕1)(北京酷来博生物技术有限公司), 充分混匀后, 10,000×g离心10 min, 吸取上清液于一支新的1.5 mL EP管中, 加入0.2倍体积7.5 mol L-1的醋酸铵和2倍体积的无水乙醇沉淀DNA, 10,000×g离心1 min, 弃上清液, 用70%无水乙醇洗涤2次, 弃上清液, 室温干燥, 用0.2 mL TE溶液溶解DNA, -20℃保存备用。
1.2.6 PCR/RNP酶切 以种子DNA为模板, 设计引物ZmWx F、ZmWx R (表1), 通过PCR扩增sgRNA靶标两侧各300~500 bp序列。PCR程序为95℃预变性3 min; 94℃变性25 s, 63℃退火25 s, 72℃延伸30 s, 35个循环; 72℃总延伸2 min; 4℃保存。将X μg Cas蛋白、X μg esgRNA、2 μL Buf 3.1 (北京NEB公司)和(12-X) μL RNase-free H2O混合均匀, 37℃孵育0.5 h进行预组装, 加入6 μL PCR产物, 37℃孵育2 h。然后加入2 μL RNaseA, 37℃孵育15 min以消化RNA, 65℃孵育20 min使蛋白质变性, 终止反应。结束后进行2%琼脂糖凝胶电泳检测。将ZmWx突变体PCR产物送上海英潍捷基公司测序, 以验证酶切检测结果。
1.2.7 引物设计 本研究所用引物除ZmWx F、ZmWx R由Primer premier软件设计外, 其他引物均由Snapgene软件设计, 并委托上海英潍捷基公司合成。引物信息见表1。
2 结果与分析
2.1 CRISPR-Cas9、Cas9NG蛋白原核表达与纯化
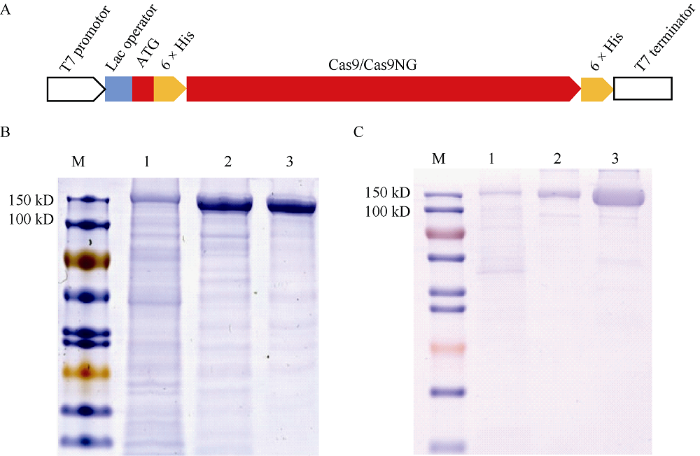
构建Cas9与Cas9NG蛋白原核表达载体, 载体的主要元件有T7启动子、Lac操纵子、His蛋白纯化标签、目标蛋白的CDS序列及T7终止子等(图1-A)。通过亲和纯化与阳离子交换纯化两步法对原核表达的Cas9与Cas9NG蛋白纯化后, 获得纯度较高的Cas9与Cas9NG蛋白制品(图1-B, C)。结果表明, 收集1 L诱导表达的大肠杆菌可纯化约4 mg的Cas9蛋白, 且经过阳离子交换柱二次纯化获得的蛋白样品, 纯度显著提高。经过一次原核表达及纯化所获得的蛋白制品在产量和纯度上可满足后续酶切实验需要。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1Cas9与Cas9NG蛋白的原核表达及纯化
A: 原核表达载体结构示意图; B: Cas9纯化蛋白10% SDS-PAGE胶图; C: Cas9-NG纯化蛋白10% SDS-PAGE胶图。M: Protein marker; 1: 上清液中蛋白; 2: Ni柱纯化蛋白; 3: 阳离子交换柱纯化蛋白。
Fig. 1Purification of Cas9 and Cas9NG protein
A: Schematics diagram of protein expression vector; B: 10% SDS-PAGE of purified Cas9; C: 10% SDS-PAGE of purified Cas9NG. M: Protein marker. 1: Supernatant protein; 2: Ni-column crude purified protein; 3: Cation exchange column purification protein.
