 ,*甘肃农业大学园艺学院, 甘肃兰州 730070
,*甘肃农业大学园艺学院, 甘肃兰州 730070Genome wide identification and expression analysis of CRK gene family in response to fungal pathogen signals in potato
ZHANG Wei-Na, FAN Yan-Ling, KANG Yi-Chen, YANG Xin-Yu, SHI Ming-Fu, YAO Kai, ZHAO Zhang-Ping, ZHANG Jun-Lian, QIN Shu-Hao,*College of Horticulture, Gansu Agricultural University, Lanzhou 730070, Gansu, China通讯作者:
收稿日期:2019-06-28接受日期:2019-12-26网络出版日期:2020-01-19
| 基金资助: |
Received:2019-06-28Accepted:2019-12-26Online:2020-01-19
| Fund supported: |
作者简介 About authors
E-mail:844741204@qq.com。

摘要
关键词:
Abstract
Keywords:
PDF (3639KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
张卫娜, 范艳玲, 康益晨, 杨昕宇, 石铭福, 要凯, 赵章平, 张俊莲, 秦舒浩. 对马铃薯类受体激酶CRK基因家族的鉴定及响应病原真菌信号的表达分析[J]. 作物学报, 2020, 46(5): 680-689. doi:10.3724/SP.J.1006.2020.94096
ZHANG Wei-Na, FAN Yan-Ling, KANG Yi-Chen, YANG Xin-Yu, SHI Ming-Fu, YAO Kai, ZHAO Zhang-Ping, ZHANG Jun-Lian, QIN Shu-Hao.
植物类受体激酶(receptor-like kinase, RLK)是一类定位在细胞膜上的单次跨膜蛋白, 由数量庞大的不同种类的蛋白质组成[1]。RLKs的分子结构和功能与动物的受体蛋白激酶相似, 在几乎所有的生命活动中起重要的调控作用。植物中的第1个RLK基因是1990年由开拓者John C Walker和Ren Zhang教授在玉米中发现的[2]。此后, 在植物中陆续鉴定到越来越多的RLKs, 在模式植物拟南芥中约有600多个[3], 而在水稻中约有1130多个, 被认为是植物体中最大的一类受体基因[4]。一个典型的RLK由N端信号肽序列、胞外受体结构域、跨膜结构域和胞内激酶区构成[5]。N端信号肽具有疏水的功能, 蛋白质合成的过程中起识别胞外信号的作用。植物RLK的胞外结构域能识别细胞外生长、发育和各种环境因子, 进而将信息传递至胞内激酶区, 使之发生磷酸化或去磷酸化等反应, 开启或关闭下游靶蛋白, 从而调节植物的生长发育和逆境响应[6,7]。根据RLKs胞外结构域氨基酸序列的差异, 可将其分为: 富含亮氨酸重复序列型(leucine-rich repeats, LRR)、Malectin、类凝集素型(lectin-like)、Lysin motif (LysM)、DUF26等, 进而其分为40余个亚家族[4]。其中, 富含半胱氨酸的类受体激酶(cysteine-rich receptor-like kinase, CRK)是一类广泛存在于动物和植物中的膜受体蛋白, 具有1个N端信号肽、1个跨膜结构域、1个C端的胞内丝氨酸/苏氨酸蛋白激酶结构域(Ser/Thr protein kinase domain)以及1个通常由1~4个拷贝的strss-antifung结构域构成的胞外结构域(extracellular domain)[8]。研究发现, 拟南芥有44个CRKs, 陆地棉中有70个[9,10]。
CRKs在植物生长发育、激素信号传导、非生物胁迫和病原体防御以及细胞过敏性死亡中发挥至关重要的作用[11,12]。拟南芥中, 过表达AtCRK4、AtCRK6、AtCRK7或AtCRK36均可增强植物防御病原菌早期和中期的免疫反应[13,14]。当拟南芥受丁香假单胞菌Pst DC3000侵染后, AtCRK5或AtCRK13过表达的植株细胞迅速发生过敏性死亡, 诱导病原菌相关防御基因上调表达[15,16,17]; 而突变体atcrk20则通过加速胞外活性氧的爆发诱导细胞过敏性死亡, 从而抑制病原菌的生长[18]。水稻中也有相似的研究, OsCRK6和OsCRK10与OsNPR1互作可以介导水稻对多种病原菌的广谱性免疫反应[19]。但是, 在大麦中, 瞬时沉默HvCRK1能够增强表皮细胞对白粉病菌Blumeria graminis f. sp. hordei的抗性而不影响R基因介导的抗病途径[20], 这些研究表明CRKs在植物免疫抗病中起重要调控作用, 但是不同CRKs的功能和作用方式不同。
CRKs作为对植物生长发育和抗逆性具有重要功能的植物类受体激酶中的一大类, 其在马铃薯基因组中的分布、表达及功能尚未见报道。本研究采用生物信息学的分析方法, 分析马铃薯CRK基因家族的成员数量、染色体定位、进化及其分类; 预测编码蛋白质的生理生化特性和顺式表达元件; 分析StCRKs对干腐病和晚疫病病菌侵染时的响应模式, 以期为进一步研究马StCRKs的功能提供理论基础。
1 材料与方法
1.1 数据搜索和CRKs鉴定
马铃薯基因组、CDS和蛋白序列及染色体位置等信息均下载自马铃薯基因组在线数据资源(http:// potato.plantbiology.msu.edu/integrated_searches.shtml)。拟南芥、香蕉、苹果、水稻、番茄和棉花蛋白序列分别下载自拟南芥在线信息资源(Arabidopsis information resource, TAIR: http://www. arabidopsis.org/)、香蕉基因组数据库(https://banana-genome-hub.southgreen. fr/)、苹果基因组数据库(Genome Database of Rosaceae species, GDR: https://www.rosaceae.org/)、水稻基因组数据库(Rice Genome Annotation Project, http://rice. plantbiologymsu.edu/)、茄科植物基因组数据库(Solanaceae Genomics Resource: http://solanaceae. plantbiology.msu.edu/index.shtml)和棉花基因组数据库(http://www.cottongen.org/)。利用HMMER3.0软件(HMMER 3.0, http://hmmer.janelia.org/)鉴定马铃薯CRK基因。利用保守域预测软件Pfam (http://pfam. janelia.org/)和SMART (http://smart.emblheidelberg. de/)初步鉴定获得的候选基因, 确保其N-末端含有一个或多个Stress-antifung结构域、C-末端Pkinase结构域和跨膜结构域。1.2 马铃薯CRKs的生物信息学分析
