 ,1,2,*
,1,2,*Identification of drought-related co-expression modules and hub genes in potato roots based on WGCNA
QIN Tian-Yuan1,2,**, SUN Chao1,2,**, BI Zhen-Zhen1,2, LIANG Wen-Jun1,2, LI Peng-Cheng1,2, ZHANG Jun-Lian1, BAI Jiang-Ping,1,2,*通讯作者:
第一联系人:
收稿日期:2019-08-27接受日期:2019-12-26网络出版日期:2020-07-12
| 基金资助: |
Received:2019-08-27Accepted:2019-12-26Online:2020-07-12
| Fund supported: |
作者简介 About authors
秦天元,E-mail:1637835362@qq.com。
孙超,E-mail:sunc@gsau.edu.cn。

摘要
关键词:
Abstract
Keywords:
PDF (5837KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
秦天元, 孙超, 毕真真, 梁文君, 李鹏程, 张俊莲, 白江平. 基于WGCNA的马铃薯根系抗旱相关共表达模块鉴定和核心基因发掘[J]. 作物学报, 2020, 46(7): 1033-1051. doi:10.3724/SP.J.1006.2020.94130
QIN Tian-Yuan, SUN Chao, BI Zhen-Zhen, LIANG Wen-Jun, LI Peng-Cheng, ZHANG Jun-Lian, BAI Jiang-Ping.
外界胁迫(生物胁迫和非生物胁迫)是农业生产面临的主要问题之一, 了解胁迫对植物的影响以及植物对胁迫信号的感知、传递和应答对解决和提高作物产量具有重要的意义。马铃薯作为四大粮食作物之一, 在中国主要种植于年平均降水量小于500 mm的干旱和半干旱地区, 长期的或季节性的干旱胁迫会严重影响马铃薯的植株长势、块茎产量和商品性[1,2]。马铃薯属浅根系作物, 对干旱胁迫较为敏感, 因此抗旱性研究一直是马铃薯产业关注的问题。研究人员从增大根系、减少蒸腾和提高水肥利用率以提高马铃薯耐旱性开展了大量研究, 也对马铃薯的根系表型和抗逆相关生理生化指标对干旱胁迫的响应情况进行了多种分析, 但针对马铃薯抗旱基因的发掘和响应干旱胁迫的系统性分子机制研究还相对较少, 有待进一步加强[3,4]。随着马铃薯双单倍体参考基因组DM的构建完成, 利用转录组、蛋白组和代谢组等生物信息学方法研究马铃薯的抗逆性已经成为一个有力的手段, 然而前人的研究大多限于单个调控因子(如顺式表达元件,反式作用因子), 鲜有利用网络分析的方法解析马铃薯抗逆机理的报道。
近年来, 随着高通量测序技术的飞速发展与成本不断降低, 已能采用多样本的转录组测序系统研究生物学问题。传统的2个样本的比较分析已经不能有效处理海量数据, 因此网络分析应运而生。其中, 权重基因共表达网络分析(weighted gene co- expression network analysis, WGCNA)应用较为广泛, 已在多种动物(如恒河猴[5])和植物(草莓[6]、油菜[7]、玉米[8]等)的多样本转录组数据的分析中发挥重要作用。与其他的网络分析不同, WGCNA可以特异地筛选出与目标性状相关联的基因, 并进行模块化分类, 得到具有高度生物学意义的共表达模块, 再从中筛选核心基因。因此, 选择一个可量化的目标性状是WGCNA分析首要考虑的问题。本研究选用清除活性氧的抗氧化酶活性等生理生化指标作为目标性状。不仅因为抗氧化酶是评价植物耐旱性的传统指标, 而且在近年来模式植物和模式作物的抗旱性研究中, 活性氧(reactive oxygen species, ROS)可作为第二信使, 促使氧化还原信号与其他多种信号通路相互作用, 直接或间接地参与植物的多种代谢过程, 共同调节植物对干旱等逆境的响应[9,10,11]。
本试验以5个时间梯度的干旱胁迫处理, 及根系转录组测序, 结合抗氧化酶活性和根活力的生理生化数据, 利用WGCNA分析构建共表达基因网络, 将基因模块与生理生化数据关联分析, 挖掘出与干旱胁迫调控通路和抗逆指标相关的核心基因, 以期为进一步研究马铃薯根系抗旱的分子遗传机制提供新的线索。
1 材料与方法
1.1 试验材料
由国际马铃薯研究中心引进2个生育期相同、根系差异明显的马铃薯栽培种C16 (CIP 397077.16)和C119 (CIP 398098.119 (表1), 其组培苗由甘肃农业大学作物遗传改良与栽培种创新重点试验室提供。Table 1
表1
表1试验材料
Table 1
| 品种 Variety | CIP编号 CIP number | 母本 Maternal parent | 父本 Paternal parent |
|---|---|---|---|
| C16 | CIP397077.16 | 392025.7=(LR93.221) | 392820.1=(C93.154) |
| C119 | CIP398098.119 | 393371.58 | 392639.31 |
新窗口打开|下载CSV
1.2 试验方法
1.2.1 试验设计 试验容器是体积250 mL的罐头瓶, 用150 mmol L-1甘露醇模拟干旱环境。将马铃薯试管苗的茎段接种在正常MS培养基上, 每瓶接生长状态基本一致的5株茎段, 为了防止污染, 每个处理接5瓶, 待长至25 d时, 取3瓶长势一致的试管苗转入含有150 mmol L-1甘露醇的液体MS培养基中, 处理0 h、2 h、6 h、12 h和24 h后, 对根系取样(其中0 h的样为对照), 样重量不少于2 g, 每样3个生物学重复, 取样后立即置液氮中速冻, -80℃冰箱保存根, 用于RNA的提取和后续转录组测序, 以及抗逆相关生理生化指标的测定。1.2.2 转录组数据的获取与分析 在不同培养条件下, 共采集30个样品[2个材料(C16、C119) 5个时间点(0、2、6、12和24 h) ×3次重复]进行转录组测序。以马铃薯基因组及其注释文件作为参考序列, 分析转录组测序数据, 所有数据均从Ensembl网站下载(http://plants.ensembl.org/Solanum_tuberosum/ Info/Index)。首先使用Hisat2 (v2.2.10)将来自干旱处理和对照文库的测序数据和马铃薯参考基因组比对, 然后利用Samtools (v0.1.19)将SAM文件转换为BAM文件, 并对BAM文件重新排序, 排序后的文件利用cufflinks (v1.2)来组装转录本, 估计这些转录本的丰度, 并且检测样本间的差异表达及可变剪切, 再利用python的一个用于处理高通量数据的HTSeq包来计算样本的表达量。根据基因长度和匹配到该基因的片段数目(readcount值)计算每一个基因的FPKM (ragments per kilobase of exon per million fragments mapped), 即每1百万个map上的reads中map到外显子的每1千个碱基上的reads的个数。一般情况下, 当基因的FPKM绝对值大于1时认为其表达, 否则认为不表达。将表达的基因用DESeq2 R package (v3.5.2)软件和WGCNA进行不同样本间的差异表达分析、基于抗逆相关生理生化数据的模块划分及核心基因筛选。为了精确定位核心基因, 本试验利用P值小于0.01的差异表达基因进行后续筛选。采用AgriGo (http://systemsbiology.cau.edu.cn/ agriGOv2/)[12,13]在线软件进行GO富集分析, 利用Cytoscape[14]软件对基因调控网络进行可视化处理, 其中显著富集的GO类别P值应小于0.05。
2 结果与分析
2.1 差异基因的分析
通过二代转录组测序, 30个样本中共获得792.64 Gb和3,801,046,731个原始测序片段, 对这些原始片段进行过滤后得到Q30大于95.09%的clean reads 3,720,269,696个, 它们的GC含量范围均在41.02%~43.49%之间。30个样本中, clean reads与基因组的匹配率皆大于85%, 说明所有试验样品的采集及测序结果高度可信。为了筛选不同处理相比于对照和不同品种间的差异表达基因, 基于每个转录本的readcount值, 利用DESeq2软件进行目的样本间的差异表达基因分析, 以校正后的P值小于0.01为阈值, 以差异倍数|log2FC| > 1为条件, 筛选出差异极显著的转录本用于后续分析。将C16和C119在不同干旱处理时间点的数据分别与未处理的数据比对, 筛选出极显著上调差异表达(图1-A, B)和极显著下调差异表达的基因(图1-C, D)。干旱处理2 h时, C16中上调差异表达基因为97个, 远远少于C119中的421个(图1-A, B)。在持续胁迫到24 h时, 上调差异表达基因数达到最大值, C16中为606个, 而C119中为843个, 在处理2 h、6 h、12 h和24 h四个时间点, 相对于未处理都上调的基因在C16中有49个, 而C119有78个, 说明在短期干旱处理后C16根系中上调表达基因数目远少于C119。而下调差异表达的基因, 胁迫2 h时 C16中为12个, 少于C119中的50个(图1-C, D)。在持续胁迫到24 h时, C16中下调表达的差异基因为1315个, 而C119中为1718个, C16在持续处理2 h、6 h、12 h和24 h四个时间点相对于0 h都下调的基因为42个, 而C119为21个, 对比发现, 在短期处理后C16根系中上下调表达基因数目远少于C119。以上结果说明, 相比于C16, C119在甘露醇模拟的干旱胁迫下有更多的基因积极上下调表达, 以调整体内代谢途径的变化来防御逆境伤害。
图1
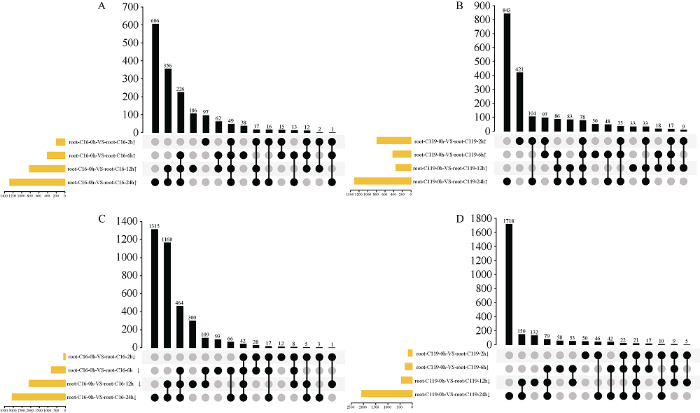 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1不同处理时间下C16与C119根系中上调和下调的差异表达基因数
A, B: C16和C119根中上调表达的差异基因数目; C, D: C16和C119根中下调表达的差异基因数目。黄色柱代表该样本的总基因数目, 黑色柱代表黑色圆点所对应的样本中的差异基因数目, 黑色圆点间的连线代表样本之间拥有的相同差异表达基因。
Fig. 1Upregulated and down-regulated genes in roots of C16 and C119 under different treatments
A, B: the number of differentially expressed genes up-regulated in C16 and C 119 roots; C, D: the down-regulated genes. The yellow column represents the total number of genes in the sample, the black column represents the number of differentially expressed genes in the sample corresponding to the black dots, and the black line between the black dots represents the same differentially expressed genes between the samples.