2.2 RNA的体外转录与基于esgRNA/Cas9-RNP和esgRNA/Cas9NG-RNP检测体系的初建立
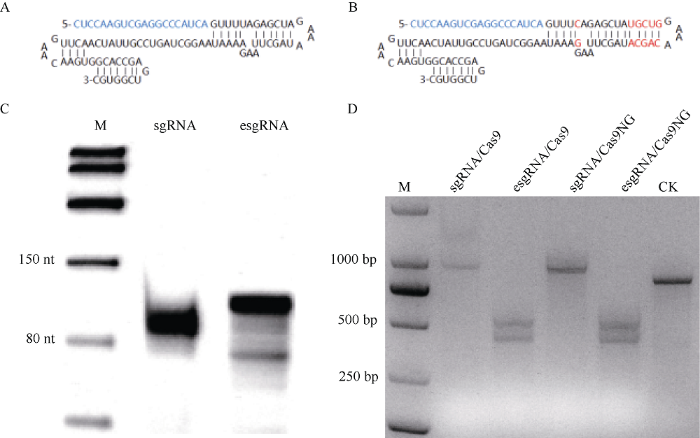
目前, 应用最为广泛的sgRNA骨架序列为81 nt (图2-A), 后续研究发现将sgRNA骨架序列的poly U位点的第4个碱基突变为C以及延长RNA双链部分5个碱基对(图2-B), 可有效提高基因编辑的效率[27,28]。因此, 本研究采用两种sgRNA骨架序列进行Cas9-RNP与Cas9NG-RNP实验体系的建立。首先, 设计体外转录靶向本实验室已发表ZmWx基因编辑位点的sgRNA或esgRNA引物, 见表1所示。通过PCR扩增获得体外转录的DNA模板后, 利用T7转录酶进行体外RNA转录, 转录产物经过纯化后, 经10% Urea-PAGE电泳鉴定。结果显示, 经过体外转录的sgRNA条带大小在100 nt左右, esgRNA条带大小在110 nt左右, 与预期RNA条带大小一致且质量较好, 纯化后esgRNA有部分杂带(图2-C), 推测可能是由体外转录DNA模板回收不纯所致。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2sgRNA、esgRNA体外转录及基于esgRNA/Cas9-RNP和esgRNA/Cas9NG-RNP检测体系的建立
A: sgRNA二级结构图; B: esgRNA二级结构图; C: RNA的10% Urea-PAGE胶图; D: PCR/RNP酶切检测; CK: 未加esgRNA和Cas蛋白的ZmWx野生型PCR产物。
Fig. 2In vitro transcription of sgRNA and esgRNA, as well as the establishment of cleavage assay via esgRNA/Cas9-RNP and esgRNA/Cas9NG-RNP
A: Predicted secondary structure of sgRNA; B: Predicted secondary structure of esgRNA; C: 10% Urea-PAGE of sgRNA and esgRNA; D: PCR/RNP cleavage assay; CK: ZmWx wild PCR product without esgRNA and Cas protein.
以野生型ZmWx种子DNA为模板, 通过PCR扩增sgRNA靶标两侧各300~500 bp序列, 作为酶切底物。然后将Cas9蛋白、Cas9NG蛋白分别与sgRNA、esgRNA预组装为4种RNP复合体, 对PCR产物进行酶切, 结束后进行琼脂糖凝胶电泳检测, 以验证RNP活性。结果显示, sgRNA/Cas9-RNP或sgRNA/Cas9NG-RNP在ZmWx位点上不具有酶切活性, 底物DNA均没有被切开。而基于esgRNA/ Cas9-RNP或基于esgRNA/Cas9NG-RNP可对ZmWx靶点底物DNA进行充分酶切, 酶切产生400 bp与500 bp两条DNA条带(图2-D)。
2.3 基于esgRNA/Cas9-RNP和esgRNA/Cas9NG- RNP检测体系的优化及其对不同基因型的区分
针对esgRNA/Cas9-RNP或esgRNA/Cas9NG- RNP两种突变体基因型检测方法进行酶切体系的进一步优化。采用控制变量法分别研究了esgRNA用量、Cas9或Cas9NG蛋白用量和酶切反应时间等对酶切效率的影响。对于esgRNA/Cas9-RNP, 当esgRNA和Cas9蛋白均为1 μg时, 酶切反应时间为0.5 h, 可实现对500 ng靶位点PCR扩增产物的充分酶切(图3-A~C)。对于esgRNA/Cas9NG-RNP, 当esgRNA和Cas9NG均为2 μg时, 酶切反应时间为4 h, 实现对500 ng 靶位点PCR扩增产物的充分酶切(图3-E~G)。采用优化后的esgRNA/ Cas9-RNP与esgRNA/Cas9NG-RNP检测体系, 分别对ZmWx基因编辑产生的3种不同基因型材料进行基因型鉴定, 以验证基于sgRNA/Cas9-RNP系统是否能够准确区分不同的基因型。优化后的esgRNA/Cas9-RNP与esgRNA/Cas9NG-RNP检测体系均具备准确区分不同基因型PCR产物的能力, 能够有效区分野生型、纯合突变体及杂合突变体材料(图3-D, H)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3基于esgRNA /Cas9-RNP及esgRNA/ Cas9NG-RNP检测体系的优化
