 ,1,*
,1,*Fine mapping of a major QTL qMES20-10 associated with deep-seeding tolerance in maize and analysis of differentially expressed genes
REN Meng-Meng1,**, ZHANG Hong-Wei1,**, WANG Jian-Hua2, WANG Guo-Ying1, ZHENG Jun,1,*通讯作者:
第一联系人:
收稿日期:2019-10-12接受日期:2020-04-15网络出版日期:2020-07-12
| 基金资助: |
Received:2019-10-12Accepted:2020-04-15Online:2020-07-12
| Fund supported: |
作者简介 About authors
任蒙蒙,E-mail:ren_mm1991@163.com。
张红伟,E-mail:zhanghongwei@caas.cn。

摘要
关键词:
Abstract
Keywords:
PDF (1693KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
任蒙蒙, 张红伟, 王建华, 王国英, 郑军. 玉米耐深播主效QTL qMES20-10的精细定位及差异表达基因分析[J]. 作物学报, 2020, 46(7): 1016-1024. doi:10.3724/SP.J.1006.2020.93054
REN Meng-Meng, ZHANG Hong-Wei, WANG Jian-Hua, WANG Guo-Ying, ZHENG Jun.
干旱胁迫严重影响玉米生长发育, 是造成玉米减产的最重要非生物胁迫因素[1]。我国有大量玉米种植在干旱半干旱地区, 通过改善栽培措施如增加播种深度, 能够减轻干旱胁迫对玉米生长发育的影响[2]。对于普通的玉米品种, 增加播种深度会导致出苗率下降, 造成苗期缺苗而影响产量[3]。具有耐深播特性的玉米材料相对于普通玉米材料, 在深播条件下具有更长的中胚轴和更强的顶土能力, 能适应较深的播种条件, 其根系能从更深层的土壤吸收水分, 有助于玉米应对干旱胁迫。因此, 研究玉米耐深播特性的遗传机制对于改良普通杂交种的耐深播特性具有重要的指导意义[4]。
目前对作物耐深播的研究主要集中在大麦、小麦和水稻等作物[5,6,7]。在深播条件下, 不同的作物通过延伸不同的组织将胚芽顶出土层。水稻、燕麦等作物主要延伸中胚轴和第一节间[8], 大麦、小麦等作物延伸其胚芽鞘和第一节间[9,10], 而玉米和高粱则主要延伸其中胚轴[11,12]。在水稻直播技术的推广中, 需要培育耐深播的水稻品种以提高出苗率。Lu等[13]用469份水稻自交系进行全基因组关联分析, 以中胚轴和根的长度作为表型指标, 挖掘了23个与耐深播相关的位点。Wu等[14]分别在水培和5 cm沙土中对270份水稻自交系进行了深播表型鉴定, 并用重测序得到的基因型数据进行了关联分析, 共挖掘出16个与耐深播相关的位点。Zhao等[15]对621个水稻品种进行深播表型分析, 发现中胚轴伸长受深层土壤覆盖诱导, 对田间耐深播能力具有重要意义, 同时利用关联分析的方法鉴定出了13个耐深播相关的QTL, 并对其中两个主效基因OsML1和OsML2进行了验证。在玉米耐深播的研究中, 赵光武等[16]对12个玉米自交系耐深播相关性状的分析表明, 中胚轴伸长是玉米种子顶土出苗的主要动力, 深播条件下的出苗率与玉米的中胚轴长度呈正相关, 因此也可以用深播条件下的中胚轴长度来评价玉米的耐深播能力。Liu等[17]用玉米优良自交系B73和Mo17组配的280份DH系(IBM Syn10)进行了深播发芽能力的QTL定位, 共发现了32个相关的QTL, 并在这些QTL的定位区间内鉴别出6个与深播发芽能力相关的候选基因。上述研究表明, 作物耐深播能力是由多个位点控制的数量性状, 需要通过QTL定位的方法挖掘相关基因。
目前, 借助分子标记辅助选择, 利用高代回交群体构建近等基因系, 是精细定位QTL并最终获得候选基因的常用方法[18]。近等基因系遗传背景相似, 只在包含QTL区段的染色体片段上存在差异, 在控制环境的情况下, 可认为表型的差异只是由于该染色体片段的不同引起, 从而能精确定位在该区段上的QTL。另外, 利用RNA-Seq的方法, 分析近等基因系中基因差异表达的情况, 结合精细定位的结果, 也是预测候选基因并对目标性状进行遗传解析的有效方法[19]。
本研究所用材料为3681-4和X178, 其中3681-4为本实验室鉴定的一个具有耐深播特性的玉米自交系[20], X178为玉米优良杂交种农大108的母本。实验室前期已通过构建F2:3家系进行了耐深播性状的初定位, 在玉米10号染色体长臂上发现了一个主效QTL qMES20-10, 并将其定位到了SSR标记umc1506与bnlg1028之间大约5.24 Mb的区间[21]。在此基础上, 构建了高代回交群体, 对qMES20-10进一步精细定位, 并分析近等基因系中差异表达基因的情况, 为该位点候选基因的挖掘提供了材料和参考依据。
1 材料与方法
1.1 实验材料
亲本材料为玉米自交系3681-4和X178在20 cm播深下的中胚轴长度(简称为MES20)有极显著差异(图1-C)。实验室前期工作用X178作为轮回亲本, 通过umc1506与bnlg1028两个分子标记筛选保留qMES20-10的杂合区段, 并对其他非目标QTL进行背景控制, 构建了170份BC3F3:4家系。图1
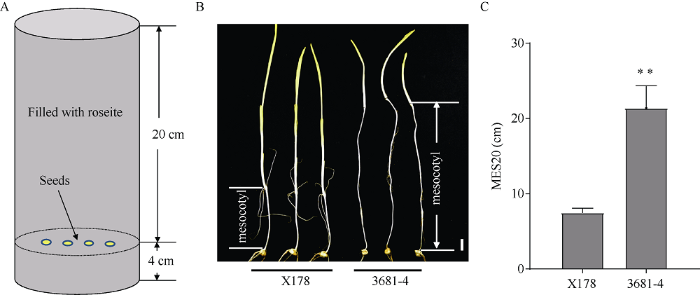 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1表型鉴定方法和亲本材料表型
A: 表型鉴定方法示意图。B: 20 cm播深条件下亲本材料的中胚轴, 标尺=1 cm。C: 亲本材料的中胚轴长度; **表示0.01水平下差异显著。
Fig. 1Phenotyping method and phenotype of the two parents
A: diagram of phenotyping method. B: mesocotyl of two parents sowing in 20 cm roseite, scale bar = 1 cm. C: mesocotyl length of two parents; ** means significant difference at P < 0.01.