分别使用在线分析工具ExPASyProtParam (https:// web.expasy.org/protparam/)、CELLO v.2.5 (http://cello. life.nctu.edu.tw/)、WoLF PSORT (https://www.genscript. com/wolf-psort.html)和MapDraw确认分析[21] 理化性质、亚细胞定位和染色体位置可视化。利用ClustalX1.83软件[22]进行多序列比对, 并用进化分析软件MEGA5.0 (http://www.megasoftware.net/), 以邻接法(Neighbor-joining method)构建进化树, 执行参数为Poission模式(Poission Model)、部分删除(Partial deletion)和1000次重复有根树(Bootstrap replicated 1000)。1.3 马铃薯CRKs结构分析
从马铃薯基因组数据库中下载马铃薯外显子和内含子分布数据的存储文件, 利用GSDS 2.0网站(http://gsds.cbi.pku.edu.cn/)绘制CRK基因结构分布图。1.4 马铃薯CRKs启动子顺式元件(cis-element)分析
从马铃薯基因组中提取CRK基因转录起始位置上游1500 bp序列查找启动子顺式作用元件, 在PlantCARE数据库(http://bioinformatics.psb.ugent.be/ webtools/plantcare/ html/)预测分析。1.5 马铃薯CRKs响应干腐病和晚疫病菌侵染的表达模式分析
采用实时荧光定量PCR (Real-time PCR)法检测马铃薯CRKs响应干腐病和晚疫病病菌侵染的表达模式。用于响应干腐病和晚疫病病菌侵染表达模式分析的材料分别是马铃薯‘陇薯7号’块茎和‘荷兰15号’叶片。‘陇薯7号’块茎由甘肃省定西市农业科学研究院提供, 硫色镰刀菌(Fusarium sulphureum)由甘肃农业大学食品科学与工程学院李永才教授课题组提供。在马铃薯的赤道部位均匀地打3个3 mm深、直径3 mm的小孔, 于每个小孔中接种提前备好的20 μL F. sulphureum的孢子悬浮液(浓度1.0×106孢子 mL-1)。 用聚乙烯袋包装后装入纸箱, 在室温25℃条件下贮藏。每个处理包括5个马铃薯块茎, 重复3次。接种不同时间(1 d、2 d、3 d和4 d)后, 取接种点向外0.5 cm的块茎组织, 以打孔后不接种块茎为对照。3次生物学试验重复。“陇薯7号”块茎种植后, 取叶片用2.5 mmol L-1水杨酸(salicylic acid, SA)和茉莉酸(jasmonic acid, JA)处理1 h和3 h用于验证试验。“荷兰15号”叶片为本实验室自己种植, 晚疫病菌(HB 09-16-2)由华中农业大学田振东教授课题组惠赠, 接种方法和培养条件参照蒋锐(2017)博士论文[23]。分别于接种1 d、4 d、6 d和8 d等不同时间点采样。每个处理重复3次, 锡箔纸包裹标记, 液氮速冻后保存于-80℃超低温冰箱备用。利用RNAout试剂盒(160906-50, Tiandz, 北京)提取组织总RNA, 经质量检测合格后分别采用试剂盒PrimeScript RT reagent Kit with gDNA Eraser (RR047, TaKaRa, 大连)和SYBR Premix Ex Taq II (TliRNaseH Plus)(RR820, TaKaRa, 大连)进行cDNA合成和qRT-PCR扩增。采用在线软件Primer 3.0 (http:// primer3.ut.ee/)设计引物, 以elongation factor 1-α (ef1α)(GenBank登录号为AB061263)作为内参基因[24], 引物序列如表1所示。反应结束后分析荧光值变化曲线及熔解曲线, 并采用2-??CT计算基因的相对表达量[25]。
Table 1
表1
表1马铃薯CRKs引物
Table 1
| 基因登录号 Gene accession number | 上游引物 Forward primer (5′-3′) | 下游引物 Reverse prime (5′-3′) |
|---|---|---|
| AB061263 | ATTGGAAACGGATATGCTCCA | TCCTTACCTGAACGCCTGTCA |
| PGSC0003DMG402000515 | ACTCTGGCTCTCTACTACAAACAGT | CTTCCACACCTGAAACCAAAATGAC |
| PGSC0003DMG400021394 | AAGAGTCCCCTTGTATACAGAACCA | ATCGAATTCATACAAGAGGTCCAGC |
| PGSC0003DMG400013525 | CAACACAACCACCATTCCTCAATTC | AAGATTGTGTAGAGGAGCTTGGTTG |
| PGSC0003DMG400018101 | CTCCTCCAGATACAAGCAGTTCATC | TCACCTGATTGAGAGCTAGAAATGC |
| PGSC0003DMG400006912 | TCGTGTTCAAGAGTTATGTCACCAG | AATTCCAAGCAAGTCGAAGTCAGAT |
| PGSC0003DMG400013524 | CATATGTAGTACGCAACCTGAGCAT | CTTCAGCATAGCATAGGACACAGTC |
| PGSC0003DMG400015170 | TCATGATCAGACTGTTGACTTCGTC | AAGCAGTGAATGGAGCAACAAAATC |
| PGSC0003DMG400015171 | TACAAAAGCCAGTGAAGGAGAAGAC | TATAATTGCCTGTTTCTTGAGCGGA |
新窗口打开|下载CSV
1.6 统计分析
利用Microsoft Excel 2010软件处理原始数据, 利用t-test (P < 0.05)法分析差异显著性, 采用软件OriginPro 8.0进行图形可视化, 所有数值均表示为“平均数±标准差”。2 结果与分析
2.1 马铃薯CRKs鉴定
以马铃薯基因组序列为参考序列, 经分析和过滤, 共获得14个蛋白序列, 通过与基因编号对应, 发现部分基因对应1个以上的蛋白序列, 所对应蛋白序列完全相同。最终获得对应的8个CRKs, 其对应蛋白的氨基酸序列大小介于459~686, 分子量和等电点分别介于50.75~77.50 kD和5.84~8.75 (表1)。利用在线软件CELLO v.2.5和WoLF PSORT对8个基因的亚细胞定位进行预测, 发现DMG400013524和DMG400015171位于质膜, 其他基因利用2种软件预测的结果有所区别, 其中4个基因位于质膜或者叶绿体, DMG400015170位于质膜或者细胞质, 而DMG400006912位于质膜或者液泡。
Table 2