A: esgRNA梯度;B: Cas9蛋白梯度; C: 酶切反应时间梯度; D: 不同基因型PCR产物酶切; E: esgRNA梯度; F: Cas9NG蛋白梯度; G: 酶切反应时间梯度; H: 不同基因型PCR产物酶切; CK: 未加esgRNA和Cas蛋白的ZmWx 野生型PCR产物。
Fig. 3Optimization of cleavage assay via esgRNA /Cas9-RNP and esgRNA/ Cas9NG-RNP
A: Effect of different gradients of esgRNA; B: Effect of different gradients of Cas9 protein; C: Effect of different gradients of digestion time; D: Digestion of PCR products of different genotypes; E: Effect of different gradients of esgRNA; F: Effect of different gradients of Cas9NG protein; G: Effect of different gradients of digestion time; H: Digestion of PCR products of different genotypes; CK: ZmWx wild PCR product without esgRNA and Cas protein.
2.4 利用Cas9NG拓宽检测位点范围
前人的研究结果表明, Cas9NG作为spCas9的变体, 由于替换了PI功能域中(PAM-interacting, PI)识别PAM的第2和第3位碱基的氨基酸, 使其不再具有识别特异性, 从而可以识别PAM为“NG”基序, 极大地拓展了基因编辑的范围[13,14]。因此, 我们探索应用Cas9NG拓宽检测位点范围。我们在ZmWx编辑位点附近重新设计了6个PAM为“NG”的靶位点(图4-A), 通过体外转录及凝胶电泳检测发现, 6个esgRNA的分子量大小正确, 转录效果较好(图4-B)。应用优化后的esgRNA/ Cas9NG-RNP体系, 将Cas9NG与6种esgRNA分别组装成RNP, 对野生型ZmWx靶位点PCR扩增产物进行酶切。从图4-C可以看出, 仅有第6组对野生型Waxy PCR产物有酶切活性, 而第6组esgRNA的PAM为“AGG”。结果表明, 利用esgRNA/Cas9NG-RNP并不能拓宽ZmWx靶位点检测的范围。
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4基于esgRNA/Cas9NG-RNP检测体系对不同ZmWx位点的检测
A: Cas9-NG的6个新的靶向位点选择; B: 6个靶点对应的esgRNA 10% Urea-PAGE鉴定; C: PCR/RNP酶切分析; CK: 未加esgRNA和Cas蛋白的ZmWx野生型PCR产物。
Fig. 4Cleavage assay of different targets for ZmWx locus via esgRNA/Cas9NG-RNP
A: Targets selection of Cas9NG; B: 10% Urea-PAGE of 6 target-related esgRNA; C: PCR/RNP cleavage assay; CK: ZmWx wild PCR product without esgRNA and Cas protein.