在此基础上利用分子标记辅助选择, 2015年冬季在海南三亚种植了目标区段为杂合基因型的BC3F3群体2600株, 通过初定位区间的两端标记umc1506和bnlg1028, 筛选出243个交换单株, 将这些交换单株与X178回交获得BC4F1群体。2016年夏季在北京顺义和2016年冬季在海南将BC4F1群体自交获得大约200份BC4F2:3家系。
1.2 表型鉴定方法
采用20 cm播种深度鉴定表型。首先在一个塑料盒(长60 cm, 宽40 cm, 高16 cm)中装上6根PVC管(直径20 cm, 高24 cm), 在PVC管的底部平铺4 cm厚混匀的蛭石(蛭石与水的质量比为1:1), 均匀点上种子后, 用蛭石填满PVC管。然后将盒子密封, 黑暗处理。环境温度25℃, 相对湿度60% (图1-A)。播种14 d后, 将幼苗取出, 用直尺测量幼苗的中胚轴长度(图1-B)。从BC3F3:4每个家系中随机选择30粒种子种到一个PVC管中鉴定表型, 用表型的平均值代表该家系的表型值。对于从BC4F2:3家系中选取的用作后代测验的交换单株上的穗子, 每个单穗按照上述方法种植180粒种子(6个PVC管)作为分离群体, 进行基因型和表型鉴定。1.3 基因型鉴定方法
取玉米苗期幼嫩叶片, 用CTAB法提取基因组DNA[23], 用于基因型鉴定。BC3F3:4家系全基因组的基因型鉴定采用了北京康普森生物技术有限公司定制的Maize iSlect6K芯片, 共5179个SNP标记, 均匀覆盖了玉米全基因组[22]。精细定位群体的基因型鉴定采用SSR分子标记或InDel标记, 所用的SSR分子标记, 来源于MaizeGDB (http://www.maizegdb. org/), InDel标记根据B73和Mo17基因组序列比对得到的InDel位点进行设计[24]。引物由上海生工生物工程有限公司合成, 引物信息如表1。Table 1
表1
表1本研究所用的引物
Table 1
| 引物 Primer | 物理位置 Position | 正向序列 Forward sequence (5'-3') | 反向序列 Reverse sequence (5'-3') |
|---|---|---|---|
| umc1506 | 133,239,898 | ATAAAGGTTGGCAAAACGTAGCCT | AAAAGAAACATGTTCAGTCGAGCG |
| DST_InD25 | 133,799,178 | TGCGCTTTATTAGGCGAAAC | TTTACGCGTTATGGGAGACC |
| DST_Ind7 | 134,830,243 | GCTTGCTGCATTGTCTTGAA | GGCAGATTGACACTGGTGAA |
| DST_Ind105 | 136,072,789 | AGAGAGACAGCCGCACTTG | TCGACCGTACTTGTTCATGG |
| DST_InD13 | 136,274,298 | GGCAACAGTTCGACGGATTA | TCCGGATGATGTTTACATGG |
| bnlg1028 | 138,503,281 | AGGAAACGAACACAGCAGCT | TGCATAGACAAAACCGACGT |
新窗口打开|下载CSV
1.4 QTL确证和精细定位
为了验证qMES20-10的真实性, 对170个BC3F3:4家系进行了表型和基因型鉴定, 表型2次重复, 基因型用umc1506和bnlg1028两对标记检测, 并用方差分析检测不同基因型家系之间的表型是否有差异。利用覆盖全基因组的芯片进一步检测了这些BC3F3:4家系的基因型, 用QTL定位软件IciMapping v4.0[25]对这些家系进行了全基因组的QTL检测, 先用bin模块去除冗余标记和偏分离标记, 再采用完备区间作图法(ICIM)进行QTL检测。表型值为两次测量结果的平均值。实验群体设置为F2, LOD阈值设置为2.5。方差分析采用R语言中的anova()函数。精细定位QTL采用两种策略。一是AB-QTL[26]分析法, 从BC4F2:3群体中直接鉴定交换单株的表型, 通过t测验比较交换单株与受体亲本之间的表型是否有差异, 从而判断QTL的位置; 二是后代测验法[27], 从BC4F2:3群体选取交换单株的穗子作为分离群体, 单株鉴定基因型和表型, 比较分别带有3681-4和X178纯合基因型材料的表型是否有差异, 从而确定目标QTL是否位于杂合区段。t测验采用SAS9.2中的Proc TTEST。
1.5 RNA-Seq和差异表达分析
从BC3F3:4家系中挑选目标QTL区段分别为X178纯合(NILX178)和3681-4纯合(NIL3681-4)基因型的材料各3份作为近等基因系, 再加上2个亲本材料X178和3681-4 (各3次重复), 共8份材料进行RNA-Seq分析。每份材料按照上述表型鉴定的方法播种, 取幼苗中胚轴部分, 用TRIol法提取总RNA[28]。RNA-Seq由北京贝瑞和康生物科技有限公司完成。差异表达分析先用TopHat2[29]将测序得到的原始Reads 锚定到B73参考基因组上, 再使用Cufflinks软件的Cuffdiff[30]组件, 通过Mapped Reads在基因上的位置信息, 对转录本和基因的表达水平进行定量。采用FPKM (fragments per kilobase of transcript per million fragments mapped)[31]作为衡量转录本或基因表达水平的指标。用DESeq2[32]进行样品组间的差异表达分析, 获得两个样品之间的差异表达基因集。在差异表达基因检测过程中, 将Fold Change≥2且FDR < 0.01作为筛选标准。其中, 差异倍数(fold change)表示两样品(组)间表达量的比值。错误发现率(false discovery rate, FDR)是通过对差异显著性P值(P-value)校正得到的。基因富集分析使用在线分析软件AgriGo (http://bioinfo.cau. edu.cn/agriGO/)[33]。
2 结果与分析
2.1 用BC3F3:4家系确证qMES20-10
在170个BC3F3:4家系中, qMES20-10的区间内X178纯合、杂合、3681-4纯合3种基因型的家系分别有34、65、35个, 符合1:2:1的分离比(χ2测验: P = 0.935)。剩余的36个家系在umc1506与bnlg1028两个标记之间发生交换, 重组率为21.2%。3种基因型材料的中胚轴长度平均值分别为7.62、8.52、9.87 cm。方差分析表明三者之间有极显著差异(图2-A)。图2
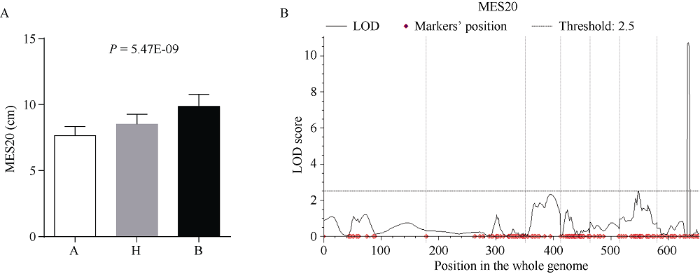 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2耐深播主效QTL qMES20-10确证
A: BC3F3:4家系中3种基因型材料的中胚轴长度。A、H、B分别表示X178纯合、杂合、3681-4纯合3种基因型。P值是对3种基因型材料的中胚轴长度作方差分析得到。B: BC3F3:4家系全基因组QTL检测。
Fig. 2Validation of the major QTL qMES20-10 for deep-seeding tolerance
A: mesocotyl length of seedlings in three different genotypes of BC3F3:4 lines. A, H, B denote homozygous X178, heterozygous, and homozygous 3681-4 genotype respectively. P-value is determined by ANOVA for mesocotyl length of different genotypic seedlings. B: QTL-mapping using BC3F3:4 lines in the whole genome.
为了排除背景对后续定位的干扰, 进一步用玉米6 K芯片鉴定了上述170份BC3F3:4家系的基因型。从6K芯片的5179个SNP标记中, 共筛选出1485个多态性SNP位点, 覆盖了玉米的全基因组。用2次表型的平均值进行全基因组的QTL检测, 发现只有10号染色体上有一个主效QTL (图2-B), 位于SNP标记PZE-110074955和chr10.S_132532921之间。2个标记在B73参考基因组(V2)的物理位置与该QTL的初定位区间基本一致。在BC3F3:4家系中, 该QTL的LOD值为11.36, 可解释的表型变异为38.66%。该位点的加性效应为0.82 cm, 显性效应为0.03 cm, 为典型的加性基因。通过以上两个实验, 进一步确证了qMES20-10的真实性。
2.2 qMES20-10的精细定位
利用组配的200份BC4F2:3家系对qMES20-10进行精细定位, 通过初定位区间内的4个分子标记DST_InD7、DST_InD105、DST_InD13、DST_InD25将这些BC4F2:3家系分成9种重组类型, 分别对不同重组类型的家系进行表型鉴定, 检验每种重组类型的表型与受体亲本X178是否有显著差异。如图3所示, 在DST_InD7和DST_InD105两个标记区段内含有3681-4染色体片段的重组类型(R1-R4), 中胚轴长度显著高于受体亲本, 而含有X178片段的重组类型(R5-R9)与受体亲本无显著差异。由此可将目标QTL定位于DST_InD7和DST_InD105两个标记之间。这2个标记在B73参考基因组(V2)中的物理位置为10号染色体133.3~136.0 Mb。图3
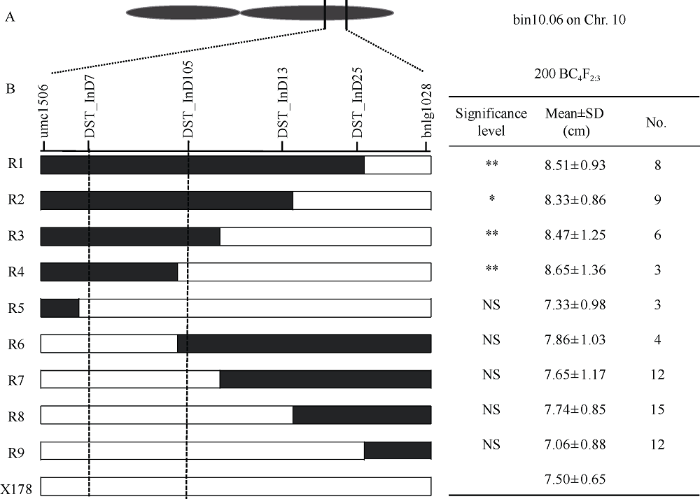 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3耐深播主效位点qMES20-10的精细定位
A: 目标QTL qMES20-10所在的染色体区段。B: 用BC4F2:3家系进行精细定位。R1~R9表示9种不同的重组类型, 黑色和白色区段分别代表来自亲本3681-4和X178的染色体片段。** 和 * 分别表示在0.01和0.05水平下差异显著, NS表示无显著差异。
Fig. 3Fine mapping of the major QTL qMES20-10 for deep-seeding tolerance
A: the locus of target QTL qMES20-1 on bin 10.06. B: fine-mapping utilizing BC4F2:3 lines. R1-R9 represent the nine recombinant types. Black and white bars represent chromosomal segments derived from 3681-4 and X178. ** and * mean significant difference at the P < 0.01 and P < 0.05, respectively; NS means no significant difference.