表2
表2马铃薯CRKs
Table 2
| 基因登录号Gene accession number | 基因编号 Genetic code | 氨基酸数 AA size | 分子量 Molecular weight (kD) | 等电点 Isoelectric point | 亚细胞定位 Subcellular localization | |
|---|---|---|---|---|---|---|
| Gene ID# | STWG 1.0b | |||||
| PGSC0003DMG402000515 | PGSC0003DMP400001023/ PGSC0003DMP400001024 | StCRK1 | 616 | 67.40 | 8.53 | PMa, Chlob |
| PGSC0003DMG400021394 | PGSC0003DMP400037082 | StCRK2 | 608 | 67.23 | 8.07 | PMa, Chlob |
| PGSC0003DMG400013525 | PGSC0003DMP400023929 | StCRK3 | 656 | 72.73 | 8.75 | PMa, Chlob |
| PGSC0003DMG400018101 | PGSC0003DMP400031539 | StCRK4 | 681 | 75.52 | 6.37 | PMa, Chlob |
| PGSC0003DMG400006912 | PGSC0003DMP400012233/ PGSC0003DMP400012234/ PGSC0003DMP400012235 | StCRK5 | 668 | 75.21 | 5.84 | PMa, Vacub |
| PGSC0003DMG400013524 | PGSC0003DMP400023928/ PGSC0003DMP400023926 | StCRK6 | 656 | 73.21 | 6.42 | PMa,b |
| PGSC0003DMG400015170 | PGSC0003DMP400026615/ PGSC0003DMP400026616/ PGSC0003DMP400026617 | StCRK7 | 459 | 50.75 | 7.27 | PMa, Cytob |
| PGSC0003DMG400015171 | PGSC0003DMP400026620 | StCRK8 | 686 | 77.50 | 7.23 | PMa, b |
新窗口打开|下载CSV
2.2 马铃薯、拟南芥、香蕉、苹果、水稻、番茄和棉花CRK家族进化树分析
从图1看出, CRKs可以分为9个亚组(图1), 其亚组Ⅲ和亚组Ⅳ中, 除Gh-A01G2043和MD09G1104200外, 其余成员均来自水稻和香蕉2种单子叶植物, 而亚组VI-Ⅸ的家族成员都来自棉花、马铃薯、苹果、拟南芥、番茄等双子叶植物。马铃薯8个CRKs分布于亚组I和VI, 其中6个分布于亚组I, 2个分布于亚组VI。其亚组I中, 基因PGSC0003DMP400037082和PGSC0003DMP400001023同源性较高, 马铃薯基因PGSC0003DMP400031539、PGSC0003DMP400023928和PGSC0003DMP400012234分别与番茄基因Solyc03g112730.3.1、Solyc05g1005070.2.1和Solyc02g067780.3.1同源性较高, 均达99%以上。图1
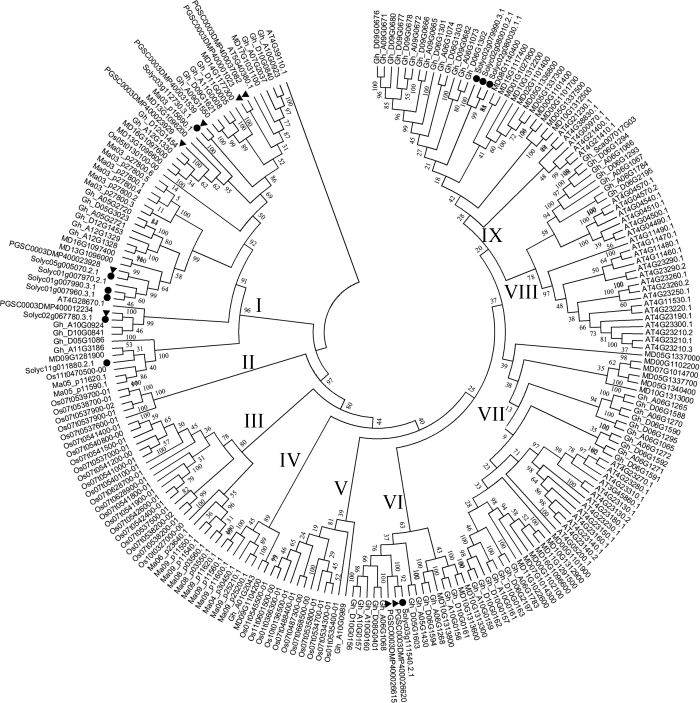 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1马铃薯、拟南芥、香蕉、苹果、水稻、番茄和棉花CRK家族进化树分析
Fig. 1Phylogenetic tree of CRKs in potato (Solanum tuberosum) (▼), apple (Malus pumila Mill.), Thale Cress (Arabidopsis thaliana), rice (Oryza sativa), cotton (G. hirsutum), banana (Musa acuminata), and tomato (Solanum lycopersicum)
2.3 马铃薯CRKs的染色体定位
通过下载马铃薯CRKs相关染色体定位信息, 利用软件MapDraw进行染色体定位并进行可视化分析。结果显示, 马铃薯的8个CRKs中, 3号和5号染色体上均分布3个CRKs, 2号染色体上分布2个CRKs。其中, PGSC0003DMG400013524和PGSC0003DMG400013525、PGSC0003DMG400015170和PGSC0003DMG400015171在染色体上的相对位置小于100 kb, 推断为串联重复基因簇。图2
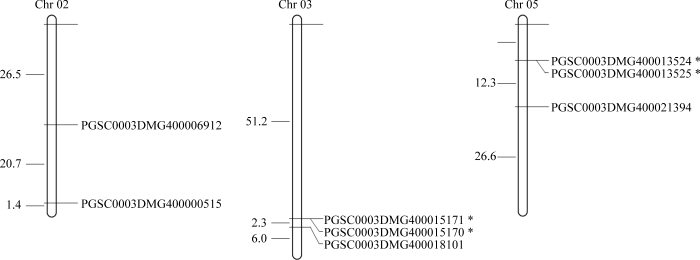 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2马铃薯CRKs染色体定位
*代表串联重复基因簇。
Fig. 2Chromosomal distribution of the StCRKs
* indicates tandem duplication genes.
2.4 马铃薯CRKs的结构和蛋白质功能结构域分析
利用CRKs家族成员单独构建的进化树和外显子-内含子结构如图3所示。CRKs家族成员单独构建的进化树和图1所示结果一致。StCRKs具有相对保守的基因结构和蛋白功能结构域(图3)。马铃薯CRKs长度介于3.2~5.1 kb, 外显子数目为6~8个, 较为接近。保守结构域分析结果显示, 马铃薯CRK家族中7个蛋白质具有2个stress-antifung结构域和1个Pkinase_Tyr结构域, 而PGSC0003DMP400026615(StCRK7)只含有1个stress-antifung结构域和1个Pkinase_Tyr 结构域, 此外, PGSC0003DMP400026615 (StCRK7)和PGSC0003DMP400026620 (StCRK8)除了具有stress-antifung和Pkinase_Tyr结构域外, 还含有1个功能未知的DUF3403 (PF11883)结构域。结合StCRK蛋白家族系统发育树分析结果, 位于进化树同一分支的同源基因具有相似的基因结构和功能结构域, 推测它们具有相似的生物学功能。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3马铃薯CRKs的基因结构和蛋白功能结构域分析
A: 马铃薯CRKs进化树; B: 马铃薯CRKs基因结构; C: 马铃薯CRK蛋白的功能结构域。
Fig. 3Gene structure and protein functional domain analysis of StCRKs
A: phylogenetic tree of StCRKs; B: gene structure of StCRKs; C: functional domains of StCRKs.