2.2 差异基因的WGCNA分析
WGCNA分析, 即权重基因共表达网络分析, 是一种多个样本基因表达模式的分析方法。通过WGCNA分析可将表达模式相似的基因聚类而形成不同的模块, 分析模块与特定性状或表型之间的关联性, 通过基因聚类和关联分析结果构建共表达调控网络。相比于只关注差异表达基因的传统分析方法, WGCNA利用数千或近万个变化最大的基因或全部基因分类为不同的基因集, 并与表型进行关联分析, 既充分利用了整体组学信息, 也把数千个基因与表型的关联转换为数个基因集与表型的关联, 免去了多重假设检验校正。位于调控网络中心的基因被称为核心基因, 即hub gene, 这类基因通常是关键的调控基因, 是值得我们深入挖掘和分析的对象, WGCNA分析在组学研究中被广泛应用。本研究将C119和C16的根系中响应干旱胁迫的差异基因结合其根系抗逆生理生化指标数据(附表1), 通过WGCNA分析和基因集-表型关联分析, 深入挖掘出马铃薯根系中抗旱调控网络的核心基因。Supplementary table 1
附表 1
附表 1C16和C119在不同处理时间下的生化指标数据
Supplementary table 1
| 处理时间Treatment time | 超氧化物歧化酶 Superoxide dismutase (U g-1) | 过氧化物酶 Peroxidase (U g-1 min-1) | 过氧化氢酶 Catalase (U g-1 min-1) | 根活力 Root vitality (mg g-1 h-1) | ||||
|---|---|---|---|---|---|---|---|---|
| 平均值 Average value | 标准差 Standard deviation | 平均值 Average value | 标准差 Standard deviation | 平均值 Average value | 标准差 Standard deviation | 平均值 Average value | 标准差 Standard deviation | |
| C16-0 h | 9.57 | 2.00 | 2637.50 | 135.95 | 116.75 | 6.72 | 457.74 | 35.21 |
| C16-2 h | 14.13 | 1.55 | 3375.00 | 187.90 | 129.93 | 7.68 | 457.75 | 30.18 |
| C16-6 h | 15.63 | 1.81 | 3862.50 | 162.71 | 144.84 | 13.26 | 496.57 | 33.10 |
| C16-12 h | 16.42 | 1.75 | 4262.50 | 149.01 | 155.83 | 15.39 | 627.09 | 30.73 |
| C16-24 h | 22.22 | 1.35 | 8575.00 | 142.27 | 209.34 | 20.20 | 815.49 | 62.11 |
| C119-0 h | 46.02 | 5.15 | 1312.50 | 190.92 | 244.25 | 15.10 | 712.44 | 36.99 |
| C119-2 h | 48.92 | 6.91 | 3200.00 | 262.15 | 274.48 | 22.32 | 811.53 | 64.44 |
| C119-6 h | 49.16 | 6.29 | 3500.00 | 218.72 | 276.74 | 14.75 | 826.29 | 81.45 |
| C119-12 h | 56.62 | 7.72 | 3687.50 | 206.59 | 341.57 | 16.84 | 901.41 | 23.47 |
| C119-24 h | 75.53 | 8.95 | 6112.50 | 210.12 | 370.75 | 19.51 | 924.88 | 70.89 |
新窗口打开|下载CSV
2.2.1 去除离群样本 由于网络模块分析结果容易受到离群样本的影响, 因此在构建网络模块之前须去除离群样本, 以保证结果的准确性。本研究共有30个样本, 通过计算各个样本表达水平的相关系数后聚类(图2-A)。相关性较低的样本, 或者在树形图上无法聚类的样本即为离群样本, 如root.C119. 6h.rep1、root.C119.12h.rep3、root.C16. 2h.rep1、root. C16.6h.rep3、root.C16.0h.rep1、root.C16. 12h.rep3。离群样本被去除后, 剩余样本的聚类树如图2-B所示。
图2
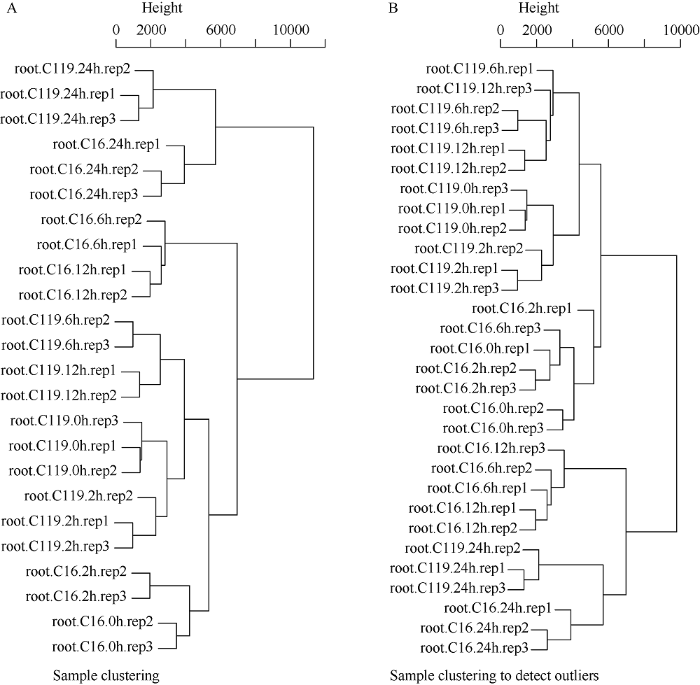 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2样本的聚类树
A: 所有的样本的聚类树; B: 去除离群样本后的聚类树。
Fig. 2Dendrogram of samples
A: dendrogram of all 30 samples; B: dendrogram of samples after removing outlier.