在上述BC4F2:3家系中, 结合分子标记辅助选择, 选取在不同位置发生交换的交换单株, 利用其穗子作为分离群体进行后代测验。如图3所示, 发现当DST_InD7和DST_InD105两个标记之间的染色体片段为杂合状态时(R1, R5-R7), 3681-4等位基因型植株的中胚轴长度显著大于X178基因型的植株, 而当染色体其他位置的片段为杂合状态时(R2-R4), 二者无显著差异。由此同样将目标QTL定位于DST_InD7和DST_InD105之间, 进一步验证了上述精细定位的结果(图4)。
图4
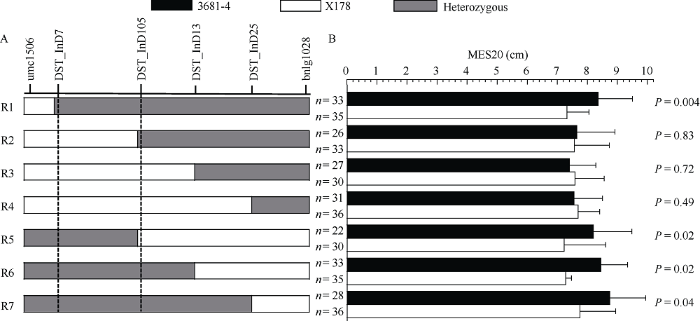 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4后代测验验证定位区间
A: 交换单株的交换位置。B: 对交换单株的后代进行基因型和表型分析。图中黑色、白色和灰色区段分别代表来自3681-4、X178和杂合的染色体片段。n代表不同基因型单株的个数, P值是不同基因型单株的中胚轴长度做t测验得到。
Fig. 4Validation of the mapping region using progeny test
A: the crossover regions of the recombinants. B: progeny test of the recombinants. Black, white, and grey bars represent homozygous regions for 3681-4, homozygous regions for X178 and heterozygous regions, respectively. n means the number of plants with different genotype; P-values were determined by Student’s t test for mesocotyl length of plants with different genotype.
2.3 近等基因系中差异表达基因分析
为了从转录组水平上解释两亲本之间以及近等基因系材料之间中胚轴长度的差异, 进而挖掘候选基因, 对近等基因系和亲本材料进行了RNA-Seq测序。各样品的Reads与参考基因组的比对效率从75.28%到80.45%不等, 大部分的Reads都能比对到参考基因组上的唯一位置, 这部分Reads所占比例从66.71%到76.59%不等。在两亲本X178和3681-4之间共检测到8800个差异表达基因, 接近玉米基因总量的1/4, 且这些基因在10条染色体上大体分布均匀, 说明两个亲本在基因表达水平上的差异很大。而在BC3F3:4家系构建的近等基因系中只检测到115个差异表达基因, 这些基因绝大部分分布在10号染色体, 表明经过连续的回交和自交后, 两个近等基因系之间基因表达的差异大大减少, 可能只在少数的染色体片段上有所差别。两组RNA-Seq数据共同检测到的差异基因共73个(图5)。图5
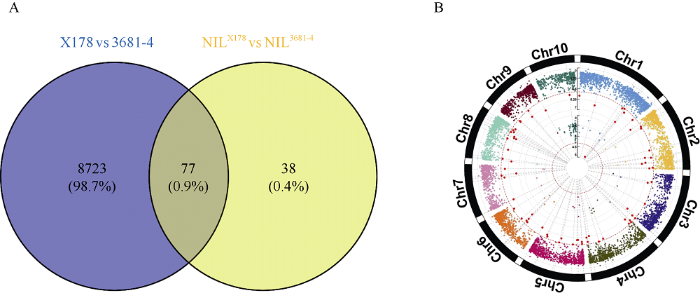 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5差异基因统计和分布图
A: 两组差异表达基因的韦恩图。B: 两组差异表达基因在染色体上的分布图, 大圆和小圆分别表示两亲本和近等基因系中的差异表达基因。P-value通过对两组基因的表达量做t测验获得, 红色的点表示P-value小于0.0001的基因。
Fig. 5Statistics and distribution of differentially expressed genes
A: Venn diagram of differentially expressed genes. B: distribution of differentially expressed genes on chromosomes. The big and small circles denote differentially expressed genes of two parents and NILs respectively. P-value is determined by students’ t-test, and red dots represent genes with P-value < 0.0001.
用AgriGo对近等基因系中的差异表达基因作基因富集分析, 发现这些基因主要参与了对化学刺激的应激反应、氧化还原反应和对氧化胁迫的应激反应等(图6-A)。这表明深播条件对于玉米来说是一种逆境, 诱导了逆境相关基因的表达变化。根据KEGG网站的注释, 这些差异表达基因中有两个与植物激素信号转导途径有关, 分别参与了细胞分裂素和脱落酸的信号转导; 一个基因参与了类胡萝卜素的合成, 在叶黄素循环中负责低光条件下的催化作用; 还有一个基因参与了卟啉和叶绿素的代谢途径, 负责将镁离子螯合到卟啉环上(图6-B)。表明候选基因可能是通过这些途径调控中胚轴伸长, 进而导致近等基因系的表型差异。
图6
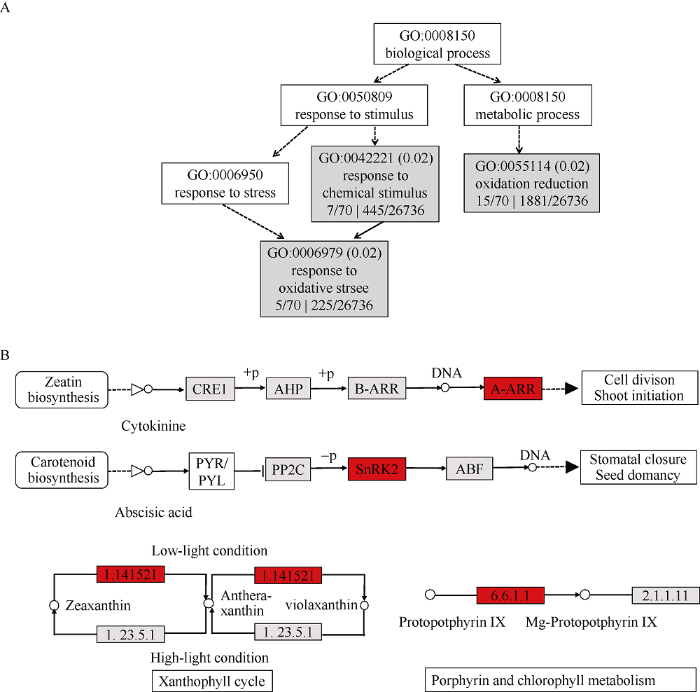 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6近等基因系中差异基因GO分析
A: 灰色方框表示能富集到的通路。B: KEGG网站中植物激素信号转导途径富集到的差异表达基因。
Fig. 6GO analysis of differentially expressed genes in near-isogenic lines
A: grey boxes show the GO terms that can be enriched. B: enriched differentially expressed genes related to plant hormone signal transduction according to KEGG.