2.5 马铃薯CRKs家族cis-element分析
为进一步理解基因的调控功能, 本研究对启动子序列中顺式元件进行了分析。通过马铃薯基因组提取CRKs起始密码子上游1500 bp启动子序列, 利用在线工具PlantCARE进行顺式元件分析, 并提取了响应激素和逆境相关的15类顺式元件。由图4可知, StCRKs启动子区域富含响应植物激素和逆境胁迫的顺式作用元件(图4)。如图所示, 与植物激素响应相关的顺式元件有5类, 分别为脱落酸响应元件(A和B)、茉莉酸甲酯响应元件(H)、水杨酸响应元件(I)、赤霉素响应元件(C、D和E)和生长素响应元件(F和G); 与生长发育相关的顺式元件有两类: 玉米醇溶蛋白代谢调控元件(J)和黄酮类代谢元件(N); 而胁迫响应相关的顺式元件主要包括: 厌氧诱导响应元件(K)、缺氧响应元件(L)、低温响应元件(O)和防御和压力响应元件(M)。这暗示了马铃薯CRK基因家族在马铃薯生长发育及多种激素和胁迫中的潜在作用。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4预测马铃薯CRKs启动子中顺式调控元件
A和B: 脱落酸响应元件; C: GARE-motif; D和E: 赤霉素响应元件; F和G: 生长素响应元件; H: 茉莉酸甲酯响应元件; I: 水杨酸响应元件; J: 玉米醇溶蛋白代谢调控元件; K: 厌氧诱导响应元件; L: 缺氧响应元件; M: 防御和压力响应元件; N: 黄酮类代谢响应元件; O: 低温响应元件。
Fig. 4Putative regulatory cis-elements in StCRKs promoters
A and B: are the abscisic acid responsive elements; C: GARE-motif; D and E: the Gibberellin responsive elements; F and G: the auxin responsive elements; H: the Methyl jasmonate responsive element; I: the Salicylic acid responsive elements; J: the regulatory element for Zein metabolism; K: the Anaerobic responsive elements; L: the anoxic responsive elements; M: the defense and stress responsive elements; N: the flavonoid responsive elements; O: the low-temperature responsive elements.
为验证所预测cis-element的准确性, 经SA和JA分别处理马铃薯叶片后检测了6个StCRKs的表达情况(图5)。StCRK4、StCRK5和StCRK8启动子区域均存在响应SA信号的cis-element。SA处理后, 以上3个StCRKs均为差异表达。其中, StCRK8上调最高, SA处理1 h和3 h后, 其表达量分别上调至对照的5.71倍和11.22倍。此外, StCRK7启动子存在响应MeJA信号的cis-element, JA处理1 h和3 h后其表达量也发生显著上调, 分别上调至对照的5.95倍和4.73倍。StCRK2和StCRK6启动子区域不存在响应SA和MeJA中任何一种激素的cis-element。尽管SA和JA处理后以上2个StCRKs也发生不同程度的变化, 但变化幅度较小。以上结果表明, 对StCRKs启动子区域cis-element预测的结果准确性较高。
图5
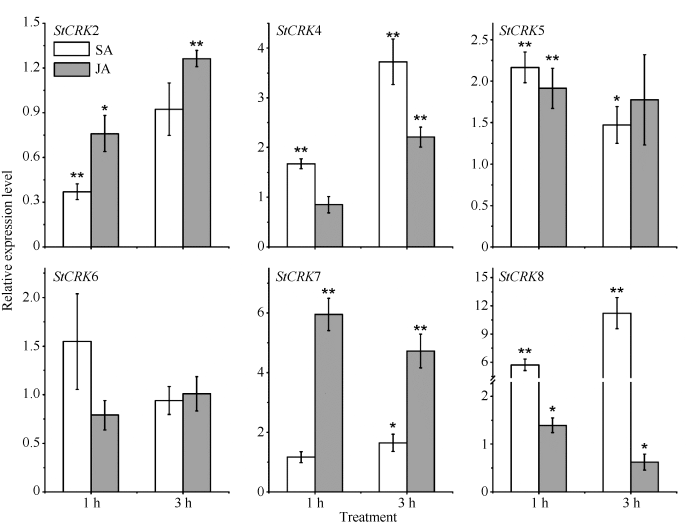 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5SA和JA处理后6个马铃薯CRKs的表达情况
Fig. 5Expression patterns of six StCRKs in response to SA and JA treatments
2.6 CRKs在晚疫病菌侵染马铃薯叶片中的表达分析
植物CRKs蛋白参与调节植物对生物胁迫和非生物胁迫的应答。生产中, 马铃薯晚疫病是由致病疫霉[Phytophthora infestans (Mont.) de Bary]引起的, 晚疫病能够导致马铃薯茎叶死亡和块茎腐烂。为进一步分析StCRKs响应病原真菌的表达模式, 我们通过对马铃薯叶片接种晚疫病菌, 利用Realtime PCR技术检测了叶片中CRKs的表达特征(图6)。通过实验发现, 8个CRKs对晚疫病菌都有响应。其中, StCRK1、StCRK4和StCRK8随着侵染时间的延长, 其相对表达量呈逐渐上调表达的趋势, 8 d时分别上调到对照的13.02±4.40、24.72±5.40和21.38±2.10倍。StCRK3、StCRK5、StCRK7和StCRK8与对照组相比, 不同时间点均有显著差异。图6
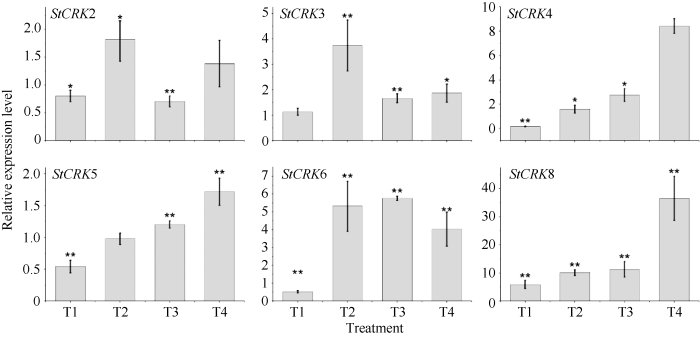 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6马铃薯CRKs响应晚疫病菌侵染的表达分析
Fig. 6Expression patter of StCRKs in response to Phytophthora infestans (Pi)
2.7 CRKs基因家族在干腐病菌侵染马铃薯块茎中的表达分析
马铃薯块茎在贮藏期间极易发生腐烂, 其中由硫色镰刀菌(Fusarium sulphureum)引起的干腐病是造成甘肃马铃薯腐烂的最主要病害, 本实验通过接种硫色镰刀菌, 检测块茎在受到病菌侵染时StCRKs的表达特征(图7)。马铃薯块茎在贮藏期受病菌侵染后, 未检测到StCRK1和StCRK7基因的表达, 而StCRK4、StCRK5和StCRK8随着病菌侵染时间的延长, 其相对表达量呈逐渐上升的趋势, 且在处理4 d时, 分别上升至对照的8.43±0.60、1.72±0.20和36.52±7.80倍; StCRK6和StCRK8与对照相比, 不同时间点均有显著差异。据此推断, 马铃薯StCRK基因家族成员 在进化过程中发生了功能分化, 分工协作参与马铃薯对病原菌的响应。图7
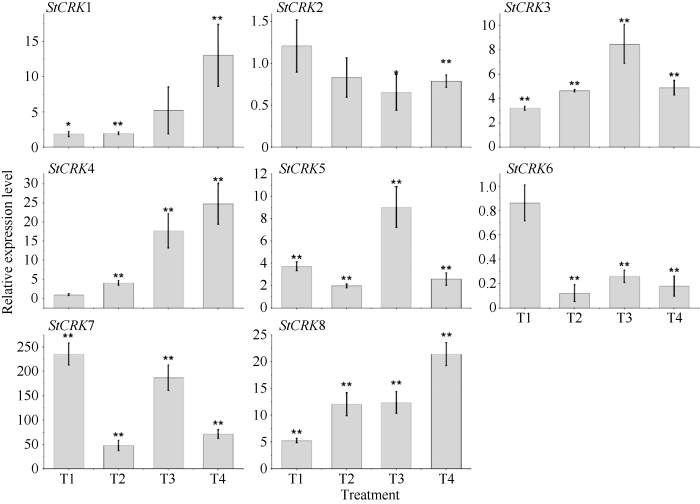 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7马铃薯CRKs响应干腐病菌侵染的表达分析
Fig. 7Expression patter of StCRKs in response to Fusarium sulphureum (Fs)
3 讨论