2.2.2 基因共表达网络中软阈值的确定 选取C16和C119两个品种的根系在未处理和短期干旱处理2、6、12和24 h得到的基因表达谱矩阵来构建差异基因的共表达网络, 需要计算出每个基因两两之间的相关系数, 进而采用公式Smn = cor(xm, xn), S = [Smn]得到基因之间的相似矩阵, 式中Smn表示基因m和基因n之间的皮尔森相关系数, S表示相似矩阵。利用R软件中的WGCNA包来构建权重基因共表达网络。为了使网络符合无尺度网络分布, 利用WGCNA包中的函数pick Soft Threshold来计算权重值。根据图3的结果, 选择软阈值β = 9来构建共表达网络。
图3
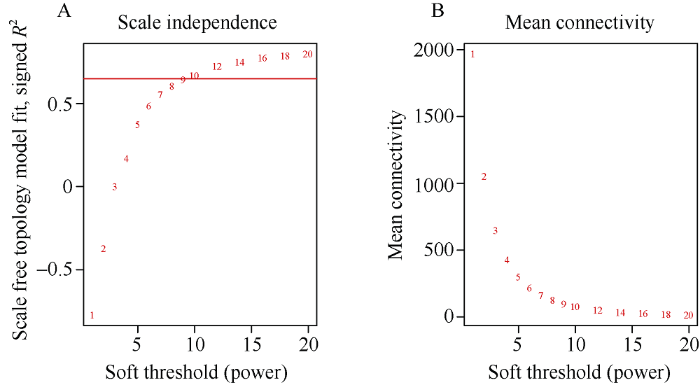 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3基因共表达网络软阈值的确定
图A和图B的横坐标均代表软阈值(β), 图A的纵坐标代表无尺度网络模型指数, 图B的纵坐标代表每一个软阈值对应的网络平均连接程度。
Fig. 3Soft threshold determination of gene co-expression network
The abscissae in both Figures A and B represent the soft threshold, the ordinate of Figure A represents the scale-free network model index, the ordinate of Figure B represents the average degree of network connectivity for each soft threshold.
2.2.3 基因共表达网络基因聚类和模块切割 确定软阈值β=9之后, 根据公式为Amn = [(1+Smn)/2]β将相似矩阵转换为邻接矩阵, 再将邻接矩阵转为拓扑重叠矩阵(Topological Overlap Matrix, TOM),同时利用函数diss Tom = 1-TOM对拓扑重叠矩阵取逆得到相异性矩阵, 这样做的目的是为了清除背景噪音和伪关联带来的误差。最后利用函数hclust对相异矩阵进行层次聚类, 采用动态切割法(Dynamic Tree Cut)将产生的聚类树切割, 该过程可以把表达模式相近的基因合并在同一个分支上, 每一个分支代表一个共表达模块, 不同的颜色代表不同的模块。差异基因根据其FPKM值进行相关度分析并聚类, 相关度较高的基因被分配到同一个模块中(图4)。
图4
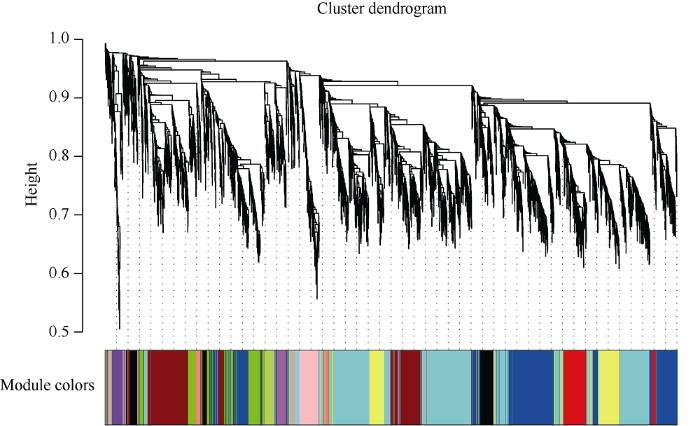 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4基因共表达网络基因聚类数和模块切割
基因聚类树的每一个分支对应一个模块。
Fig. 4Gene co-expression network gene clustering number and modular cutting
Each branch of the gene clustering tree corresponds to one module.
2.2.4 基因共表达网络中模块与生理生化性状矩阵的关联分析 将上一步所获模块中按照75%的相关性兼并融合, 结合样本的4个生理生化性状[超氧化物歧化酶(superoxide dismutase, SOD)、过氧化物酶(peroxidase, POD)、过氧化氢酶(catalase, CAT)和根活力(root vigor, RV)]在不同处理时间下的矩阵得到15个与这些性状相关联的模块, 不同的模块分别以不同的颜色表示(图5)。其中, 部分模块与生理生化性状高度关联。例如, Yellow模块内的基因表达模式随着处理时间的增加与生理生化性状的相关度存在由负到正的趋势, 并在24 h时与SOD、POD、CAT和RV均呈极显著正相关(分别为r=0.97, P=7E-15; r=0.95, P=2E-12; r=0.87, P=4E-08和r=0.98, P=3E-16), Red模块与与生理生化性状的关联情况与Yellow模块类似; Turquoise模块内的基因表达模式则随着处理时间的增加与生理生化性状的相关度存在由正到负的趋势, 并在与处理24 h时与SOD、POD、CAT和根活力(RV)均呈极显著负相关(分别为r= -0.82, P=1E-06; r= -0.81, P=1E-06; r= -0.65, P=6E-04和r= -0.82, P=1E-06); Blue模块与生理生化性状的关联情况与Turquoise模块类似; Pink模块与处理2 h时的SOD、POD、CAT和根活力(RV)存在高度正相关(分别为r=0.80, P=2E-06; r=0.81, P=2E-06; r=0.95, P=3E-12和r=0.80, P=3E-06)(图5)。
图5
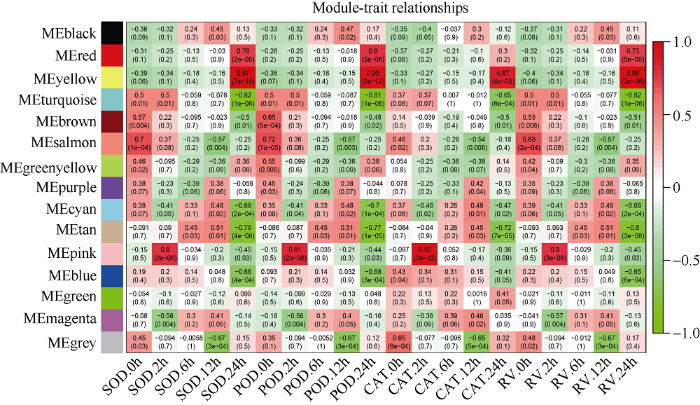 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5基因共表达网络模块与生理生化性状的关联热图
横轴表示不同处理时间下的生理生化性状, 纵轴表示每一个模块的特征向量。红色的格子代表性状与模块具有正相关性, 绿色的格子代表性状与模块具有负相关性。0 h表示未进行干旱处理。
Fig. 5Association analysis of gene co-expression network modules with physiological and biochemical traits
The horizontal axis represents different characteristics, and the vertical axis represents the eigenvectors of each module. The red lattice represents a positive correlation between the physiological traits with the module, while the green lattice represents a negative correlation. 0 h means no drought treatment, as the control.