3 讨论
目前玉米中已报道的耐深播相关的QTL相对较少。Troyer[9]曾经定位到3个深播条件下与中胚轴长度相关的QTL, 分别位于3号、6号、9号染色体上, Liu等[17]检测到32个与深播发芽能力相关的QTL, 在玉米10条染色体上均有分布。尽管本实验中的耐深播QTL qMES20-10与他们报道的QTL并没有重合, 但我们首先利用170份BC3F3:4家系的基因型和表型, 对QTL区段不同基因型家系的表型做了方差分析, 初步确证了qMES20-10的真实性(图2-A); 并利用覆盖玉米全基因组的芯片对该群体进行了全基因组的QTL检测, 进一步确证了qMES20-10的真实性, 实验结果中仅10号染色体上存在一个主效QTL (LOD>2.5) (图2-B), 说明经过多代自交和回交后, 基本排除了其他位点的干扰, 所以本研究中构建的群体是对目标位点精细定位的理想材料。在精细定位的研究中, 利用BC4F2:3家系以两种策略都将qMES20-10定位到10号染色体133.3~136.0 Mb的区间(图3和图4), 为该位点的进一步图位克隆打下基础。对近等基因系进行差异表达分析, 进而挖掘候选基因, 是QTL定位中寻找候选基因的常用方法[34]。同时差异表达分析还能预测候选基因通过怎样的代谢通路影响表型[35]。本研究的RNA-Seq结果有助于本研究和其他后续研究确定与耐深播相关的候选基因。根据近等基因系中差异表达基因的GO分析结果, 二者之间的差异表达基因主要影响了对化学刺激的应激反应、氧化还原反应和对氧化胁迫的应激反应等(图6-A)。深播条件下, 作物处于缺氧状态, 细胞内可能会通过氧化还原反应产生较多的氧来维持平衡, 进而造成氧化胁迫, 诱导相关基因的表达。因此, 这些差异表达基因很好地解释了近等基因系中胚轴长度的差异。
赵光武等[16]研究表明, 深播条件下玉米中胚轴部位GA3和IAA含量均显著提高, 且外源的GA3和IAA均能促进中胚轴伸长, 这说明深播条件下对两种激素有响应的基因被诱导表达, 从而促进中胚轴伸长。而根据拟南芥中的报道, 细胞分裂素和脱落酸等植物激素也会影响下胚轴伸长[36,37], 说明植物激素在中胚轴伸长中有重要的调控作用。本研究根据KEGG网站的注释信息, 差异表达基因影响了细胞分裂素和脱落酸的信号转导, 说明候选基因可能通过参与这些植物激素的调控影响中胚轴伸长。另外, 光信号通路也会影响拟南芥的下胚轴伸长[38], 本研究中的差异表达基因还参与了叶绿素合成和类胡萝卜素合成, 表明候选基因也可能通过光信号通路调控中胚轴伸长(图6-B)。总之, 这些差异表达基因为进一步克隆qMES20-10位点和解释基因功能奠定了良好的基础。
4 结论
构建了X178作为受体亲本的高代回交群体, 将耐深播主效QTL qMES20-10进一步精细定位到10号染色体上133.3~136.0 Mb的区间, 并分析了近等基因系中基因差异表达情况。研究结果为该QTL的克隆奠定了基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1126/science.1251423URLPMID:24786079 [本文引用: 1]

A key question for climate change adaptation is whether existing cropping systems can become less sensitive to climate variations. We use a field-level data set on maize and soybean yields in the central United States for 1995 through 2012 to examine changes in drought sensitivity. Although yields have increased in absolute value under all levels of stress for both crops, the sensitivity of maize yields to drought stress associated with high vapor pressure deficits has increased. The greater sensitivity has occurred despite cultivar improvements and increased carbon dioxide and reflects the agronomic trend toward higher sowing densities. The results suggest that agronomic changes tend to translate improved drought tolerance of plants to higher average yields but not to decreasing drought sensitivity of yields at the field scale.
[本文引用: 1]
DOI:10.1007/s11104-004-4040-8URL [本文引用: 1]
Crop residues protect soils from erosion, reduce soil water evaporation and increase soil organic matter. Yet management of stubbles for cropping can be difficult. Surface-retained residue can act as a mechanical barrier to slow emergence and reduce seedling biomass. Longer coleoptiles improve seedling emergence with deep sowing and may assist where stubble load is large. In a glasshouse study, six wheat and barley genotypes were sown at 30 and 50
 mm depth into pots containing pasteurised soil. Unweathered sorghum, canola and wheat stubble were added at 0, 3 and 6t/ha equivalents to the soil surface and pots watered above or below the stubble. Stubble species and watering regime had little effect on seedling growth. However, deeper sowing and increased stubble mass adversely affected most seedling characteristics particularly slowing seedling emergence and reducing tiller number to decrease plant biomass (environmental correlations (re) of –0.98** and 0.88**, respectively). Shorter coleoptile Rht-B1b wheats
mm depth into pots containing pasteurised soil. Unweathered sorghum, canola and wheat stubble were added at 0, 3 and 6t/ha equivalents to the soil surface and pots watered above or below the stubble. Stubble species and watering regime had little effect on seedling growth. However, deeper sowing and increased stubble mass adversely affected most seedling characteristics particularly slowing seedling emergence and reducing tiller number to decrease plant biomass (environmental correlations (re) of –0.98** and 0.88**, respectively). Shorter coleoptile Rht-B1b wheats  Banks
Banks and Janz, and barley Beecher emerged slower and abnormally with thicker stubble, and had more sterile tillers to reduce total tiller number and biomass. Deeper crowns for these genotypes also resulted in proportionally less biomass located above the stubble. The converse was true of long coleoptile Vigour 18, Halberd, and its Rht8 progeny, HM14bS which were less affected by stubble mass and sowing depth. In a corresponding field study, increasing wheat stubble mass from 0 to 3 and 6 t/ha delayed seedling emergence and decreased plant number to reduce biomass. Short coleoptile wheat genotypes Hartog and Janz emerged slower and produced less biomass at 3 and 6t/ha of stubble than long coleoptile wheat genotypes Halberd and HM14bS. Emergence of seedlings sown at 50mm depth with 6t/ha overlying stubble was similar to that sown at 120mm with no stubble, reflecting the similar impact of retained residues to deep sowing. Genetic variation for coleoptile length and availability of gibberellin-responsive dwarfing genes such as Rht8 will allow development of long coleoptile wheats for deep sowing or where stubble retention is practiced.