2.5 基于esgRNA /Cas9-RNP检测体系筛选基因编辑突变体
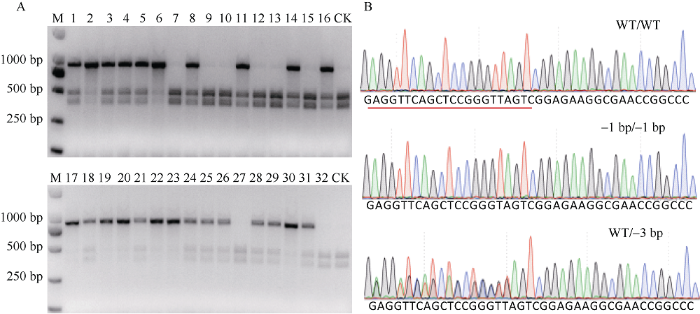
由于esgRNA/Cas9-RNP在检测反应中RNP用量、酶切反应时间及剪切效率均优于esgRNA/Cas9NG- RNP, 本研究利用esgRNA/Cas9-RNP系统对ZmWx基因编辑突变群体进行基因型检测。结果表明, 对随机选择的32个ZmWx基因编辑材料的靶位点进行酶切后, ZmWx野生型、纯合突变体及杂合突变体均得到有效区分(图5-A)。为进一步确定利用esgRNA/ Cas9-RNP进行突变体基因型鉴定的可靠性, 我们随机选择3种基因型的一个样品进行Sanger测序。通过峰图判读与序列比对可知, 测序结果与检测结果一致(图5-B)。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5基于esgRNA/Cas9-RNP的ZmWx基因编辑突变体检测
A: 32个ZmWx的基因编辑材料的esgRNA/Cas9-RNP酶切检测; B: Sanger测序峰图。WT/WT为野生型, -1 bp/-1 bp为缺失1 bp的纯合突变体, WT/-3 bp为一个等位基因缺失3 bp的杂合突变体; CK: 未加esgRNA和Cas蛋白的ZmWx野生型PCR产物。
Fig. 5Genotyping genome-edited ZmWx mutant via esgRNA/Cas9-RNP
A: Genotyping genome-edited ZmWx mutant via esgRNA/Cas9-RNP; B: Sanger sequencing peak graph of different genotypes. WT/WT is a wild type, -1 bp/-1 bp is a homozygous mutant with 1 bp deleted, and WT/-3 bp is a heterozygous mutant with 3 bp allele deleted; CK: ZmWx wild PCR product without esgRNA and Cas protein.
3 讨论
建立一种快速、经济、高效与高通量的突变体基因型鉴定技术对作物基因功能鉴定等基础研究与分子育种研究等方面具有广泛的应用价值。因此, 此类技术具有重要研发价值。此前已研发了多种基因型鉴定方法, 本方法与其相比, 在应用范围与简便高效性上具有重要的优势。以当前应用较广的基于限制性内切酶的PCR/RE检测和基于DNA双链错配原理的T7E I或CEL I酶切检测为例。PCR/RE方法严重依赖于突变位点处具有合适的II型限制性内切酶位点, 识别位点为6 bp的限制性内切酶约4K DNA区间才能有一个合适的位点, 而依赖CRISPR/ Cas9 PAM-NGG基序的特征, 约16 bp即能有一个合适的位点, 约为前者的260倍。基于DNA双链错配原理的T7E I或CEL I酶切检测无法有效区分纯合突变体与野生型, 双等位基因突变体和杂合突变体, 商业化的酶价格高昂, 检测体系还依赖PAGE胶, 成本高昂, 实验体系操作繁琐。而本研究采用的方法, 经过一次原核表达及纯化获得的Cas9蛋白(4 L原核表达菌液)可用于至少10,000个酶切体系的检测, 不同位点突变体材料的检测只需替换与靶位点匹配的sgRNA即可, 体外转录1个反应的esgRNA可用于至少200个酶切体系检测, 合计1个反应的成本在1元以下。整个检测反应可在1~3 h内完成, 检测都是基于简易的琼脂糖凝胶分析, 能够满足对突变体进行早期高通量基因型的快速高效分析。此外, 基于sgRNA/Cas9-RNP的检测体系还具有应用范围广的特点, 不仅适用于由ZFN、TALEN、CRISPR-Cas9、CRISPR-Cpf1等序列特异性核酸酶定点编辑突变体的基因型分析, 还适用于自然变异、人工诱变突变体在特定位点上的基因型分析。我们在本实验积累的经验还表明, ZmWx位点对应的sgRNA无法介导Cas蛋白活性, 推测原因是通过T7启动子体外转录靶向ZmWx位点的sgRNA, 在体外酶切缓冲液中不能形成稳定的二级结构, 从而不能与Cas蛋白形成有剪切活性的核糖核蛋白体(RNP)。因此, 将sgRNA替换为骨架序列优化的esgRNA是必要的, 该优化会显著提高检测体系对位点的兼容性, 优化的esgRNA分别与Cas9及Cas9NG蛋白组装形成的RNP复合体可实现底物DNA的充分酶切, 这与前人研究的结论是一致的, 即通过对sgRNA骨架序列优化, 可以提高sgRNA结合靶位点的能力, 从而提高CRISPR-Cas9的切割效率[27,28]。此外, 本研究发现, 基于esgRNA/ Cas9NG-RNP的检测体系, 除在PAM为“NGG”序列的6号靶位点有较强的剪切活性外, 在PAM为非“NGG”的靶位点均没有检测到明显的剪切活性。推测原因可能是Cas9NG蛋白变体对于靶向位点具有一定的偏好性, 不同的靶位点由于GC含量的不同、碱基的分布差异, sgRNA识别结合靶位点的能力有所不同, 基于esgRNA/Cas9NG-RNP的检测体系还有待于蛋白活性的进一步优化。该数据对利用Cas9NG的NG-PAM基序特征扩大体内CRISPR/ Cas9基因编辑设计位点自由度具有重要参考价值。
4 结论
利用Cas9或Cas9NG变体蛋白与sgRNA核糖核蛋白复合体(sgRNA/Cas9-RNP或sgRNA/ Cas9NG-RNP)体外DNA定点内切酶活性, 建立并优化了一种简便、高效与低成本的基因型分析技术。esgRNA/Cas9形成的RNP复合体核酸酶活性比esgRNA/Cas9NG高, 酶切等量底物需要的RNP复合体的量少且时间更短, 可用于突变体基因型的高通量鉴定。利用esgRNA/Cas9NG拓宽目标靶位点的检测数据为CRISPR/Cas9NG活体基因编辑技术研发提供了重要参考数据。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:22745249 [本文引用: 1]
URLPMID:23287718 [本文引用: 1]
URLPMID:24336571 [本文引用: 1]
URLPMID:25263330 [本文引用: 1]
URLPMID:27096365 [本文引用: 1]
URLPMID:25337876 [本文引用: 1]
URLPMID:29160308 [本文引用: 1]
DOI:10.1038/nature14136URLPMID:25494202 [本文引用: 1]