2.2.5 通过特征向量基因分析确定目标基因模块
基因模块是指一系列高度相关基因的集合, 同一个模块中的基因可能具有相似的生物学功能。由于具有相似表达模式的基因可能具有相似的生物学功能, 通过WGCNA构建的模块在很大程度上与特定的性状相联系, 而特定的性状又与特定的生命活动存在高度协同性。因此, 为了进一步解析生理生化性状特异性模块的生物学功能, 根据基因共表达网络模块与时间性状的关联热图, 挑选出与性状关联度较高的模块, 并进行下一步研究。
由于模块中的基因数目庞大, 可利用降维的思路来研究基因模块, 即把模块中具有代表性的基因—特征向量基因(module eigengene, ME)挑选出来。用ME代表模块中成千上万的基因进行相关性分析, 利用这种方法可以进一步理清基因模块与模块之间的相互关系及与不同处理时间的表型性状的相关性, 筛选出目标基因模块。对所有模块的ME进行聚类分析后发现, 部分ME之间存在很高的相关性(图6)。ME之间的相关性越高, 其所在的模块的相关性也就越高, 通过两两之间的ME相关性分析, 发现Red模块与Blue模块中ME之间的相关性达到0.87, Yellow模块与Turquoise模块中ME之间的相关性达到0.88 (图7), 且Red、Yellow模块内基因随着干旱处理时间的增加, 基因表达模式与抗逆相关生理生化性状由负相关逐渐转为极显著正相关, Tuqurise、Blue模块内基因随着处理时间的增加, 基因表达模式与目标性状由正相关逐渐转为极显著负相关, 因此这4个模块可作为目标基因模块。
图6
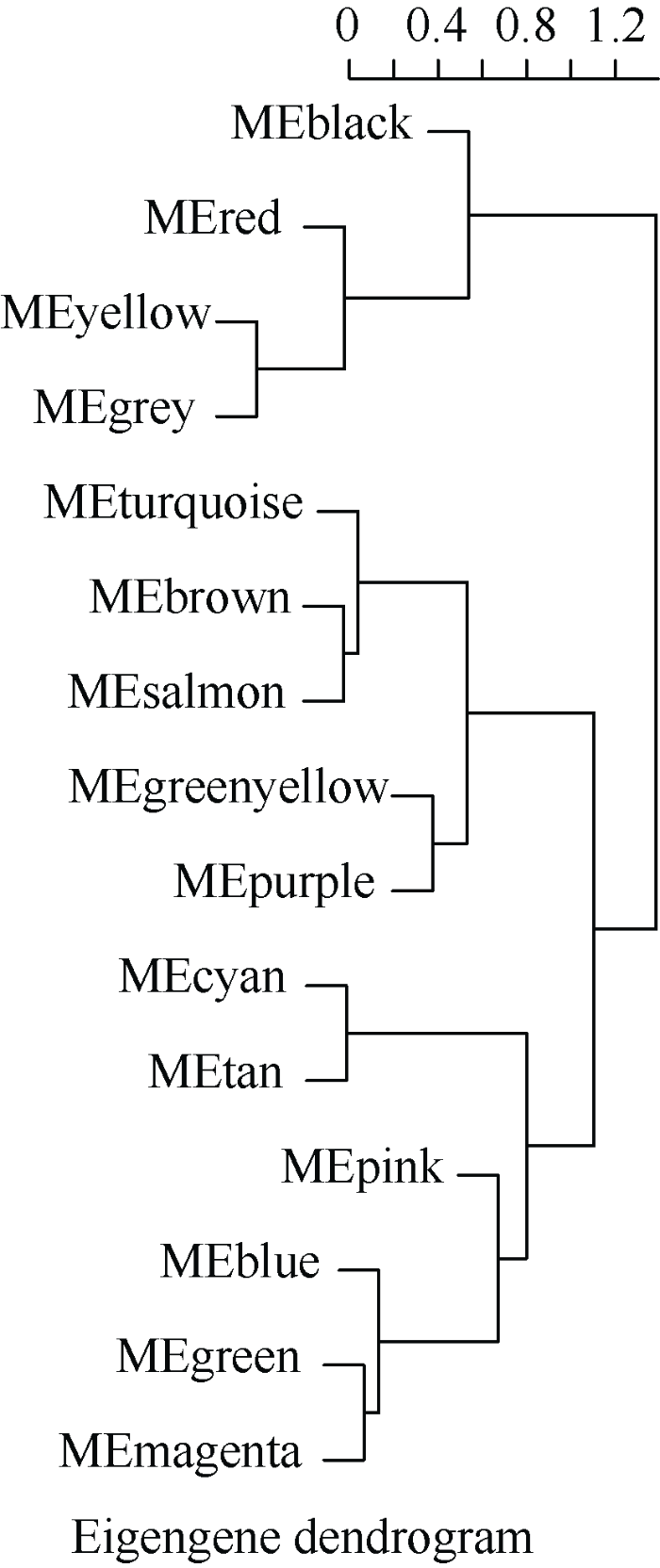 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6ME聚类树
Fig. 6ME cluster tree
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7不同模块两两之间ME的相关性
Fig. 7ME correlation between different modules
将这4个模块中的所有基因和ME基因在所有样本中分别进行了表达水平分析, 发现每个模块内的基因表达水平高度相关, ME表达水平与模块整体表达水平也高度相关, 说明目标模块的ME可充分代表其模块中的整体基因(图8)。
图8
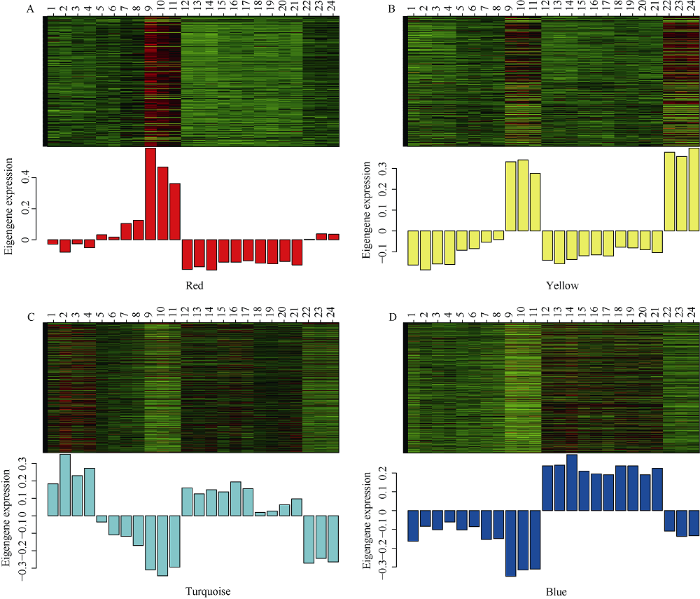 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8各样本中不同模块的所有基因与相应ME的表达水平
上图(热图)为各个样本在模块中所有基因的表达水平。A: Red模块; B: Yellow模块; C: Turquoise模块; D: Blue模块。行代表该模块内的所有基因, 列代表不同的样本。下图(柱状图)为对应的样本中该模块ME的表达水平。
Fig. 8Expression levels of all genes and corresponding ME in different modules of each sample
The above figure (heat map) shows the expression level of all genes in modules. A: Red module; B: Yellow module; C: Turquoise module; D: Blue module. The row represents all genes in the module, and the column represents each sample. The below figure shows the ME expression level of the module in the sample.
2.2.6 目标基因模块的GO注释 为进一步挖掘目标基因模块的功能, 利用AgriGo在线工具对筛选出的4个目标基因模块Red、Yellow、Turquoise和Blue进行GO功能富集分析。结果表明这4个模块都可以显著富集到生物学过程、分子功能以及细胞组分三大分类下的若干GO通路(图9)。如表2所示, Red模块可以富集到氧化还原相关基因(GO: 0016705)和内肽酶抑制剂活性基因(GO:0004866)等调控通路; Yellow模块可以富集到与蛋白质结合相关的通路(GO:0005515); Turquoise模块可以富集到光合作用(GO:0015979), 也富集到与氧化还原酶活性相关的通路(GO:0016705), 还富集到类固醇合成途径中催化类固醇脱氢反应(以类固醇羟基为氢供体, 以NAD或NADP为氢受体)的脱氢酶及相关通路(GO:0006694、GO:0003854、GO: 0033764、GO: 0016229、GO: 0016614、GO: 0016616和GO:0051287); Blue模块可以富集到与氧化还原酶活性相关的基因(GO:0016705)和与细胞骨架相关的基因(GO: 0007010和GO:0015629)。总体来看, 4个模块都富集到了氧化还原酶活性及细胞骨架组织相关的通路, 说明WGCNA可以有效构建到具有生物学意义的共表达模块, 这些模块可成为本研究的重点关注对象。
图9
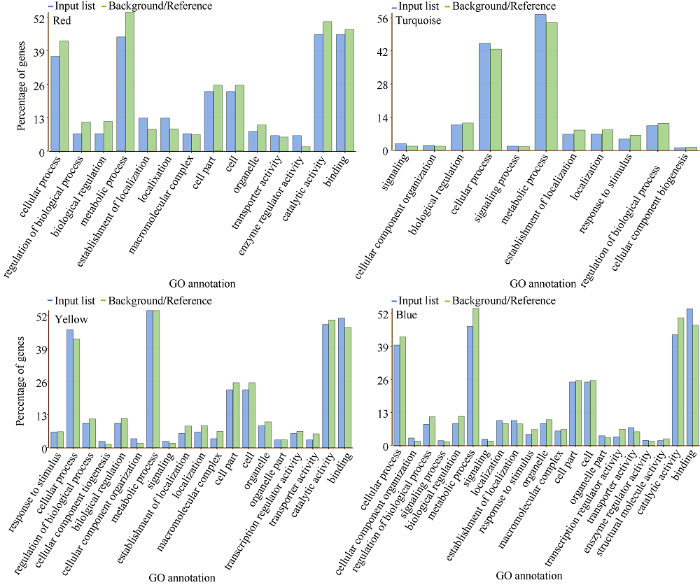 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9目标模块内基因GO注释
Fig. 9Gene GO annotation in target module
Table 2
表2
表2目标模块的部分GO富集分析结果
Table 2
| 模块 Module | GO条目 GO term | 基因本体 Ontology | 描述 Description | P值 P-value |
|---|---|---|---|---|
| Red | GO:0016705 | F | 氧化还原酶活性, 作用于成对供体, 与分子氧结合或还原。 Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen. | 2.90E-09 |
| Red | GO:0004866 | F | 内肽酶抑制剂活性。 Endopeptidase inhibitor activity. | 0.00062 |
| Red | GO:0030414 | F | 肽酶抑制剂活性。 Peptidase inhibitor activity. | 0.00062 |
| Red | GO:0005515 | F | 蛋白质结合。 Protein binding. | 0.0012 |
| Yellow | GO:0005515 | F | 蛋白质结合。 Protein binding. | 2.20E-13 |
| Turquoise | GO:0015979 | P | 光合作用。 Photosynthesis. | 7.10E-07 |
| 模块 Module | GO条目 GO term | 基因本体 Ontology | 描述 Description | P值 P-value |
| Turquoise | GO:0019684 | P | 光合作用, 光反应。 Photosynthesis, light reaction. | 5.90E-06 |
| Turquoise | GO:0006694 | P | 类固醇生物合成过程。 Steroid biosynthetic process. | 5.60E-05 |
| Turquoise | GO:0010033 | P | 对有机物质的反应。 Response to organic substance. | 0.00023 |
| Turquoise | GO:0005515 | F | 蛋白结合。 Protein binding. | 4.00E-27 |
| Turquoise | GO:0016705 | F | 氧化还原酶活性, 作用于成对供体, 与分子氧结合或还原。 Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen. | 3.00E-07 |
| Turquoise | GO:0003854 | F | 3-β-羟基-Δ5-类固醇脱氢酶活性。 3-beta-hydroxy-delta5-steroid dehydrogenase activity. | 3.30E-05 |
| Turquoise | GO:0033764 | F | 类固醇脱氢酶活性, 作用于供体的羟基, NAD或NADP作为受体。 Steroid dehydrogenase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor. | 3.30E-05 |
| Turquoise | GO:0016229 | F | 类固醇脱氢酶活性。 Steroid dehydrogenase activity. | 3.30E-05 |
| Turquoise | GO:0016614 | F | 氧化还原酶活性, 作用于供体的羟基。 Ooxidoreductase activity, acting on CH-OH group of donors. | 7.10E-05 |
| Turquoise | GO:0016616 | F | 氧化还原酶活性, 作用于供体的羟基, NAD或NADP作为受体。 Oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor. | 0.00026 |
| Turquoise | GO:0051287 | F | NAD或NADH结合。 NAD or NADH binding. | 0.00039 |
| Turquoise | GO:0008092 | F | 细胞骨架蛋白结合。 Cytoskeletal protein binding. | 0.00062 |