and Janz, and barley Beecher emerged slower and abnormally with thicker stubble, and had more sterile tillers to reduce total tiller number and biomass. Deeper crowns for these genotypes also resulted in proportionally less biomass located above the stubble. The converse was true of long coleoptile Vigour 18, Halberd, and its Rht8 progeny, HM14bS which were less affected by stubble mass and sowing depth. In a corresponding field study, increasing wheat stubble mass from 0 to 3 and 6 t/ha delayed seedling emergence and decreased plant number to reduce biomass. Short coleoptile wheat genotypes Hartog and Janz emerged slower and produced less biomass at 3 and 6t/ha of stubble than long coleoptile wheat genotypes Halberd and HM14bS. Emergence of seedlings sown at 50mm depth with 6t/ha overlying stubble was similar to that sown at 120mm with no stubble, reflecting the similar impact of retained residues to deep sowing. Genetic variation for coleoptile length and availability of gibberellin-responsive dwarfing genes such as Rht8 will allow development of long coleoptile wheats for deep sowing or where stubble retention is practiced.DOI:10.1016/j.fcr.2006.08.003URL [本文引用: 1]
DOI:10.1007/s00122-007-0540-2URL [本文引用: 1]
Wheat crops with greater early vigour shade the soil surface more rapidly and reduce water loss. Evaporative losses affect water-use efficiency particularly in drier regions where most of the rainfall occurs early in the growing season before canopy closure. Greater seedling leaf area and longer coleoptiles are major determinants of increased vigour and better crop establishment. A previously developed high vigour breeding line ‘Vigour 18’ was used to establish a large recombinant inbred family and framework map to identify a QTL on chromosome 6A that accounted for up to 8% of the variation for coleoptile length, 14% of seedling leaf width and was associated with increased plant height. The SSR marker NW3106, nearest to the 6A QTL, was also associated with greater leaf width in a breeding population that was also derived from a cross involving the high vigour donor line ‘Vigour18’. The association between the NW3106 marker and coleoptile length was validated in a second breeding population which was developed using an unrelated long coleoptile donor line. The ‘Vigour18’ allele of the QTL on chromosome 6A promoted coleoptile length and leaf width during early plant growth but was also associated with increased plant height at maturity. Markers linked to the QTL are being used to increase the frequency of increased vigour and long coleoptile alleles in early generations of breeding populations.
DOI:10.1016/j.fcr.2004.06.004URL [本文引用: 1]
DOI:10.5897/AJARURL [本文引用: 1]
DOI:10.1080/09670870050206019URL [本文引用: 1]
URLPMID:9093865 [本文引用: 2]
Knowing breeding behavior and cytological location of traits helps breeders. My objective was to locate dominant genes for long first internode of corn (Zea mays L.). I determined that Hopi Indian corn PI213733 (variety Komona) displayed the trait and grew well in the U.S. Corn Belt. I crossed PI213733 to 26 translocation tester stocks in Minnesota inbred A188 background, backcrossed semi-sterile plants carrying the translocation to A188 the next generation, and grew the segregating generation planted in trenches 15 cm deep with ridges of dirt 10 cm high one year, in trenches 25 cm deep the other year and also at normal (6 cm) depth. Emerged plants were classified for semi-sterility or for normal pollen. I concluded from multiple testers for each chromosome arm that dominant genes for long first internode are located (chromosome and region) on 3S, on 6 near the centromere, and on 9S; spurious associations occurred for two testers. Measurement of cell lengths indicated that PI213733 had more cells than A188 both in upper and in lower mesocotyl sections and that lower, older cells elongated sooner. I found a normal-sized kernel with twin embryos that developed two long first internode seedlings indicating that the amount of endosperm did not limit mesocotyl growth.
DOI:10.2134/agronj1950.00021962004200050010xURL [本文引用: 1]
DOI:10.1104/pp.19.3.537URLPMID:16653935 [本文引用: 1]
DOI:10.1016/j.fcr.2006.05.001URL [本文引用: 1]
DOI:10.1007/s00425-015-2434-xURLPMID:26612069 [本文引用: 1]
MAIN CONCLUSION: Totally, 23 loci were detected, and 383 candidate genes were identified, and four of these candidate genes, Os01g0392100, Os04g0630000, Os01g0904700 and Os07g0615000, were regarded as promising targets. Direct-seeding cultivation is becoming popular in rice (Oryza sativa L.)-planting countries because it is labor- and time-efficient. However, low seedling establishment and slow seedling emergence have restricted the application and popularity of the technique. Mesocotyl elongation and shoot length are two important traits that can enhance rice seedling emergence. A single nucleotide polymorphism (SNP) is a genome sequence variation caused by a single base within a population, and SNPs evenly distributed throughout the genomes of plant species. In this study, a genome-wide association study (GWAS), based on 4136 SNPs, was performed using a compressed mixed linear model that accounted for population structure and relative kinship to detect novel loci for the two traits. Totally, 23 loci were identified, including five loci located known QTLs region. For the mesocotyl elongation, 17 major loci were identified, explaining ~19.31 % of the phenotypic variation. For the shoot length, six major loci were detected, explaining ~ 39.79 % of the phenotypic variation. In total, 383 candidate genes were included in a 200-kb genomic region (+/- 100 kb of each locus). Additionally, 32 SNPs were identified in 30 candidate genes. Relative expression level analyses indicated that four candidate genes containing SNP variations, Os01g0392100, Os04g0630000, Os01g0904700 and Os07g0615000, represented promising targets. Finally, eight elite accessions with long mesocotyl and shoot lengths were chosen as breeding donors for further rice direct-seeding variety modifications.
DOI:10.1186/s12870-015-0608-0URLPMID:26362270 [本文引用: 1]
BACKGROUND: Mechanized dry seeded rice can save both labour and water resources. Rice seedling establishment is sensitive to sowing depth while mesocotyl elongation facilitates the emergence of deeply sown seeds. RESULTS: A set of 270 rice accessions, including 170 from the mini-core collection of Chinese rice germplasm (C Collection) and 100 varieties used in a breeding program for drought resistance (D Collection), was screened for mesocotyl lengths of seedlings grown in water (MLw) in darkness and in 5 cm sand culture (MLs). Twenty six accessions (10.53 %) have MLw longer than 1.0 cm. Eleven accessions had the highest mesocotyl lengths, i.e. 1.4 - 5.05 cm of MLw and 3.0 - 6.4 cm in 10 cm sand culture, including 7 upland landraces or varieties. The genotypic data of 1,019,883 SNPs were developed by re-sequencing of those accessions. A whole-genome SNP array (Rice SNP50) was used to genotype 24 accessions as a validation panel, giving 98.41 % of consistent SNPs with the re-sequencing data in average. GWAS based on compressed mixed linear model was conducted using GAPIT. Based on a threshold of -log(P) >/=8.0, 13 loci were associated to MLw on rice chromosome 1, 3, 4, 5, 6 and 9, respectively. Three associated loci, on chromosome 3, 6, and 10, were detected for MLs. A set of 99 associated SNPs for MLw, based on a compromised threshold (-log(P) >/=7.0), located in intergenic regions or different positions of 36 annotated genes, including one cullin and one growth regulating factor gene. CONCLUSIONS: Higher proportion and extension of elongated mesocotyls were observed in the mini-core collection of rice germplasm and upland rice landraces or varieties, possibly causing the correlation between mesocotyl elongation and drought resistance. GWAS found 13 loci for mesocotyl length measured in dark germination that confirmed the previously reported co-location of two QTLs across populations and experiments. Associated SNPs hit 36 annotated genes including function-matching candidates like cullin and GRF. The germplasm with elongated mesocotyl, especially upland landraces or varieties, and the associated SNPs could be useful in further studies and breeding of mechanized dry seeded rice.
DOI:10.3389/fpls.2018.00332URLPMID:29616055 [本文引用: 1]
Dry direct-seeding of rice is rapidly increasing in China, but variable planting depth associated with machine sowing can lead to low seedling emergence rates. Phenotype analysis of 621 rice accessions showed that mesocotyl length (ML) was induced by deep soil covering and was important in deep-sowing tolerance in the field. Here, we performed and compared GWAS using three types of SNPs (non-synonymous SNP, non-synonymous SNPs and SNPs within promoters and 3 million randomly selected SNPs from the entire set of SNPs) and found that Non-Syn GWAS (GWAS using non-synonyomous SNP) decreased computation time and eliminated confounding by other loci relative to GWAS using randomly selected SNPs. Thirteen QTLs were finally detected, and two new major-effect genes, named OsML1 and OsML2, were identified by an integrated analysis. There were 2 and 7 non-synonymous SNPs in OsML1 and OsML2, respectively, from which 3 and 4 haplotypes were detected in cultivated rice. Combinations of superior haplotypes of OsML1 and OsML2 increased ML by up to 4 cm, representing high emergence rate (85%) in the field with 10 cm of soil cover. The studies provide key loci and naturally occurring alleles of ML that can be used in improving tolerance to dry direct-seeding.