Systematic interrogation of gene function requires the ability to perturb gene expression in a robust and generalizable manner. Here we describe structure-guided engineering of a CRISPR-Cas9 complex to mediate efficient transcriptional activation at endogenous genomic loci. We used these engineered Cas9 activation complexes to investigate single-guide RNA (sgRNA) targeting rules for effective transcriptional activation, to demonstrate multiplexed activation of ten genes simultaneously, and to upregulate long intergenic non-coding RNA (lincRNA) transcripts. We also synthesized a library consisting of 70,290 guides targeting all human RefSeq coding isoforms to screen for genes that, upon activation, confer resistance to a BRAF inhibitor. The top hits included genes previously shown to be able to confer resistance, and novel candidates were validated using individual sgRNA and complementary DNA overexpression. A gene expression signature based on the top screening hits correlated with markers of BRAF inhibitor resistance in cell lines and patient-derived samples. These results collectively demonstrate the potential of Cas9-based activators as a powerful genetic perturbation technology.
URLPMID:24797424 [本文引用: 1]
DOI:10.1038/nbt.3199URLPMID:25849900 [本文引用: 1]
Technologies that enable targeted manipulation of epigenetic marks could be used to precisely control cell phenotype or interrogate the relationship between the epigenome and transcriptional control. Here we describe a programmable, CRISPR-Cas9-based acetyltransferase consisting of the nuclease-null dCas9 protein fused to the catalytic core of the human acetyltransferase p300. The fusion protein catalyzes acetylation of histone H3 lysine 27 at its target sites, leading to robust transcriptional activation of target genes from promoters and both proximal and distal enhancers. Gene activation by the targeted acetyltransferase was highly specific across the genome. In contrast to previous dCas9-based activators, the acetyltransferase activates genes from enhancer regions and with an individual guide RNA. We also show that the core p300 domain can be fused to other programmable DNA-binding proteins. These results support targeted acetylation as a causal mechanism of transactivation and provide a robust tool for manipulating gene regulation.
URLPMID:28084328 [本文引用: 1]
URLPMID:24906146 [本文引用: 1]
DOI:10.1038/nature26155URLPMID:29512652 [本文引用: 2]
A key limitation of the use of the CRISPR-Cas9 system for genome editing and other applications is the requirement that a protospacer adjacent motif (PAM) be present at the target site. For the most commonly used Cas9 from Streptococcus pyogenes (SpCas9), the required PAM sequence is NGG. No natural or engineered Cas9 variants that have been shown to function efficiently in mammalian cells offer a PAM less restrictive than NGG. Here we use phage-assisted continuous evolution to evolve an expanded PAM SpCas9 variant (xCas9) that can recognize a broad range of PAM sequences including NG, GAA and GAT. The PAM compatibility of xCas9 is the broadest reported, to our knowledge, among Cas9 proteins that are active in mammalian cells, and supports applications in human cells including targeted transcriptional activation, nuclease-mediated gene disruption, and cytidine and adenine base editing. Notably, despite its broadened PAM compatibility, xCas9 has much greater DNA specificity than SpCas9, with substantially lower genome-wide off-target activity at all NGG target sites tested, as well as minimal off-target activity when targeting genomic sites with non-NGG PAMs. These findings expand the DNA targeting scope of CRISPR systems and establish that there is no necessary trade-off between Cas9 editing efficiency, PAM compatibility and DNA specificity.