| Blue | GO:0007010 | P | 细胞骨架组织。 Cytoskeleton organization. | 1.20E-05 |
| Blue | GO:0005515 | F | 蛋白结合。 Protein binding. | 2.70E-29 |
| Blue | GO:0016705 | F | 氧化还原酶活性, 作用于成对供体, 与分子氧结合或还原。 Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen. | 2.50E-10 |
| Blue | GO:0016021 | C | 膜结合。 Integral to membrane. | 7.30E-05 |
| Blue | GO:0015629 | C | 肌动蛋白细胞骨架。 Actin cytoskeleton. | 0.00021 |
| Blue | GO:0031224 | C | 膜固有的。 Intrinsic to membrane. | 0.00036 |
新窗口打开|下载CSV
2.2.7 目标模块中核心基因的筛选和功能分析
为了获得上述4个模块中的核心基因, 利用Cytoscape软件对基因调控网络进行可视化处理, 筛选出模块中高连通性的基因, 这些基因则作为该模块中的核心基因(图10和图11)。在这些网络中, 每一个节点代表一个基因, 节点通过线相联系, 处于连线两端的基因通常被认为具有相同的生物功能。其中在Red模块中筛选出了5个核心基因, Yellow模块中筛选出了9个核心基因, Turquoise模块中筛选出4个核心基因, Blue模块中筛选出了2个核心基因,为了获取这些核心基因的功能信息, 我们利用马铃薯基因数据库(http://solanaceae.plantbiology.msu. edu/index.shtml)和NCBI数据库(https://www.ncbi. nlm.nih.gov/)查询这些核心基因的相关报道, 并借助TAIR数据库(https://www.arabidopsis.org/)注释这些核心基因在拟南芥中的同源基因的功能(表3), 通过已报道的文献, 本研究发现这些筛选出的核心基因基本都与干旱胁迫相关, 其中PGSC0003 DMG 400026572在拟南芥中的同源基因AT3G51730基因参与了植物液泡的形成, 而液泡在植物生长、发育和应激反应中起着中心作用。PGSC0003DMG 4000 29350在拟南芥中的同源基因AT1G05680基因通过调节活性氧和氧化还原信号与植物激素发生协同或拮抗作用, 引起植物对生物和非生物胁迫的保护反应。PGSC0003DMG400006704在拟南芥中的同源基因AT5G35200基因通过诱导启动蛋白磷酸化调节下游的信号包括穿过质膜的离子通量、MAPK的活化和活性氧的形成。PGSC0003 DMG400024687在拟南芥中的同源基因AT1G55000基因与泛素化的特异性相关, 能促进泛素转移到合适的靶点进而对细胞活动进行调控。PGSC0003 DMG400003792在拟南芥中的同源基因AT3G14110基因与植物体内的活性氧的变化相关, 在逆境环境下, 该基因可激活多种应激反应。
图10
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10Red和Yellow模块内的基因共表达网络及其核心基因
Fig. 10Gene co-expression network and hub genes in Red and Yellow modules
图11
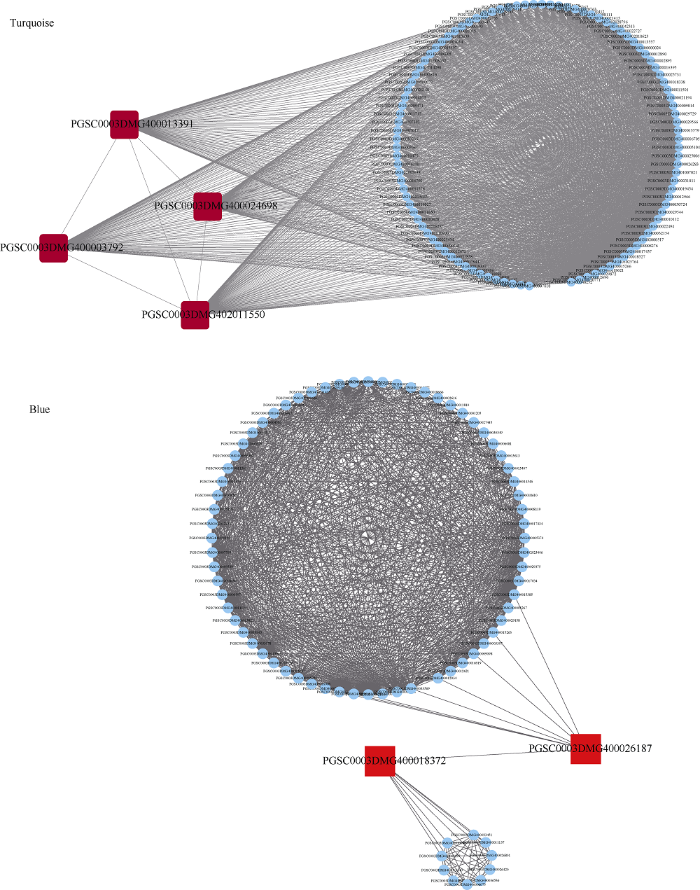 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图11Turquoise和Blue模块内的基因共表达网络及其核心基因
Fig. 11Gene co-expression network and hub genes in Turquoise and Blue modules
Table 3
表3
表3不同模块中核心基因的功能注释
Table 3
| 模块 Module | 核心基因 Hub gene | 核心基因在 拟南芥同源基因 Hub gene in A. thaliana | 基因功能 Gene function |
|---|---|---|---|
| Red | PGSC0003DMG400030368 | AT1G07140 | 编码假定的RAN结合蛋白。 Encodes a putative RAN-binding protein (siRANBP). |
| Red | PGSC0003DMG400003662 | AT1G20270; AT5G66060 | 2-氧戊二酸和铁依赖性加氧酶超家族蛋白; 铁离子结合/氧化还原酶/氧化还原酶蛋白。 2-oxoglutarate and Fe-dependent oxygenase superfamily protein; Ironion binding/oxidoreductase/oxidoreductase protein. |
| Red | PGSC0003DMG402020462 | AT5G25360 | 假设性蛋白质。 Hypothetical protein (source: Araport 11). |
| Red | PGSC0003DMG400005154 | X | 未知功能。Unknown. |
| Red | PGSC0003DMG400020562 | AT2G40470 | 含LOB结构域的蛋白质。参与木质部分化的调节——作为VND7的调节器, VND7是木质部细胞分化的主要调节器。 LOB-domain containing protein. Involved in regulation of xylem differentiation—acts as a regulator of VND7 which is a master regulator of xylem cell differentiation. |
| Yellow | PGSC0003DMG400029350 | AT1G05680[15] | 编码一个UDP-葡萄糖基转移酶, ugt74e2, 作用于IBA (吲哚-3-丁酸)并影响生长素的稳态。这种酶的转录和蛋白质水平被H2O2强烈诱导, 可能允许ROS (活性氧物种)和生长素信号的整合。 Encodes a UDP-glucosyltransferase, ugt74e2, that acts on IBA (indole-3-butyric acid) and affects auxin homeostasis. The transcript and protein levels of this enzyme are strongly induced by H2O2 and mayallow integration of ROS (reactive oxygen species) and auxin signaling. |
| Yellow | PGSC0003DMG400024717 | AT3G05545 | 超家族蛋白。 RING/U-box superfamily protein (source: Araport 11). |
| Yellow | PGSC0003DMG400026572 | AT3G51730[16] | 含皂苷B结构域的蛋白质。 Saposin B domain-containing protein (source: Araport 11). |
| Yellow | PGSC0003DMG400006704 | AT5G35200[17] | 超家族蛋白。 ENTH/ANTH/VHS superfamily protein (source: Araport 11). |
| Yellow | PGSC0003DMG400015386 | AT2G42610 | 光依赖性短下胚轴样蛋白。 LIGHT-DEPENDENT SHORT HYPOCOTYLS-like protein (DUF640) (source: Araport 11). |
| Yellow | PGSC0003DMG400026238 | AT3G60410 | 假设性蛋白质。 Hypothetical protein (DUF1639) (source: Araport 11). |
| 模块 Module | 核心基因 Hub gene | 核心基因在 拟南芥同源基因 Hub gene in A. thaliana | 基因功能 Gene function |
| Yellow | PGSC0003DMG400020683 | AT3G25910 | MAP激酶 Mitogen-activated protein kinases (DUF1644)(source: Araport 11). |
| Yellow | PGSC0003DMG400024687 | AT1G55000[18] | 含蛋白的肽聚糖结合赖氨酸域。 Peptidoglycan-binding LysM domain-containing protein (source: Araport 11). |
| Yellow | PGSC0003DMG400006516 | AT1G55570 | SKU5 similar 12 (source: Araport 11). |
| Turquoise | PGSC0003DMG400013391 | AT3G22990 | 含有蛋白质的犰狳重复序列, 位于核内, 广泛表达于植物。 Armadillo-repeat containing protein, located in nucleus, broadly expressed throughout vegetative. |
| Turquoise | PGSC0003DMG400003792 | AT3G14110[19] | 编码一种新的盘绕线圈, 含有TPR结构域的蛋白质, 定位于叶绿体膜并参与叶绿素的生物合成。 Encodes a novel coiled-coil, TPR domain containing protein that is localized to the chloroplast membrane and is involved in chlorophyll biosynthesis. |