[本文引用: 2]
[本文引用: 2]
DOI:10.3389/fpls.2017.00813URLPMID:28588594 [本文引用: 2]
Deep-sowing is an effective measure to ensure seeds absorbing water from deep soil layer and emerging normally in arid and semiarid regions. However, existing varieties demonstrate poor germination ability in deep soil layer and some key quantitative trait loci (QTL) or genes related to deep-sowing germination ability remain to be identified and analyzed. In this study, a high-resolution genetic map based on 280 lines of the intermated B73 x Mo17 (IBM) Syn10 doubled haploid (DH) population which comprised 6618 bin markers was used for the QTL analysis of deep-sowing germination related traits. The results showed significant differences in germination related traits under deep-sowing condition (12.5 cm) and standard-germination condition (2 cm) between two parental lines. In total, 8, 11, 13, 15, and 18 QTL for germination rate, seedling length, mesocotyl length, plumule length, and coleoptile length were detected for the two sowing conditions, respectively. These QTL explained 2.51-7.8% of the phenotypic variance with LOD scores ranging from 2.52 to 7.13. Additionally, 32 overlapping QTL formed 11 QTL clusters on all chromosomes except for chromosome 8, indicating the minor effect genes have a pleiotropic role in regulating various traits. Furthermore, we identified six candidate genes related to deep-sowing germination ability, which were co-located in the cluster regions. The results provide a basis for molecular marker assisted breeding and functional study in deep-sowing germination ability of maize.
DOI:10.1093/jxb/eru506URLPMID:25680791 [本文引用: 1]
Characterizing the physiological mechanisms behind major-effect drought-yield quantitative trait loci (QTLs) can provide an understanding of the function of the QTLs-as well as plant responses to drought in general. In this study, we characterized rice (Oryza sativa L.) genotypes with QTLs derived from drought-tolerant traditional variety AdaySel that were introgressed into drought-susceptible high-yielding variety IR64, one of the most popular megavarieties in South Asian rainfed lowland systems. Of the different combinations of the four QTLs evaluated, genotypes with two QTLs (qDTY 2.2 + qDTY 4.1 ) showed the greatest degree of improvement under drought compared with IR64 in terms of yield, canopy temperature, and normalized difference vegetation index (NDVI). Furthermore, qDTY 2.2 and qDTY 4.1 showed a potential for complementarity in that they were each most effective under different severities of drought stress. Multiple drought-response mechanisms were observed to be conferred in the genotypes with the two-QTL combination: higher root hydraulic conductivity and in some cases greater root growth at depth. As evidenced by multiple leaf water status and plant growth indicators, these traits affected transpiration but not transpiration efficiency or harvest index. The results from this study highlight the complex interactions among major-effect drought-yield QTLs and the drought-response traits they confer, and the need to evaluate the optimal combinations of QTLs that complement each other when present in a common genetic background.
URL [本文引用: 1]
应用cDNA微阵列对来源于中156/谷梅2号重组自交系的水稻抗稻瘟病近等基因系G205和G71的稻瘟病菌胁迫基因表达谱进行了分析,发现有3个cDNA克隆的表达仅在抗病基因系G205接种病原菌12h后受到诱导,其中两个为功能已知基因,另一个为功能未知的新基因.另有35个差异表达克隆在两个近等基因系中均检测到,其中17个克隆的表达在G205和G71均受到病原菌的诱导,另外18个克隆的表达则在G205和G71均受到病原菌的抑制.序列分析表明,这些稻瘟病菌应答基因分别与防卫反应、信号传递、逆境胁迫和光合作用及糖代谢等功能相关,为植物抗病机制提供了相关信息.另外,Northern还证实了编码富含甘氨酸蛋白基因(Glycinerich protein Grp)的表达受稻瘟病病原菌的诱导,是一个稻瘟病诱导相关基因.
URL [本文引用: 1]
应用cDNA微阵列对来源于中156/谷梅2号重组自交系的水稻抗稻瘟病近等基因系G205和G71的稻瘟病菌胁迫基因表达谱进行了分析,发现有3个cDNA克隆的表达仅在抗病基因系G205接种病原菌12h后受到诱导,其中两个为功能已知基因,另一个为功能未知的新基因.另有35个差异表达克隆在两个近等基因系中均检测到,其中17个克隆的表达在G205和G71均受到病原菌的诱导,另外18个克隆的表达则在G205和G71均受到病原菌的抑制.序列分析表明,这些稻瘟病菌应答基因分别与防卫反应、信号传递、逆境胁迫和光合作用及糖代谢等功能相关,为植物抗病机制提供了相关信息.另外,Northern还证实了编码富含甘氨酸蛋白基因(Glycinerich protein Grp)的表达受稻瘟病病原菌的诱导,是一个稻瘟病诱导相关基因.
DOI:10.1111/pbr.2010.129.issue-1URL [本文引用: 1]
DOI:10.1007/s00122-011-1700-yURL [本文引用: 1]
Deep-seeding tolerant seeds can emerge from deep soil where the moisture is suitable for seed germination. Breeding deep-seeding tolerant cultivars is becoming increasingly important in arid and semi-arid regions. To dissect the quantitative trait loci (QTL) controlling deep-seeding tolerance traits, we selected a tolerant maize inbred line 3681-4 and crossed it with the elite inbred line-X178 to generate an F-2 population and the derivative F-2:3 families. A molecular linkage map composed of 179 molecular markers was constructed, and 25 QTL were detected including 10 QTL for sowing at 10 cm depth and 15 QTL for sowing at 20 cm depth. The QTL analysis results confirmed that deep-seeding tolerance was mainly caused by mesocotyl elongation and also revealed considerable overlap among QTL for different traits. To confirm a major QTL on chromosome 10 for mesocotyl length measured at 20 cm depth, we selected and self-pollinated a BC3F2 plant that was heterozygous at the markers around the target QTL and homozygous at other QTL to generate a BC3F3 population. We found that this QTL explained more phenotypic variance in the BC3F3 population than that in the F-2 population, which laid the foundation for fine mapping and NIL (near-isogenic line) construction.
DOI:10.1534/genetics.111.133066URL [本文引用: 1]
Haploids and doubled haploid (DH) inbred lines have become an invaluable tool for maize genetic research and hybrid breeding, but the genetic basis of in vivo induction of maternal haploids is still unknown. This is the first study reporting comparative quantitative trait locus (QTL) analyses of this trait in maize. We determined haploid induction rates (HIR) in testcrosses of a total of 1061 progenies of four segregating populations involving two temperate haploid inducers, UH400 (HIR = 8%) and CAUHOI (HIR = 2%), one temperate and two tropical inbreds with HIR = 0%, and up to three generations per population. Mean HIR of the populations ranged from 0.6 to 5.2% and strongly deviated from the midparent values. One QTL (qhir1) explaining up to (p) over cap = 66% of the genetic variance was detected in bin 1.04 in the three populations involving a noninducer parent and the HIR-enhancing allele was contributed by UH400. Segregation ratios of loci in bin 1.04 were highly distorted against the UH400 allele in these three populations, suggesting that transmission failure of the inducer gamete and haploid induction ability are related phenomena. In the CAUHOI x UH400 population, seven QTL were identified on five chromosomes, with qhir8 on chromosome 9 having (p) over cap > 20% in three generations of this cross. The large-effect QTL qhir1 and qhir8 will likely become fixed quickly during inducer development due to strong selection pressure applied for high HIR. Hence, marker-based pyramiding of small-effect and/or modifier QTL influencing qhir1 and qhir8 may help to further increase HIR in maize. We propose a conceptual genetic framework for inheritance of haploid induction ability, which is also applicable to other dichotomous traits requiring progeny testing, and discuss the implications of our results for haploid inducer development.
DOI:10.1023/A:1007585532036URL [本文引用: 1]
DOI:10.1534/g3.114.010454URLPMID:24747759 [本文引用: 1]
Positional cloning in maize (Zea mays) requires development of markers in the region of interest. We found that primers designed to amplify annotated insertion-deletion polymorphisms of seven base pairs or greater between B73 and Mo17 produce polymorphic markers at a 97% frequency with 49% of the products showing co-dominant fragment length polymorphisms. When the same polymorphisms are used to develop markers for B73 and W22 or Mo17 and W22 mapping populations, 22% and 31% of markers are co-dominant, respectively. There are 38,223 Indel polymorphisms that can be converted to markers providing high-density coverage throughout the maize genome. This strategy significantly increases the efficiency of marker development for fine-mapping in maize.