不同物种中CRKs的数目不尽相同, 水稻中有39个CRKs, 而拟南芥中有44个[9], 不同遗传背景的棉花中CRKs数量不同, G. hirsutum中有70个而G. barbadense中有54个[10]。拟南芥44个CRKs中19个成员是以串联成簇的形式排列于4号染色体上, 而且序列之间高度相似[26]。本研究首次基于转录组数据和结构域保守序列, 对马铃薯CRKs成员进行鉴定及生物信息学分析, 共获得8个家族成员, 无规律地分布于2号、3号和5号染色体上, 进化分析将其分为2个进化分支。荧光实时定量PCR分析表明CRKs对干腐病菌和晚疫病菌侵染的响应情况说明CRKs可能在调控马铃薯对病原真菌的抗性反应中发挥作用。部分马铃薯CRKs基因结构和保守结构域特征与其他基因有显著差异。其中, PGSC0003DMP400026615 (StCRK7)和PGSC0003DMP400026620 (StCRK8)除含有典型的stress-antifung结构域和Pkinase_Tyr结构域外还含有1个功能未知的DUF3403结构域(PF11883); CRKs在进化过程中基因结构和保守结构域的差异, 为成员之间的功能分化提供遗传变异的基础。通过对外显子和内含子的研究, 有助于我们分析了解基因结构和功能上的差异[27]。马铃薯CRKs的外显子为6个至8个, 而且位于进化树同一亚族内的CRKs具有类似的内含子/外显子数目和排列模式, 同时它们的保守结构域也具有类似的数量和位置顺序, 说明亚族内成员间在进化过程中较为保守。
植物启动子要正确调控基因的表达水平和表达模式, 需要核心启动子以及上下游的顺式作用元件协同作用。Xu等[28]对中国华东野生葡萄中受白粉病和赤星病诱导表达的VpSTS诱导型启动子的序列分析发现, 该启动子含有Box-W1、TC-rich repeat element、ABRE、MBS和LTR等顺式作用元件。其中, Box-W1 (TTGACC)与欧芹中真菌激发子响应元件的保守核心序列相似, 特异性地被SA诱导的转录因子WRKY结合蛋白识别[29]。TC-rich repeat element最早是从烟草中识别出来的与防御和胁迫相关的顺式作用元件[30]。MBS是MYB转录因子同源物的结合位点[31], ABRE是ABA响应的元件[32], 而LTR是低温响应顺式元件[33]。本研究发现, 马铃薯的8个CRKs中, 有4个StCRKs (StCRK1、StCRK2、StCRK3、StCRK4)含有TC-rich repeat element顺式元件, 7个StCRKs (StCRK1、StCRK2、StCRK3、StCRK4、StCRK5、StCRK7和StCRK8)具有ABRE元件, 1个StCRK (StCRK1)具有MBS元件, 2个StCRKs (StCRK4和StCRK6)含有LTR元件。研究表明, 植物激素包括IAA、ET、SA、ABA、GA、JA等, 在植物对多种病原菌和真菌的抗性反应中起至关重要的作用[34]。马铃薯CRKs中含有与SA、MeJA、IAA、GA等逆境激素响应元件的基因分别为3、1、1和4个, 推测马铃薯CRKs可能通过响应不同的激素信号参与对病原菌的防卫反应。而SA和JA在植物抗病中的重要作用已被证实, 为进一步验证所预测cis-elements的准确性和StCRKs对生物胁迫的响应情况, 本研究用激素SA和JA对叶片进行了处理, 实验结果表明对StCRKs启动子区域cis-element预测的结果准确性较高。而当块茎和叶片分别受到干腐病菌和晚疫病菌侵染后, StCRK4和StCRK8基因的表达随着侵染时间的延长而逐渐增强, 推断这2个基因在马铃薯响应病原真菌的过程中起重要的作用, 可作为后续功能分析的候选基因。
4 结论
鉴定出8个马铃薯CRKs, 并对其理化性质、染色体定位、基因结构和蛋白功能结构域进行分析, 预测了CRKs启动子区域响应多种激素和胁迫的调控元件, 并检测了CRKs响应马铃薯干腐病菌和晚疫病菌侵染的表达模式, 为后续研究StCRKs感受并传递内外环境刺激, 调控其生理机能的分子机制提供了理论基础, 为研究StCRKs的功能及其作用机制提供了参考。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1186/1471-2229-12-229URLPMID:23198823 [本文引用: 1]

Receptor-like kinases (RLKs) play key roles during development and in responses to the environment. Despite the relevance of the RLK family and the completion of the tomato genome sequencing, the tomato RLK family has not yet been characterized, and a framework for functional predictions of the members of the family is lacking.
DOI:10.1038/345743a0URLPMID:2163028 [本文引用: 1]
The protein kinase family of enzymes mediates the responses of eukaryotic cells to both inter- and intracellular signals. These enzymes are either serine/threonine-specific or tyrosine-specific. Many of the latter are transmembrane receptors and are important in transduction of extracellular signals across the plasma membrane, whereas few examples of receptor serine kinases have been reported. We have now identified a complementary DNA clone from Zea mays (L.) encoding a putative serine/threonine-specific protein kinase structurally related to the receptor tyrosine kinases. This structural similarity is evidence for a previously undescribed class of transmembrane receptor in higher plants likely to be involved in signal reception and transduction. Furthermore, the catalytic domain of this protein kinase is linked through a transmembrane domain to an extracellular domain similar to that of glycoproteins encoded in the self-incompatibility locus of Brassica which are involved in the self-recognition system between pollen and stigma.