| Turquoise | PGSC0003DMG400024698 | AT5G27620 | 核心细胞周期基因的mRNA是细胞间流动的。 Core cell cycle genes the mRNA is cell-to-cell mobile. |
| Turquoise | PGSC0003DMG402011550 | AT5G23200 | C5orf35 (source: Araport 11). |
| Blue | PGSC0003DMG400018372 | X | 未知功能。Unknown. |
| Blue | PGSC0003DMG400026187 | AT3G50950 | 编码一种典型的CC型NLR蛋白, 该蛋白是识别病原菌丁香中T3SE-Hopz1a所必需的。 Encodes a canonical CC-type NLR protein that is required for the recognition of the T3SE HopZ1a from the pathogenic bacteria P. syringae |
新窗口打开|下载CSV
2.2.8 核心基因的RT-qPCR验证 从相关性最高的4个基因模块中筛选到20个可能与干旱胁迫相关的核心基因(表3), 从中挑选了14个基因进行实时荧光定量PCR验证。将用于转录组测序的备份RNA反转录, 通过q-PCR检测这些基因在C16和C119中对干旱胁迫的响应情况。最终结果的变化趋势与转录组结果基本一致(表4和图12), 进一步证明了转录组测序结果的可靠性, 并验证了这些核心基因确实响应干旱胁迫。
Table 4
表4
表4字母与基因的对应表
Table 4
| 字母 Letter | 基因 Gene | 字母 Letter | 基因 Gene | 字母 Letter | 基因 Gene |
|---|---|---|---|---|---|
| A | PGSC0003DMG400026572 | E | PGSC0003DMG400030368 | I | PGSC0003DMG400006704 |
| B | PGSC0003DMG400005154 | F | PGSC0003DMG400003792 | J | PGSC0003DMG400020683 |
| C | PGSC0003DMG400029350 | G | PGSC0003DMG402011550 | K | PGSC0003DMG400024687 |
| D | PGSC0003DMG400020462 | H | PGSC0003DMG400013391 | L | PGSC0003DMG400026238 |
新窗口打开|下载CSV
图12
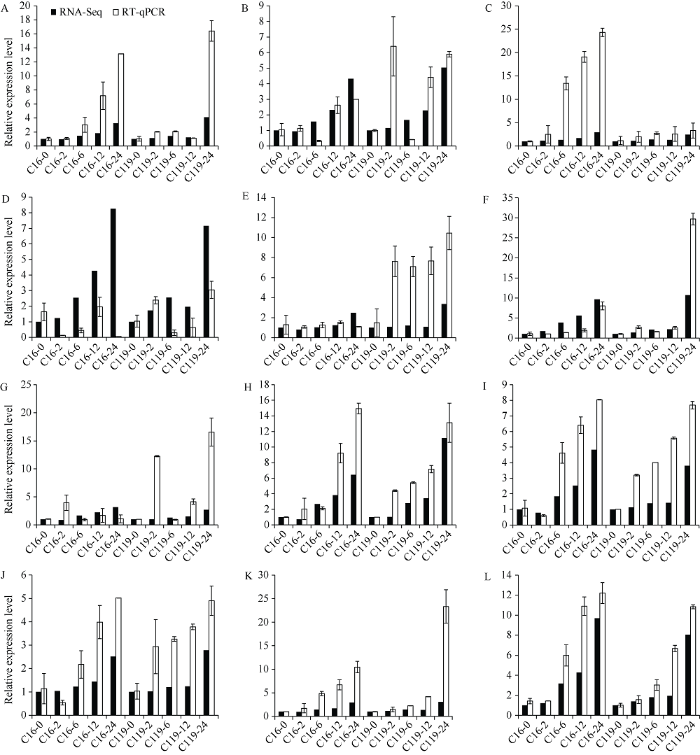 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图12核心基因的RT-qPCR验证
Fig. 12RT-qPCR validation of Hub genes
3 讨论
干旱胁迫是影响自然界植物地理分布, 限制农业生产力和威胁粮食安全的主要环境因素之一。全球气候变化更加剧了干旱胁迫的不利影响, 预计还将导致极端天气的频率增加[20,21]。马铃薯作为第四大粮食作物, 在我国的主产区也处在气候变化的高风险区域, 干旱胁迫是主要的风险之一[22]。因此, 提高马铃薯的抗旱性, 解析其在干旱胁迫下的感知和适应机制对提高农业生产力和节约环境资源均具有重要意义。植物根系是植物汲取和转化水分和营养的主要器官, 也是植物面临土壤胁迫环境时首先感知胁迫信号的器官, 在植物应对非生物逆境尤其是干旱胁迫时起关键作用[23,24,25]。虽然马铃薯根系抗逆遗传机理的解析取得了一定的进展, 例如通过数量性状位点定位和关联分析等方法鉴定了许多重要的根系抗
逆基因[26,27], 但是这些研究手段都有一定的局限性, 例如数量性状定位不能反映遗传背景复杂群体的遗传异质性, 此外, 大多数的农艺性状还与整个群体结构存在很高的相关性, 而利用现在生物信息技术和数据分析方法则可以通过共表达模块化处理, 将成千上万具有相似生物学功能的基因分配到同一个模块中, 通过研究模块的生物学意义来进一步挖掘模块内基因的功能, 较好地解决了对遗传背景复杂群体的性状解析不足的问题, 因此被广泛地应用于大数据组学的研究之中[28,29,30]。因此, 本研究干旱处理马铃薯幼苗后, 收集其根系材料进行转录组分析, 以期获得马铃薯根系抗逆相关的新的研究线索。与传统的仅对比处理前后的转录组差异分析不同, 本研究针对2个品种, 分别设置了4个处理时间梯度, 加上未处理的对照, 每样3个生物学重复, 共计30个样本进行了转录组测序。与此同时, 本研究还检测了所有处理时间下每个样本的胁迫相关生理生化指标(SOD、POD、CAT和RV)。
大量的转录组数据和生理数据获得后, 如何从中挖掘蕴含的生物学意义成为首要考虑的问题。网络分析在基因组、转录组和代谢组等高通量组学数据的发掘中应用较为广泛。为了发掘与抗旱相关的基因或调控通路, 本研究采用了WGCNA分析方法, 可特异地筛选出与目标性状相关的基因, 并进行模块化分类, 得到具有高度生物学意义的共表达模块, 已经被证明是一种高效的数据挖掘手段[31]。ROS的产生是植物受到干旱胁迫后最早的反应之一, ROS是许多胁迫相关生物合成、信号转导和代谢相关途径的重要调节剂, 其过量积累对细胞具有毒害作用[32], 因此维持其动态平衡是植物应对胁迫的重要调节方式, 而SOD、POD和CAT作为清除ROS的抗氧化酶, 可较为直接地反映植物对胁迫的响应情况[33], 可作为WGCNA分析中的目标性状。因此, 我们利用WGCNA分析方法, 富集到了15个与生理性状相关联的共表达模块, 其中4个模块与这些抗逆性状高度相关, 通过进一步的网络分析, 找到了这4个模块的核心基因。表明WGCNA可以构建到具有生物学意义的共表达模块和通路, 为下一步深入研究打下基础。
本研究通过WGCNA富集到的15个与胁迫相关生理生化指标(SOD、POD、CAT和RV)相关联的共表达模块, 由于模块较多, 因此挑选了4个与生理性状关联度最高的模块进行下一步研究。模块内的基因表达模式随着处理时间的增加, 存在与生理性状由负相关逐渐转为极显著正相关的Yellow模块和Red模块, 以及存在相反趋势的Turquoise模块和Blue模块。由于每个模块的基因数量庞大, 本研究利用各个模块的特征向量基因对模块两两之间进行相关性分析, 结果表明, 这4个模块间的确存在极显著的相关性且关联度最高, 进一步印证这4个模块可作为根系抗逆性状机理研究的目标模块。通过对这4个模块的GO功能富集分析发现, 均可以得到与抗逆相关的具有生物学意义的调控途径。例如, Red模块、Turquoise模块和Blue模块都富集到与氧化还原活性相关的通路(GO:0016705), Turquoise模块还富集到类固醇合成途径中催化类固醇脱氢反应的脱氢酶及相关通路(GO:0006694)。有研究表明, 在拟南芥和玉米中过量表达油菜素固醇合成途径的脱氢酶基因, 可导致ABA的积累, 增强植物抗性, 提高玉米在逆境下的产量[34,35]。说明WGCNA分析的确可富集到与目标性状相关的具有生物学意义的共表达模块。
本研究针对4个与目标性状关联度最高的模块分析表明, 其核心基因包含干旱胁迫相关基因, 例如Red模块中的参与木质部分化的基因(PGSC0003 DMG400020562)、Yellow模块中的MAP激酶(PGSC0003DMG400020683)、Turquoise模块可能参与叶绿素合成的基因(PGSC0003DMG40000 3792)。其中Yellow模块中PGSC0003DMG4000 29350基因在拟南芥中的同源基因AT1G05680可编码一种UDP-葡萄糖基转移酶UGT74E2, 在拟南芥中的研究表明, 其可被过氧化氢或非生物胁迫诱导表达, 通过对生长素吲哚-3-丁酸(IBA)和吲哚-3-乙酸(IAA)的动态平衡调节, 参与氧化还原信号与植物激素信号的整合互作, 增加植物在非生物逆境中的保护反应[36]。这些基因在模式植物拟南芥和其他作物如玉米、水稻等中有较多的研究, 在马铃薯中的报道还相对较少, 可进行深入探究。
以上结果表明, 利用WGCNA分析, 可挖掘到与目标性状的生物学意义高度关联的基因模块和核心基因, 为挖掘目标基因提供新的研究思路, 为解析复杂的农艺性状提供参考。在本研究中, 重点关注了Red、Yellow、Turquoise和Blue四个相关性高的调控模块, 其余的基因模块虽然没有被详细论述, 但也可能包含与根系抗逆相关的通路, 可进一步挖掘其蕴含的生物学意义。
4 结论
本研究构建了与抗逆生理性状相关联的权重基因共表达网络, 得到了15个与根系抗旱密切相关的基因模块并进行了GO通路富集。挑选出4个与目标性状关联度最佳的基因模块进行深入分析, 其中在Red模块中挑选出了5个核心基因, Yellow模块中挑选出了9个核心基因, Turquoise模块中挑选出4个核心基因, Blue模块中挑选出了2个核心基因, 通过功能注释, 发现部分核心基因与已报道非生物胁迫调控通路紧密相关, 还有一部分功能仅在模式植物中有一些研究。本研究结果可为马铃薯根系抗旱的分子机制研究提供线索, 也为耐旱性马铃薯新品种培育提供理论支持。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1023/A:1026140122848URL [本文引用: 1]

The mechanical and physiological bases for root growth against high mechanical impedance are reviewed. The best estimates of maximum axial root growth pressure (