DOI:10.1007/s00122-007-0663-5URL [本文引用: 1]
It has long been recognized that epistasis or interactions between non-allelic genes plays an important role in the genetic control and evolution of quantitative traits. However, the detection of epistasis and estimation of epistatic effects are difficult due to the complexity of epistatic patterns, insufficient sample size of mapping populations and lack of efficient statistical methods. Under the assumption of additivity of QTL effects on the phenotype of a trait in interest, the additive effect of a QTL can be completely absorbed by the flanking marker variables, and the epistatic effect between two QTL can be completely absorbed by the four marker-pair multiplication variables between the two pairs of flanking markers. Based on this property, we proposed an inclusive composite interval mapping (ICIM) by simultaneously considering marker variables and marker-pair multiplications in a linear model. Stepwise regression was applied to identify the most significant markers and marker-pair multiplications. Then a two-dimensional scanning (or interval mapping) was conducted to identify QTL with significant digenic epistasis using adjusted phenotypic values based on the best multiple regression model. The adjusted values retain the information of QTL on the two current mapping intervals but exclude the influence of QTL on other intervals and chromosomes. Epistatic QTL can be identified by ICIM, no matter whether the two interacting QTL have any additive effects. Simulated populations and one barley doubled haploids (DH) population were used to demonstrate the efficiency of ICIM in mapping both additive QTL and digenic interactions.
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/j.1744-7909.2012.01108.xURL [本文引用: 1]
A thorough understanding of the quantitative trait loci (QTLs) that underlie agronomically important traits in crops would greatly increase agricultural productivity. Although advances have been made in QTL cloning, the majority of QTLs remain unknown because of their low heritability and minor contributions to phenotypic performance. Here we summarize the key advantages and disadvantages of current QTL fine-mapping methodologies, and then introduce a sequential QTL fine-mapping strategy based on both genotypes and phenotypes of progeny derived from recombinants. With this mapping strategy, experimental errors could be dramatically diminished so as to reveal the authentic genetic effect of target QTLs. The number of progeny required to detect QTLs at various R2 values was calculated, and the backcross generation suitable to start QTL fine-mapping was also estimated. This mapping strategy has proved to be very powerful in narrowing down QTL regions, particularly minor-effect QTLs, as revealed by fine-mapping of various resistance QTLs in maize. Application of this sequential QTL mapping strategy should accelerate cloning of agronomically important QTLs, which is currently a substantial challenge in crops.
Yang Q, Zhang D, Xu M (2012) A sequential QTL fine-mapping strategy using recombinant-derived progeny. J. Integr. Plant Biol. 54(4), 228–237.
DOI:10.1007/s00122-006-0223-4URL [本文引用: 1]
The objective of the present study was to identify favourable exotic Quantitative Trait Locus (QTL) alleles for the improvement of agronomic traits in the BC2DH population S42 derived from a cross between the spring barley cultivar Scarlett and the wild barley accession ISR42-8 (Hordeum vulgare ssp. spontaneum). QTLs were detected as a marker main effect and/or a marker × environment interaction effect (M×E) in a three-factorial ANOVA. Using field data of up to eight environments and genotype data of 98 SSR loci, we detected 86 QTLs for nine agronomic traits. At 60 QTLs the marker main effect, at five QTLs the M×E interaction effect, and at 21 QTLs both the effects were significant. The majority of the M×E interaction effects were due to changes in magnitude and are, therefore, still valuable for marker assisted selection across environments. The exotic alleles improved performance in 31 (36.0%) of 86 QTLs detected for agronomic traits. The exotic alleles had favourable effects on all analysed quantitative traits. These favourable exotic alleles were detected, in particular on the short arm of chromosome 2H and the long arm of chromosome 4H. The exotic allele on 4HL, for example, improved yield by 7.1%. Furthermore, the presence of the exotic allele on 2HS increased the yield component traits ears per m2 and thousand grain weight by 16.4% and 3.2%, respectively. The present study, hence, demonstrated that wild barley does harbour valuable alleles, which can enrich the genetic basis of cultivated barley and improve quantitative agronomic traits.
DOI:10.1007/978-1-4939-3167-5_18URLPMID:26519415 [本文引用: 1]
The recent advances in high throughput RNA sequencing (RNA-Seq) have generated huge amounts of data in a very short span of time for a single sample. These data have required the parallel advancement of computing tools to organize and interpret them meaningfully in terms of biological implications, at the same time using minimum computing resources to reduce computation costs. Here we describe the method of analyzing RNA-seq data using the set of open source software programs of the Tuxedo suite: TopHat and Cufflinks. TopHat is designed to align RNA-seq reads to a reference genome, while Cufflinks assembles these mapped reads into possible transcripts and then generates a final transcriptome assembly. Cufflinks also includes Cuffdiff, which accepts the reads assembled from two or more biological conditions and analyzes their differential expression of genes and transcripts, thus aiding in the investigation of their transcriptional and post transcriptional regulation under different conditions. We also describe the use of an accessory tool called CummeRbund, which processes the output files of Cuffdiff and gives an output of publication quality plots and figures of the user's choice. We demonstrate the effectiveness of the Tuxedo suite by analyzing RNA-Seq datasets of Arabidopsis thaliana root subjected to two different conditions.
DOI:10.1038/nbt.1621URLPMID:20436464 [本文引用: 1]
High-throughput mRNA sequencing (RNA-Seq) promises simultaneous transcript discovery and abundance estimation. However, this would require algorithms that are not restricted by prior gene annotations and that account for alternative transcription and splicing. Here we introduce such algorithms in an open-source software program called Cufflinks. To test Cufflinks, we sequenced and analyzed >430 million paired 75-bp RNA-Seq reads from a mouse myoblast cell line over a differentiation time series. We detected 13,692 known transcripts and 3,724 previously unannotated ones, 62% of which are supported by independent expression data or by homologous genes in other species. Over the time series, 330 genes showed complete switches in the dominant transcription start site (TSS) or splice isoform, and we observed more subtle shifts in 1,304 other genes. These results suggest that Cufflinks can illuminate the substantial regulatory flexibility and complexity in even this well-studied model of muscle development and that it can improve transcriptome-based genome annotation.
DOI:10.1186/gb-2010-11-10-r106URLPMID:20979621 [本文引用: 1]
High-throughput sequencing assays such as RNA-Seq, ChIP-Seq or barcode counting provide quantitative readouts in the form of count data. To infer differential signal in such data correctly and with good statistical power, estimation of data variability throughout the dynamic range and a suitable error model are required. We propose a method based on the negative binomial distribution, with variance and mean linked by local regression and present an implementation, DESeq, as an R/Bioconductor package.
DOI:10.1093/nar/gkx382URLPMID:28472432 [本文引用: 1]
The agriGO platform, which has been serving the scientific community for >10 years, specifically focuses on gene ontology (GO) enrichment analyses of plant and agricultural species. We continuously maintain and update the databases and accommodate the various requests of our global users. Here, we present our updated agriGO that has a largely expanded number of supporting species (394) and datatypes (865). In addition, a larger number of species have been classified into groups covering crops, vegetables, fish, birds and insects closely related to the agricultural community. We further improved the computational efficiency, including the batch analysis and P-value distribution (PVD), and the user-friendliness of the web pages. More visualization features were added to the platform, including SEACOMPARE (cross comparison of singular enrichment analysis), direct acyclic graph (DAG) and Scatter Plots, which can be merged by choosing any significant GO term. The updated platform agriGO v2.0 is now publicly accessible at http://systemsbiology.cau.edu.cn/agriGOv2/.