DOI:10.1126/science.aas9129URLPMID:30166441 [本文引用: 2]
The RNA-guided endonuclease Cas9 cleaves its target DNA and is a powerful genome-editing tool. However, the widely used Streptococcus pyogenes Cas9 enzyme (SpCas9) requires an NGG protospacer adjacent motif (PAM) for target recognition, thereby restricting the targetable genomic loci. Here, we report a rationally engineered SpCas9 variant (SpCas9-NG) that can recognize relaxed NG PAMs. The crystal structure revealed that the loss of the base-specific interaction with the third nucleobase is compensated by newly introduced non-base-specific interactions, thereby enabling the NG PAM recognition. We showed that SpCas9-NG induces indels at endogenous target sites bearing NG PAMs in human cells. Furthermore, we found that the fusion of SpCas9-NG and the activation-induced cytidine deaminase (AID) mediates the C-to-T conversion at target sites with NG PAMs in human cells.
DOI:10.1007/s13205-019-1787-4URLPMID:31192079 [本文引用: 1]
Proline-rich proteins (PRPs) play multiple physiological and biochemical roles in plant growth and stress response. In this study, we reported that the knockout of OsPRP1 induced cold sensitivity in rice. Mutant plants were generated by CRISPR/Cas9 technology to investigate the role of OsPRP1 in cold stress and 26 mutant plants were obtained in T0 generation with the mutation rate of 85% including 15% bi-allelic, 53.3% homozygous, and 16.7% heterozygous and 16 T-DNA-free lines in T1 generation. The conserved amino acid sequence was changed and the expression level of OsPRP1 was reduced in mutant plants. The OsPRP1 mutant plants displayed more sensitivity to cold stress and showed low survival rate with decreased root biomass than wild-type (WT) and homozygous mutant line with large fragment deletion was more sensitive to low temperature. Mutant lines accumulated less antioxidant enzyme activity and lower levels of proline, chlorophyll, abscisic acid (ABA), and ascorbic acid (AsA) content relative to WT under low-temperature stress. The changes of antioxidant enzymes were examined in the leaves and roots with exogenous salicylic acid (SA) treatment which resulted in increased activity of superoxide dismutase (SOD), peroxidase (POD), and catalase (CAT) under cold stress, while enzyme antioxidant activity was lower in untreated seedlings which showed that exogenous SA pretreatment could alleviate the low-temperature stress in rice. Furthermore, the expression of three genes encoding antioxidant enzyme activities (SOD4, POX1, and OsCAT3) was significantly down-regulated in the mutant lines as compared to WT. These results suggested that OsPRP1 enhances cold tolerance by modulating antioxidants and maintaining cross talk through signaling pathways. Therefore, OsPRP1 gene could be exploited for improving cold tolerance in rice and CRISPR/Cas9 technology is helpful to study the function of a gene by analyzing the phenotypes of knockout mutants generated.