DOI:10.1073/pnas.181141598URLPMID:11526204 [本文引用: 1]
Plant receptor-like kinases (RLKs) are proteins with a predicted signal sequence, single transmembrane region, and cytoplasmic kinase domain. Receptor-like kinases belong to a large gene family with at least 610 members that represent nearly 2.5% of Arabidopsis protein coding genes. We have categorized members of this family into subfamilies based on both the identity of the extracellular domains and the phylogenetic relationships between the kinase domains of subfamily members. Surprisingly, this structurally defined group of genes is monophyletic with respect to kinase domains when compared with the other eukaryotic kinase families. In an extended analysis, animal receptor kinases, Raf kinases, plant RLKs, and animal receptor tyrosine kinases form a well supported group sharing a common origin within the superfamily of serine/threonine/tyrosine kinases. Among animal kinase sequences, Drosophila Pelle and related cytoplasmic kinases fall within the plant RLK clade, which we now define as the RLK/Pelle family. A survey of expressed sequence tag records for land plants reveals that mosses, ferns, conifers, and flowering plants have similar percentages of expressed sequence tags representing RLK/Pelle homologs, suggesting that the size of this gene family may have been close to the present-day level before the diversification of land plant lineages. The distribution pattern of four RLK subfamilies on Arabidopsis chromosomes indicates that the expansion of this gene family is partly a consequence of duplication and reshuffling of the Arabidopsis genome and of the generation of tandem repeats.
DOI:10.1105/tpc.020834URLPMID:15105442 [本文引用: 2]
Receptor-like kinases (RLKs) belong to the large RLK/Pelle gene family, and it is known that the Arabidopsis thaliana genome contains &gt;600 such members, which play important roles in plant growth, development, and defense responses. Surprisingly, we found that rice (Oryza sativa) has nearly twice as many RLK/Pelle members as Arabidopsis does, and it is not simply a consequence of a larger predicted gene number in rice. From the inferred phylogeny of all Arabidopsis and rice RLK/Pelle members, we estimated that the common ancestor of Arabidopsis and rice had &gt;440 RLK/Pelles and that large-scale expansions of certain RLK/Pelle members and fusions of novel domains have occurred in both the Arabidopsis and rice lineages since their divergence. In addition, the extracellular domains have higher nonsynonymous substitution rates than the intracellular domains, consistent with the role of extracellular domains in sensing diverse signals. The lineage-specific expansions in Arabidopsis can be attributed to both tandem and large-scale duplications, whereas tandem duplication seems to be the major mechanism for recent expansions in rice. Interestingly, although the RLKs that are involved in development seem to have rarely been duplicated after the Arabidopsis-rice split, those that are involved in defense/disease resistance apparently have undergone many duplication events. These findings led us to hypothesize that most of the recent expansions of the RLK/Pelle family have involved defense/resistance-related genes.
[本文引用: 1]
DOI:10.1007/bf00016492URLPMID:7858206 [本文引用: 1]
Cell surface receptors located in the plasma membrane have a prominent role in the initiation of cellular signalling. Recent evidence strongly suggests that plant cells carry cell surface receptors with intrinsic protein kinase activity. The plant receptor-like protein kinases (RLKs) are structurally related to the polypeptide growth factor receptors of animals which consist of a large extracytoplasmic domain, a single membrane spanning segment and a cytoplasmic domain of the protein kinase gene family. Most of the animal growth factor receptor protein kinases are tyrosine kinases; however, the plant RLKs all appear to be serine/threonine protein kinases. Based on structural similarities in their extracellular domains the RLKs fall into three categories: the S-domain class, related to the self-incompatibility locus glycoproteins of Brassica; the leucine-rich repeat class, containing a tandemly repeated motif that has been found in numerous proteins from a variety of eukaryotes; and a third class that has epidermal growth factor-like repeats. Distinct members of these putative receptors have been found in both monocotyledonous plants such as maize and in members of the dicotyledonous Brassicaceae. The diversity among plant RLKs, reflected in their structural and functional properties, has opened up a broad new area of investigation into cellular signalling in plants with far-reaching implications for the mechanisms by which plant cells perceive and respond to extracellular signals.
DOI:10.1104/pp.108.119487URLPMID:18434605 [本文引用: 1]
Receptor-like proteins (RLPs) are cell surface receptors that typically consist of an extracellular leucine-rich repeat domain, a transmembrane domain, and a short cytoplasmatic tail. In several plant species, RLPs have been found to play a role in disease resistance, such as the tomato (Solanum lycopersicum) Cf and Ve proteins and the apple (Malus domestica) HcrVf2 protein that mediate resistance against the fungal pathogens Cladosporium fulvum, Verticillium spp., and Venturia inaequalis, respectively. In addition, RLPs play a role in plant development; Arabidopsis (Arabidopsis thaliana) TOO MANY MOUTHS (TMM) regulates stomatal distribution, while Arabidopsis CLAVATA2 (CLV2) and its functional maize (Zea mays) ortholog FASCINATED EAR2 regulate meristem maintenance. In total, 57 RLP genes have been identified in the Arabidopsis genome and a genome-wide collection of T-DNA insertion lines was assembled. This collection was functionally analyzed with respect to plant growth and development and sensitivity to various stress responses, including susceptibility toward pathogens. A number of novel developmental phenotypes were revealed for our CLV2 and TMM insertion mutants. In addition, one AtRLP gene was found to mediate abscisic acid sensitivity and another AtRLP gene was found to influence nonhost resistance toward Pseudomonas syringae pv phaseolicola. This genome-wide collection of Arabidopsis RLP gene T-DNA insertion mutants provides a tool for future investigations into the biological roles of RLPs.
DOI:10.1104/pp.126.2.473URLPMID:11402176 [本文引用: 1]
DOI:10.1186/1471-2229-10-95URLPMID:20500828 [本文引用: 2]
Plant Receptor-like/Pelle kinases (RLK) are a group of conserved signalling components that regulate developmental programs and responses to biotic and abiotic stresses. One of the largest RLK groups is formed by the Domain of Unknown Function 26 (DUF26) RLKs, also called Cysteine-rich Receptor-like Kinases (CRKs), which have been suggested to play important roles in the regulation of pathogen defence and programmed cell death. Despite the vast number of RLKs present in plants, however, only a few of them have been functionally characterized.