 max) in completely impeded pea roots appear to be from 0.5 to 0.6 MPa, which results from a turgor pressure of about 0.8 MPa. When roots are incompletely impeded, a range of responses has been reported. Roots do not change elongation rate in a simple mechanical way in response to changes in mechanical impedance. Instead, ethylene might play a key role in mediating an increase in root diameter and a decrease in elongation rate. These changes persist for some hours or days after impedance is removed. Differences between species in their ability to penetrate strong soil layers are not related to differences in max, but appear to be due to differences in root diameter. In rice, differences between cultivars in the ability of their roots to penetrate strong wax layers are not related to their elongation rates through uniformly strong media. Differences between species or cultivars in their ability to penetrate strong layers may be due to differences in the tendency of roots to deflect or buckle when they grow from a weak to a strong environment.
max) in completely impeded pea roots appear to be from 0.5 to 0.6 MPa, which results from a turgor pressure of about 0.8 MPa. When roots are incompletely impeded, a range of responses has been reported. Roots do not change elongation rate in a simple mechanical way in response to changes in mechanical impedance. Instead, ethylene might play a key role in mediating an increase in root diameter and a decrease in elongation rate. These changes persist for some hours or days after impedance is removed. Differences between species in their ability to penetrate strong soil layers are not related to differences in max, but appear to be due to differences in root diameter. In rice, differences between cultivars in the ability of their roots to penetrate strong wax layers are not related to their elongation rates through uniformly strong media. Differences between species or cultivars in their ability to penetrate strong layers may be due to differences in the tendency of roots to deflect or buckle when they grow from a weak to a strong environment.DOI:10.1101/gr.217463.116URLPMID:28687705 [本文引用: 1]
Long noncoding RNAs (lncRNAs) mediate important epigenetic regulation in a wide range of biological processes and diseases. We applied comprehensive analyses of RNA-seq and CAGE-seq (cap analysis of gene expression and sequencing) to characterize the dynamic changes in lncRNA expression in rhesus macaque (Macaca mulatta) brain in four representative age groups. We identified 18 anatomically diverse lncRNA modules and 14 mRNA modules representing spatial, age, and sex specificities. Spatiotemporal- and sex-biased changes in lncRNA expression were generally higher than those observed in mRNA expression. A negative correlation between lncRNA and mRNA expression in cerebral cortex was observed and functionally validated. Our findings offer a fresh insight into spatial-, age-, and sex-biased changes in lncRNA expression in macaque brain and suggest that the changes represent a previously unappreciated regulatory system that potentially contributes to brain development and aging.
DOI:10.7554/eLife.29655URLPMID:28826479 [本文引用: 1]
The dynamics of local climates make development of agricultural strategies challenging. Yield improvement has progressed slowly, especially in drought-prone regions where annual crop production suffers from episodic aridity. Underlying drought responses are circadian and diel control of gene expression that regulate daily variations in metabolic and physiological pathways. To identify transcriptomic changes that occur in the crop Brassica rapa during initial perception of drought, we applied a co-expression network approach to associate rhythmic gene expression changes with physiological responses. Coupled analysis of transcriptome and physiological parameters over a two-day time course in control and drought-stressed plants provided temporal resolution necessary for correlation of network modules with dynamic changes in stomatal conductance, photosynthetic rate, and photosystem II efficiency. This approach enabled the identification of drought-responsive genes based on their differential rhythmic expression profiles in well-watered versus droughted networks and provided new insights into the dynamic physiological changes that occur during drought.