DOI:10.1007/s00122-017-3023-0URLPMID:29170790 [本文引用: 1]
KEY MESSAGE: This study demonstrates how identification of genes underpinning disease-resistance QTL based on differential expression and SNPs can be improved by performing transcriptomic analysis on multiple near isogenic lines. Transcriptomic analysis has been widely used to understand the genetic basis of a trait of interest by comparing genotypes with contrasting phenotypes. However, these approaches identify such large sets of differentially expressed genes that it proves difficult to isolate which genes underpin the phenotype of interest. This study tests whether using multiple near isogenic lines (NILs) can improve the resolution of RNA-seq-based approaches to identify genes underpinning disease-resistance QTL. A set of NILs for a major effect Fusarium crown rot-resistance QTL in barley on the 4HL chromosome arm were analysed under Fusarium crown rot using RNA-seq. Differential gene expression and single nucleotide polymorphism detection analyses reduced the number of putative candidates from thousands within individual NIL pairs to only one hundred and two genes, which were differentially expressed or contained SNPs in common across NIL pairs and occurred on 4HL. Our findings support the value of performing RNA-seq analysis using multiple NILs to remove genetic background effects. The enrichment analyses indicated conserved differences in the response to infection between resistant and sensitive isolines suggesting that sensitive isolines are impaired in systemic defence response to Fusarium pseudograminearum.
DOI:10.1186/s12870-017-1124-1URLPMID:29143606 [本文引用: 1]
BACKGROUND: Some plant species have 'melanin-like' black seed pigmentation. However, the chemical and genetic nature of this 'melanin-like' black pigment have not yet been fully explored due to its complex structure and ability to withstand almost all solvents. Nevertheless, identification of genetic networks participating in trait formation is key to understanding metabolic processes involved in the expression of 'melanin-like' black seed pigmentation. The aim of the current study was to identify differentially expressed genes (DEGs) in barley near-isogenic lines (NILs) differing by allelic state of the Blp (black lemma and pericarp) locus. RESULTS: RNA-seq analysis of six libraries (three replicates for each line) was performed. A total of 957 genome fragments had statistically significant changes in expression levels between lines BLP and BW, with 632 fragments having increased expression levels in line BLP and 325 genome fragments having decreased expression. Among identified DEGs, 191 genes were recognized as participating in known pathways. Among these were metabolic pathways including 'suberin monomer biosynthesis', 'diterpene phytoalexins precursors biosynthesis', 'cutin biosynthesis', 'cuticular wax biosynthesis', and 'phenylpropanoid biosynthesis, initial reactions'. Differential expression was confirmed by real-time PCR analysis of selected genes. CONCLUSIONS: Metabolic pathways and genes presumably associated with black lemma and pericarp colour as well as Blp-associated resistance to oxidative stress and pathogens, were revealed. We suggest that the black pigmentation of lemmas and pericarps is related to increased level of phenolic compounds and their oxidation. The effect of functional Blp on the synthesis of ferulic acid and other phenolic compounds can explain the increased antioxidant capacity and biotic and abiotic stress tolerance of black-grained cereals. Their drought tolerance and resistance to diseases affecting the spike may also be related to cuticular wax biosynthesis. In addition, upregulated synthesis of phytoalexins, suberin and universal stress protein (USP) in lemmas and pericarps of the Blp carriers may contribute to their increased disease resistance. Further description of the DEGs haplotypes and study of their association with physiological characteristics may be useful for future application in barley pre-breeding.
DOI:10.1007/s00425-004-1421-4URLPMID:15843964 [本文引用: 1]
Cytokinins inhibit hypocotyl elongation in darkness but have no obvious effect on hypocotyl length in the light. However, we found that cytokinins do promote hypocotyl elongation in the light when ethylene action is blocked. A 50% increase in Arabidopsis thaliana (L.) Heynh. hypocotyl length was observed in response to N6-benzyladenine (BA) treatment in the presence of Ag+. The level of the ethylene precursor 1-aminocyclopropane-1-carboxylic acid was strongly increased, indicating that ethylene biosynthesis was up-regulated by treatment with cytokinin. Furthermore, the effects of cytokinins on hypocotyl elongation were also tested using a series of mutants in the cascade of the ethylene-signal pathway. In the ethylene-insensitive mutants etr1-3 and ein2-1, cytokinin treatment resulted in hypocotyl lengths comparable to those of wild-type seedlings treated with both Ag+ and BA. A similar phenotypical response to cytokinin was observed when auxin transport was blocked by
 -naphthylphthalamic acid (NPA). Applied cytokinin largely restored cell elongation in the basal and middle parts of the hypocotyls of NPA-treated seedlings and at the same time abolished the NPA-induced decrease in indole-3-acetic acid levels. Our data support the hypothesis that, in the light, cytokinins interact with the ethylene-signalling pathway and conditionally up-regulate ethylene and auxin synthesis.
-naphthylphthalamic acid (NPA). Applied cytokinin largely restored cell elongation in the basal and middle parts of the hypocotyls of NPA-treated seedlings and at the same time abolished the NPA-induced decrease in indole-3-acetic acid levels. Our data support the hypothesis that, in the light, cytokinins interact with the ethylene-signalling pathway and conditionally up-regulate ethylene and auxin synthesis.DOI:10.1093/pcp/pcu028URLPMID:24492258 [本文引用: 1]
Plasma membrane H+-ATPase is thought to mediate hypocotyl elongation, which is induced by the phytohormone auxin through the phosphorylation of the penultimate threonine of H+-ATPase. However, regulation of the H+-ATPase during hypocotyl elongation by other signals has not been elucidated. Hypocotyl elongation in etiolated seedlings of Arabidopsis thaliana was suppressed by the H+-ATPase inhibitors vanadate and erythrosine B, and was significantly reduced in aha2-5, which is a knockout mutant of the major H+-ATPase isoform in etiolated seedlings. Application of the phytohormone ABA to etiolated seedlings suppressed hypocotyl elongation within 30 min at the half-inhibitory concentration (4.2 mu M), and induced dephosphorylation of the penultimate threonine of H+-ATPase without affecting the amount of H+-ATPase. Interestingly, an ABA-insensitive mutant, abi1-1, did not show ABA inhibition of hypocotyl elongation or ABA-induced dephosphorylation of H+-ATPase. This indicates that ABI1, which is an early ABA signaling component through the ABA receptor PYR/PYL/RCARs (pyrabactin resistance/pyrabactin resistance 1-like/regulatory component of ABA receptor), is involved in these responses. In addition, we found that the fungal toxin fusiccocin (FC), an H+-ATPase activator, induced hypocotyl elongation and phosphorylation of the penultimate threonine of H+-ATPase, and that FC-induced hypocotyl elongation and phosphorylation of H+-ATPase were significantly suppressed by ABA. Taken together, these results indicate that ABA has an antagonistic effect on hypocotyl elongation through, at least in part, dephosphorylation of H+-ATPase in etiolated seedlings.
DOI:10.1105/tpc.113.121657URLPMID:24951480 [本文引用: 1]
In Arabidopsis thaliana, the cryptochrome and phytochrome photoreceptors act together to promote photomorphogenic development. The cryptochrome and phytochrome signaling mechanisms interact directly with CONSTITUTIVELY PHOTOMORPHOGENIC1 (COP1), a RING motif-containing E3 ligase that acts to negatively regulate photomorphogenesis. COP1 interacts with and ubiquitinates the transcription factors that promote photomorphogenesis, such as ELONGATED HYPOCOTYL5 and LONG HYPOCOTYL IN FAR-RED1 (HFR1), to inhibit photomorphogenic development. Here, we show that COP1 physically interacts with PIF3-LIKE1 (PIL1) and promotes PIL1 degradation via the 26S proteasome. We further demonstrate that phyB physically interacts with PIL1 and enhances PIL1 protein accumulation upon red light irradiation, probably through suppressing the COP1-PIL1 association. Biochemical and genetic studies indicate that PIL1 and HFR1 form heterodimers and promote photomorphogenesis cooperatively. Moreover, we demonstrate that PIL1 interacts with PIF1, 3, 4, and 5, resulting in the inhibition of the transcription of PIF direct-target genes. Our results reveal that PIL1 stability is regulated by phyB and COP1, likely through physical interactions, and that PIL1 coordinates with HFR1 to inhibit the transcriptional activity of PIFs, suggesting that PIL1, HFR1, and PIFs constitute a subset of antagonistic basic helix-loop-helix factors acting downstream of phyB and COP1 to regulate photomorphogenic development.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}