DOI:10.3864/j.issn.0578-1752.2018.13.002URL [本文引用: 2]
【目的】富含半胱氨酸类受体激酶(CRK)是植物中最大的类受体激酶家族之一,在植物生长发育、激素信号传导和抗逆境胁迫中发挥重要作用。从全基因组水平鉴定陆地棉CRK基因家族并进行生物信息学和表达模式分析,为研究和利用陆地棉CRK基因家族奠定基础。【方法】从Pfam数据库下载stress-antifung结构域氨基酸序列,应用BLASTp程序搜索棉花基因组数据库,鉴定棉花CRK基因家族;利用Compute pI/Mw tool、SignalP、TMHMM Server V2.0、WoLF POSRT等在线工具预测陆地棉CRK家族蛋白的分子量、信号肽、跨膜结构域和亚细胞定位等;用ClustalX1.8软件对棉花和拟南芥CRK蛋白质进行氨基酸序列比对,MEGA5.0分析棉花和拟南芥CRK蛋白的系统进化关系;使用TBtools制作陆地棉CRK基因家族的染色体定位、基因结构和蛋白质结构域示意图;应用植物顺式调控元件数据库PlantCARE分析棉花启动子序列;通过植物磷酸化位点数据库PlantPhos预测陆地棉CRK家族蛋白的磷酸化位点;从NCBI数据库下载RNA-Seq数据,利用转录组定量工具Kallisto计算TPM值,通过在线工具Morpheus绘制陆地棉CRK家族基因表达热图。【结果】陆地棉基因组中有70个CRK基因,分布于14条染色体,其中52个基因(74.3%)集中串联成簇分布于A6/D6、A9/D9、A10/D10染色体,且在A/D染色体组之间呈现高度共线性关系。编码302—901个氨基酸,58个蛋白质(82.9%)具有跨膜结构域,主要定位于叶绿体、质膜和胞外。磷酸化位点预测结果表明,陆地棉和拟南芥CRK有5个相同的磷酸化位点基序,包括3种丝氨酸磷酸化位点基序和2种苏氨酸磷酸化位点基序。65个陆地棉CRK基因的启动子区(92.9%)至少含有一种逆境激素响应元件,69个基因启动子区(98.6%)至少含有一种生物或非生物胁迫响应元件。根据RNA-Seq数据分析结果,陆地棉CRK基因可分为3种不同的组织表达特征类型;盐、干旱、冷、热胁迫以及接种大丽轮枝菌均可以导致部分陆地棉CRK基因表达水平的改变。GhCRK25在根、茎、叶和胚珠中优势表达,在纤维中几乎不表达,ABA、GA3、SA、PEG-6000、氯化钠和大丽轮枝菌Vd991处理均能刺激GhCRK25迅速上调表达。应用病毒诱导的基因沉默技术(VIGS)沉默GhCRK25可导致棉花对大丽轮枝菌Vd991更为敏感。【结论】陆地棉CRK基因家族有70个成员,具有保守的基因结构和功能结构域,多样化的组织表达特征,大多数基因受激素和逆境调控。
DOI:10.3864/j.issn.0578-1752.2018.13.002URL [本文引用: 2]
【目的】富含半胱氨酸类受体激酶(CRK)是植物中最大的类受体激酶家族之一,在植物生长发育、激素信号传导和抗逆境胁迫中发挥重要作用。从全基因组水平鉴定陆地棉CRK基因家族并进行生物信息学和表达模式分析,为研究和利用陆地棉CRK基因家族奠定基础。【方法】从Pfam数据库下载stress-antifung结构域氨基酸序列,应用BLASTp程序搜索棉花基因组数据库,鉴定棉花CRK基因家族;利用Compute pI/Mw tool、SignalP、TMHMM Server V2.0、WoLF POSRT等在线工具预测陆地棉CRK家族蛋白的分子量、信号肽、跨膜结构域和亚细胞定位等;用ClustalX1.8软件对棉花和拟南芥CRK蛋白质进行氨基酸序列比对,MEGA5.0分析棉花和拟南芥CRK蛋白的系统进化关系;使用TBtools制作陆地棉CRK基因家族的染色体定位、基因结构和蛋白质结构域示意图;应用植物顺式调控元件数据库PlantCARE分析棉花启动子序列;通过植物磷酸化位点数据库PlantPhos预测陆地棉CRK家族蛋白的磷酸化位点;从NCBI数据库下载RNA-Seq数据,利用转录组定量工具Kallisto计算TPM值,通过在线工具Morpheus绘制陆地棉CRK家族基因表达热图。【结果】陆地棉基因组中有70个CRK基因,分布于14条染色体,其中52个基因(74.3%)集中串联成簇分布于A6/D6、A9/D9、A10/D10染色体,且在A/D染色体组之间呈现高度共线性关系。编码302—901个氨基酸,58个蛋白质(82.9%)具有跨膜结构域,主要定位于叶绿体、质膜和胞外。磷酸化位点预测结果表明,陆地棉和拟南芥CRK有5个相同的磷酸化位点基序,包括3种丝氨酸磷酸化位点基序和2种苏氨酸磷酸化位点基序。65个陆地棉CRK基因的启动子区(92.9%)至少含有一种逆境激素响应元件,69个基因启动子区(98.6%)至少含有一种生物或非生物胁迫响应元件。根据RNA-Seq数据分析结果,陆地棉CRK基因可分为3种不同的组织表达特征类型;盐、干旱、冷、热胁迫以及接种大丽轮枝菌均可以导致部分陆地棉CRK基因表达水平的改变。GhCRK25在根、茎、叶和胚珠中优势表达,在纤维中几乎不表达,ABA、GA3、SA、PEG-6000、氯化钠和大丽轮枝菌Vd991处理均能刺激GhCRK25迅速上调表达。应用病毒诱导的基因沉默技术(VIGS)沉默GhCRK25可导致棉花对大丽轮枝菌Vd991更为敏感。【结论】陆地棉CRK基因家族有70个成员,具有保守的基因结构和功能结构域,多样化的组织表达特征,大多数基因受激素和逆境调控。
DOI:10.1371/journal.pgen.1005373URLPMID:26197346 [本文引用: 1]
Cysteine-rich receptor-like kinases (CRKs) are transmembrane proteins characterized by the presence of two domains of unknown function 26 (DUF26) in their ectodomain. The CRKs form one of the largest groups of receptor-like protein kinases in plants, but their biological functions have so far remained largely uncharacterized. We conducted a large-scale phenotyping approach of a nearly complete crk T-DNA insertion line collection showing that CRKs control important aspects of plant development and stress adaptation in response to biotic and abiotic stimuli in a non-redundant fashion. In particular, the analysis of reactive oxygen species (ROS)-related stress responses, such as regulation of the stomatal aperture, suggests that CRKs participate in ROS/redox signalling and sensing. CRKs play general and fine-tuning roles in the regulation of stomatal closure induced by microbial and abiotic cues. Despite their great number and high similarity, large-scale phenotyping identified specific functions in diverse processes for many CRKs and indicated that CRK2 and CRK5 play predominant roles in growth regulation and stress adaptation, respectively. As a whole, the CRKs contribute to specificity in ROS signalling. Individual CRKs control distinct responses in an antagonistic fashion suggesting future potential for using CRKs in genetic approaches to improve plant performance and stress tolerance.