DOI:10.1186/s12870-019-1746-6URLPMID:30961525 [本文引用: 1]
BACKGROUND: The plant architecture has significant effects on grain yield of various crops, including soybean (Glycine max), but the knowledge on optimization of plant architecture in order to increase yield potential is still limited. Recently, CRISPR/Cas9 system has revolutionized genome editing, and has been widely utilized to edit the genomes of a diverse range of crop plants. RESULTS: In the present study, we employed the CRISPR/Cas9 system to mutate four genes encoding SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) transcription factors of the SPL9 family in soybean. These four GmSPL9 genes are negatively regulated by GmmiR156b, a target for the improvement of soybean plant architecture and yields. The soybean Williams 82 was transformed with the binary CRISPR/Cas9 plasmid, assembled with four sgRNA expression cassettes driven by the Arabidopsis thaliana U3 or U6 promoter, targeting different sites of these four SPL9 genes via Agrobacterium tumefaciens-mediated transformation. A 1-bp deletion was detected in one target site of the GmSPL9a and one target site of the GmSPL9b, respectively, by DNA sequencing analysis of two T0-generation plants. T2-generation spl9a and spl9b homozygous single mutants exhibited no obvious phenotype changes; but the T2 double homozygous mutant spl9a/spl9b possessed shorter plastochron length. In T4 generation, higher-order mutant plants carrying various combinations of mutations showed increased node number on the main stem and branch number, consequently increased total node number per plants at different levels. In addition, the expression levels of the examined GmSPL9 genes were higher in the spl9b-1 single mutant than wild-type plants, which might suggest a feedback regulation on the expression of the investigated GmSPL9 genes in soybean. CONCLUSIONS: Our results showed that CRISPR/Cas9-mediated targeted mutagenesis of four GmSPL9 genes in different combinations altered plant architecture in soybean. The findings demonstrated that GmSPL9a, GmSPL9b, GmSPL9c and GmSPL9 function as redundant transcription factors in regulating plant architecture in soybean.
DOI:10.1007/s00299-019-02378-1URLPMID:30684023 [本文引用: 1]
KEY MESSAGE: The analysis of 93 mutant alleles in 18 genes demonstrated that CRISPR-Cas9 is a robust tool for targeted mutagenesis in maize, permitting efficient generation of single and multiple knockouts. CRISPR-Cas9 technology is a simple and efficient tool for targeted mutagenesis of the genome. It has been implemented in many plant species, including crops such as maize. Here we report single- and multiple-gene mutagenesis via stably transformed maize plants. Two different CRISPR-Cas9 vectors were used allowing the expression of multiple guide RNAs and different strategies to knockout either independent or paralogous genes. A total of 12 plasmids, representing 28 different single guide RNAs (sgRNAs), were generated to target 20 genes. For 18 of these genes, at least one mutant allele was obtained, while two genes were recalcitrant to sequence editing. 19% (16/83) of mutant plants showed biallelic mutations. Small insertions or deletions of less than ten nucleotides were most frequently observed, regardless of whether the gene was targeted by one or more sgRNAs. Deletions of defined regions located between the target sites of two guide RNAs were also reported although the exact deletion size was variable. Double and triple mutants were created in a single step, which is especially valuable for functional analysis of genes with strong genetic linkage. Off-target effects were theoretically limited due to rigorous sgRNA design and random experimental checks at three potential off-target sites did not reveal any editing. Sanger chromatograms allowed to unambiguously class the primary transformants; the majority (85%) were fully edited plants transmitting systematically all detected mutations to the next generation, generally following Mendelian segregation.
DOI:10.1016/j.molp.2017.06.006URLPMID:28645639 [本文引用: 1]
[本文引用: 2]
DOI:10.1111/pbi.13144URLPMID:31050154 [本文引用: 1]
DOI:10.1111/pbi.2017.15.issue-12URLPMID:28379609 [本文引用: 1]
DOI:10.1016/j.molp.2018.06.011URLPMID:30010025 [本文引用: 1]
DOI:10.1007/s40291-016-0195-2URLPMID:26993322 [本文引用: 1]
BACKGROUND: Polymerase chain reaction (PCR) is widely used in biological research and diagnostics because of its high sensitivity and specificity. However, the sensitivity of PCR is strongly influenced by topological characteristics of the template. Supercoiled templates are known to inhibit PCR, whereas linearized forms of the same supercoiled templates facilitate PCR. OBJECTIVES: This study was conducted to compare the PCR efficiency of circular supercoiled DNA templates to their restriction endonuclease (RE)-mediated linearized forms. Additionally, we also evaluated the possibility of RE digestion of the circular supercoiled templates within the complete PCR buffer. METHODS: Following a systematic approach, we demonstrated that circular supercoiled templates could be efficiently linearized by RE in the complete PCR buffer itself. This allowed linearization of circular supercoiled templates and their subsequent amplification in the PCR buffer in a single-tube format. RESULTS: Using this extremely simple RE-PCR approach, we documented up to tenfold increases in detection efficiency of PCR with two different circular supercoiled templates of clinical origin, including an international calibration standard. CONCLUSIONS: This inexpensive and easy approach to increasing PCR sensitivity can be easily adapted to any standard PCR protocol aimed at amplifying circular supercoiled genomes. Apart from its application in the development of sensitive clinical diagnostic PCR assays for a large number of organisms, this method could also prove to be very useful in simplifying the existing protocols for other applications where pre-PCR restriction digestion is required, such as mutation detection, genotyping, and selective template amplification.