URLPMID:24828307 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/j.1365-3040.2009.02041.xURLPMID:19712065 [本文引用: 1]
Water deficit and salinity, especially under high light intensity or in combination with other stresses, disrupt photosynthesis and increase photorespiration, altering the normal homeostasis of cells and cause an increased production of reactive oxygen species (ROS). ROS play a dual role in the response of plants to abiotic stresses functioning as toxic by-products of stress metabolism, as well as important signal transduction molecules. In this review, we provide an overview of ROS homeostasis and signalling in response to drought and salt stresses and discuss the current understanding of ROS involvement in stress sensing, stress signalling and regulation of acclimation responses.
DOI:10.1016/j.pbi.2013.07.002URLPMID:23876676 [本文引用: 1]
Reactive oxygen species are recognized as important signaling components in a wide range of processes in plants and most other organisms. Reactive oxygen species are produced in different subcellular compartments in response to specific stimuli and the production is under tight control in order to avoid detrimental side-effects. Calcium signaling, protein phosphorylation and other signaling pathways are intimately involved in the control and coordination of reactive oxygen production. Any signal that should result in a specific response must eventually be perceived. Direct redox modification of transcription factors and other proteins are critical for the perception of intracellular reactive oxygen species; however, sensing of their extracellular counterparts awaits elucidation.
DOI:10.1093/jxb/ert375URLPMID:24253197 [本文引用: 1]
Reactive oxygen species (ROS) play an integral role as signalling molecules in the regulation of numerous biological processes such as growth, development, and responses to biotic and/or abiotic stimuli in plants. To some extent, various functions of ROS signalling are attributed to differences in the regulatory mechanisms of respiratory burst oxidase homologues (RBOHs) that are involved in a multitude of different signal transduction pathways activated in assorted tissue and cell types under fluctuating environmental conditions. Recent findings revealed that stress responses in plants are mediated by a temporal-spatial coordination between ROS and other signals that rely on production of stress-specific chemicals, compounds, and hormones. In this review we will provide an update of recent findings related to the integration of ROS signals with an array of signalling pathways aimed at regulating different responses in plants. In particular, we will address signals that confer systemic acquired resistance (SAR) or systemic acquired acclimation (SAA) in plants.
[本文引用: 1]
[本文引用: 1]
URLPMID:14597658 [本文引用: 1]
DOI:10.1105/tpc.109.071316URLPMID:20798329 [本文引用: 1]
Reactive oxygen species and redox signaling undergo synergistic and antagonistic interactions with phytohormones to regulate protective responses of plants against biotic and abiotic stresses. However, molecular insight into the nature of this crosstalk remains scarce. We demonstrate that the hydrogen peroxide-responsive UDP-glucosyltransferase UGT74E2 of Arabidopsis thaliana is involved in the modulation of plant architecture and water stress response through its activity toward the auxin indole-3-butyric acid (IBA). Biochemical characterization of recombinant UGT74E2 demonstrated that it strongly favors IBA as a substrate. Assessment of indole-3-acetic acid (IAA), IBA, and their conjugates in transgenic plants ectopically expressing UGT74E2 indicated that the catalytic specificity was maintained in planta. In these transgenic plants, not only were IBA-Glc concentrations increased, but also free IBA levels were elevated and the conjugated IAA pattern was modified. This perturbed IBA and IAA homeostasis was associated with architectural changes, including increased shoot branching and altered rosette shape, and resulted in significantly improved survival during drought and salt stress treatments. Hence, our results reveal that IBA and IBA-Glc are important regulators of morphological and physiological stress adaptation mechanisms and provide molecular evidence for the interplay between hydrogen peroxide and auxin homeostasis through the action of an IBA UGT.
URLPMID:15539469 [本文引用: 1]
DOI:10.1074/mcp.M600429-MCP200URLPMID:17317660 [本文引用: 1]
Perception of general elicitors by plant cells initiates signal transduction cascades that are regulated by protein phosphorylation. The earliest signaling events occur within minutes and include ion fluxes across the plasma membrane, activation of MAPKs, and the formation of reactive oxygen species. The phosphorylation events that regulate these signaling cascades are largely unknown. Here we present a mass spectrometry-based quantitative phosphoproteomics approach that identified differentially phosphorylated sites in signaling and response proteins from Arabidopsis cells treated with either flg22 or xylanase. Our approach was sensitive enough to quantitate phosphorylation on low abundance signaling proteins such as calcium-dependent protein kinases and receptor-like kinase family members. With this approach we identified one or more differentially phosphorylated sites in 76 membrane-associated proteins including a number of defense-related proteins. Our data on phosphorylation indicate a high degree of complexity at the level of post-translational modification as exemplified by the complex modification patterns of respiratory burst oxidase protein D. Furthermore the data also suggest that protein translocation and vesicle traffic are important aspects of early signaling and defense in response to general elicitors. Our study presents the largest quantitative Arabidopsis phosphoproteomics data set to date and provides a new resource that can be used to gain novel insight into plant defense signal transduction and early defense response.
URLPMID:12169662 [本文引用: 1]
URLPMID:16720605 [本文引用: 1]
DOI:10.1126/science.1186834URLPMID:20150494 [本文引用: 1]
Population growth, arable land and fresh water limits, and climate change have profound implications for the ability of agriculture to meet this century's demands for food, feed, fiber, and fuel while reducing the environmental impact of their production. Success depends on the acceptance and use of contemporary molecular techniques, as well as the increasing development of farming systems that use saline water and integrate nutrient flows.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s11032-018-0907-xURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.lfs.2016.02.074URLPMID:26916823 [本文引用: 1]
AIMS: Occupational exposure to chloroprene via inhalation may lead to acute toxicity and chronic pulmonary diseases, including lung cancer. Currently, most research is focused on epidemiological studies of chloroprene production workers. The specific molecular mechanism of carcinogenesis by chloroprene in lung tissues still remains obscure, and specific candidate therapeutic targets for lung cancer are lacking. The present study identifies specific gene modules and valuable hubs associated with lung cancer. MAIN METHODS: We downloaded the dataset GSE40795 from the Gene Expression Omnibus (GEO) and divided the dataset into the non-carcinogenic dose chloroprene exposed mice group and the carcinogenic dose chloroprene exposed mice group. With a systemic biological view, we discovered significantly altered gene modules between the two groups and identified hub genes in the carcinogenic dose exposed group using weighted co-expression network analysis (WGCNA). KEY FINDINGS: A total of 2434 differentially expressed genes were identified. Twelve gene modules with multiple biological activities were related to the carcinogenesis of chloroprene in lung tissue. Seven hub genes that were critical for the carcinogenesis of chloroprene in lung tissue were ultimately identified, including Cftr, Hip1, Tbl1x, Ephx1, Cbr3, Antxr2 and Ccnd2. They were implicated in inflammatory response, cell transformation, gene transcription regulation, phase II detoxification, angiogenesis, cell adhesion, motility and the cell cycle. SIGNIFICANCE: The seven hub genes may become valuable candidates for risk assessment biomarkers and therapeutic targets in lung cancer.
DOI:10.3389/fphys.2017.00001URLPMID:28154536 [本文引用: 1]
DOI:10.1007/s11802-018-3656-xURL [本文引用: 1]
[本文引用: 1]
URLPMID:20028478 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1105/tpc.109.071316URLPMID:20798329 [本文引用: 1]
Reactive oxygen species and redox signaling undergo synergistic and antagonistic interactions with phytohormones to regulate protective responses of plants against biotic and abiotic stresses. However, molecular insight into the nature of this crosstalk remains scarce. We demonstrate that the hydrogen peroxide-responsive UDP-glucosyltransferase UGT74E2 of Arabidopsis thaliana is involved in the modulation of plant architecture and water stress response through its activity toward the auxin indole-3-butyric acid (IBA). Biochemical characterization of recombinant UGT74E2 demonstrated that it strongly favors IBA as a substrate. Assessment of indole-3-acetic acid (IAA), IBA, and their conjugates in transgenic plants ectopically expressing UGT74E2 indicated that the catalytic specificity was maintained in planta. In these transgenic plants, not only were IBA-Glc concentrations increased, but also free IBA levels were elevated and the conjugated IAA pattern was modified. This perturbed IBA and IAA homeostasis was associated with architectural changes, including increased shoot branching and altered rosette shape, and resulted in significantly improved survival during drought and salt stress treatments. Hence, our results reveal that IBA and IBA-Glc are important regulators of morphological and physiological stress adaptation mechanisms and provide molecular evidence for the interplay between hydrogen peroxide and auxin homeostasis through the action of an IBA UGT.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}