[本文引用: 1]
DOI:10.1007/s11427-018-9402-9URLPMID:30446870 [本文引用: 1]
The CRISPR/Cas system has been extensively applied to make precise genetic modifications in various organisms. Despite its importance and widespread use, large-scale mutation screening remains time-consuming, labour-intensive and costly. Here, we developed Hi-TOM (available at https://doi.org/www.hi-tom.net/hi-tom/ ), an online tool to track the mutations with precise percentage for multiple samples and multiple target sites. We also described a corresponding next-generation sequencing (NGS) library construction strategy by fixing the bridge sequences and barcoding primers. Analysis of the samples from rice, hexaploid wheat and human cells reveals that the Hi-TOM tool has high reliability and sensitivity in tracking various mutations, especially complex chimeric mutations frequently induced by genome editing. Hi-TOM does not require special design of barcode primers, cumbersome parameter configuration or additional data analysis. Thus, the streamlined NGS library construction and comprehensive result output make Hi-TOM particularly suitable for high-throughput identification of all types of mutations induced by CRISPR/Cas systems.
DOI:10.1111/pbi.12938URLPMID:29723918 [本文引用: 1]
Despite the great achievements in genome editing, accurately detecting mutations induced by sequence-specific nucleases is still a challenge in plants, especially in polyploidy plants. An efficient detection method is particularly vital when the mutation frequency is low or when a large population needs to be screened. Here, we applied purified CRISPR ribonucleoprotein complexes to cleave PCR products for genome-edited mutation detection in hexaploid wheat and diploid rice. We show that this mutation detection method is more sensitive than Sanger sequencing and more applicable than PCR/RE method without the requirement for restriction enzyme site. We also demonstrate that this detection method is especially useful for genome editing in wheat, because target sites are often surrounded by single nucleotide polymorphisms. Using this screening method, we were also able to detect foreign DNA-free tagw2 mutations induced by purified TALEN protein. Finally, we show that partial base editing mutations can also be detected using high-fidelity SpCas9 variants or FnCpf1. The PCR/RNP method is low-cost and widely applicable for rapid detection of genome-edited mutation in plants.
DOI:10.1186/s13059-015-0846-3URLPMID:26671237 [本文引用: 2]
BACKGROUND: Single-guide RNA (sgRNA) is one of the two key components of the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 genome-editing system. The current commonly used sgRNA structure has a shortened duplex compared with the native bacterial CRISPR RNA (crRNA)-transactivating crRNA (tracrRNA) duplex and contains a continuous sequence of thymines, which is the pause signal for RNA polymerase III and thus could potentially reduce transcription efficiency. RESULTS: Here, we systematically investigate the effect of these two elements on knockout efficiency and showed that modifying the sgRNA structure by extending the duplex length and mutating the fourth thymine of the continuous sequence of thymines to cytosine or guanine significantly, and sometimes dramatically, improves knockout efficiency in cells. In addition, the optimized sgRNA structure also significantly increases the efficiency of more challenging genome-editing procedures, such as gene deletion, which is important for inducing a loss of function in non-coding genes. CONCLUSIONS: By a systematic investigation of sgRNA structure we find that extending the duplex by approximately 5 bp combined with mutating the continuous sequence of thymines at position 4 to cytosine or guanine significantly increases gene knockout efficiency in CRISPR-Cas9-based genome editing experiments.
DOI:10.1038/nbt.4005URLPMID:29131148 [本文引用: 2]
Efficient genome editing with Cas9-sgRNA in vivo has required the use of viral delivery systems, which have limitations for clinical applications. Translational efforts to develop other RNA therapeutics have shown that judicious chemical modification of RNAs can improve therapeutic efficacy by reducing susceptibility to nuclease degradation. Guided by the structure of the Cas9-sgRNA complex, we identify regions of sgRNA that can be modified while maintaining or enhancing genome-editing activity, and we develop an optimal set of chemical modifications for in vivo applications. Using lipid nanoparticle formulations of these enhanced sgRNAs (e-sgRNA) and mRNA encoding Cas9, we show that a single intravenous injection into mice induces >80% editing of Pcsk9 in the liver. Serum Pcsk9 is reduced to undetectable levels, and cholesterol levels are significantly lowered about 35% to 40% in animals. This strategy may enable non-viral, Cas9-based genome editing in the liver in clinical settings.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}