DOI:10.1007/s00122-015-2644-4URLPMID:26660669 [本文引用: 1]
A dominantly inherited major-effect QTL for powdery mildew resistance in cucumber was fine mapped. Two tandemly arrayed cysteine-rich receptor-like protein kinase genes were identified as the most possible candidates. Powdery mildew (PM) is one of the most severe fungal diseases of cucumber (Cucumis sativus L.) and other cucurbit crops, but the molecular genetic mechanisms of powdery mildew resistance in cucurbits are still poorly understood. In this study, through marker-assisted backcrossing with an elite cucumber inbred line, D8 (PM susceptible), we developed a single-segment substitution line, SSSL0.7, carrying 95 kb fragment from PM resistance donor, Jin5-508, that was defined by two microsatellite markers, SSR16472 and SSR16881. A segregating population with 3600 F2 plants was developed from the SSSL0.7 × D8 mating; segregation analysis confirmed a dominantly inherited major-effect QTL, Pm1.1 in cucumber chromosome 1 underlying PM resistance in SSSL0.7. New molecular markers were developed through exploring the next generation resequenced genomes of Jin5-508 and D8. Linkage analysis and QTL mapping in a subset of the F2 plants delimited the Pm1.1 locus into a 41.1 kb region, in which eight genes were predicted. Comparative gene expression analysis revealed that two concatenated genes, Csa1M064780 and Csa1M064790 encoding the same function of a cysteine-rich receptor-like protein kinase, were the most likely candidate genes. GFP fusion protein-aided subcellular localization indicated that both candidate genes were located in the plasma membrane, but Csa1M064780 was also found in the nucleus. This is the first report of dominantly inherited PM resistance in cucumber. Results of this study will provide new insights into understanding the phenotypic and genetic mechanisms of PM resistance in cucumber. This work should also facilitate marker-assisted selection in cucumber breeding for PM resistance.
DOI:10.3389/fpls.2015.00322URLPMID:26029224 [本文引用: 1]
Upon recognition of microbe-associated molecular patterns (MAMPs) such as the bacterial flagellin (or the derived peptide flg22) by pattern-recognition receptors (PRRs) such as the FLAGELLIN SENSING2 (FLS2), plants activate the pattern-triggered immunity (PTI) response. The L-type lectin receptor kinase-VI.2 (LecRK-VI.2) is a positive regulator of Arabidopsis thaliana PTI. Cysteine-rich receptor-like kinases (CRKs) possess two copies of the C-X8-C-X2-C (DUF26) motif in their extracellular domains and are thought to be involved in plant stress resistance, but data about CRK functions are scarce. Here, we show that Arabidopsis overexpressing the LecRK-VI.2-responsive CRK4, CRK6, and CRK36 demonstrated an enhanced PTI response and were resistant to virulent bacteria Pseudomonas syringae pv. tomato DC3000. Notably, the flg22-triggered oxidative burst was primed in CRK4, CRK6, and CRK36 transgenics and up-regulation of the PTI-responsive gene FLG22-INDUCED RECEPTOR-LIKE 1 (FRK1) was potentiated upon flg22 treatment in CRK4 and CRK6 overexpression lines or constitutively increased by CRK36 overexpression. PTI-mediated callose deposition was not affected by overexpression of CRK4 and CRK6, while CRK36 overexpression lines demonstrated constitutive accumulation of callose. In addition, Pst DC3000-mediated stomatal reopening was blocked in CRK4 and CRK36 overexpression lines, while overexpression of CRK6 induced constitutive stomatal closure suggesting a strengthening of stomatal immunity. Finally, bimolecular fluorescence complementation and co-immunoprecipitation analyses in Arabidopsis protoplasts suggested that the plasma membrane localized CRK4, CRK6, and CRK36 associate with the PRR FLS2. Association with FLS2 and the observation that overexpression of CRK4, CRK6, and CRK36 boosts specific PTI outputs and resistance to bacteria suggest a role for these CRKs in Arabidopsis innate immunity.
DOI:10.3389/fpls.2017.01856URLPMID:29163585 [本文引用: 1]
Receptor-like kinases are important signaling components that regulate a variety of cellular processes. In this study, an Arabidopsis cDNA microarray analysis led to the identification of the cysteine-rich receptor-like kinase CRK36 responsive to the necrotrophic fungal pathogen, Alternaria brassicicola. To determine the function of CRK36 in plant immunity, T-DNA-insertion knockdown (crk36) and overexpressing (CRK36OE) plants were prepared. CRK36OE plants exhibited increased hypersensitive cell death and ROS burst in response to avirulent pathogens. Treatment with a typical pathogen-associated molecular pattern, flg22, markedly induced pattern-triggered immune responses, notably stomatal defense, in CRK36OE plants. The immune responses were weakened in crk36 plants. Protein-protein interaction assays revealed the in vivo association of CRK36, FLS2, and BIK1. CRK36 enhanced flg22-triggered BIK1 phosphorylation, which showed defects with Cys mutations in the DUF26 motifs of CRK36. Disruption of BIK1 and RbohD/RbohF genes further impaired CRK36-mediated stomatal defense. We propose that CRK36, together with BIK1 and NADPH oxidases, may form a positive activation loop that enhances ROS burst and leads to the promotion of stomatal immunity.
DOI:10.1007/s11103-004-3381-2URLPMID:15604743 [本文引用: 1]
In Arabidopsis, there is a family of receptor-like protein kinases (RLKs) containing novel cysteine-rich repeats in their extracellular domains. Genes encoding many of these cysteine-rich RLKs (CRKs) are induced by pathogen infection, suggesting a possible role in plant defense responses. We have previously generated Arabidopsis plants expressing four pathogen-regulated CRK genes (CRK5, 6, 10 and 11) under control of a steroid-inducible promoter and found that induced expression of CRK5, but not the other three CRK genes, triggered hypersensitive response-like cell death in transgenic plants. In the present study, we have analyzed the structural relationship of the CRK family and identified three CRKs (CRK4, 19 and 20) that are structurally closely related to CRK5. Genes encoding these three CRKs are all induced by salicylic acid and pathogen infection. Furthermore, induced expression of CRK4, 19 and 20 all activates rapid cell death in transgenic plants. Thus, the activity of inducing rapid cell death is shared by these structurally closely related CRKs. We have also performed yeast two-hybrid screens and identified proteins that interact with the kinase domains of CRKs. One of the identified CRK-interacting proteins is the kinase-associated type 2C protein phospohatase known to interact with a number of other RLKs through its kinase-interacting FHA domain. Other CRK-interacting proteins include a second protein with a FHA domain and another type 2C protein phosphatase. Interactions of CRKs with these three proteins in vivo were demonstrated through co-immunoprecipitation. These CRK-interacting proteins may play roles in the regulation and signaling of CRKs.
DOI:10.1111/j.1365-313X.2007.03064.xURLPMID:17419849 [本文引用: 1]
Protein kinases play important roles in relaying information from perception of a signal to the effector genes in all organisms. Cysteine-rich receptor-like kinases (CRKs) constitute a sub-family of plant receptor-like kinases (RLKs) with more than 40 members that contain the novel C-X8-C-X2-C motif (DUF26) in the extracellular domains. Here we report molecular characterization of one member of this gene family, CRK13. Expression of this gene is induced more quickly and strongly in response to the avirulent compared with the virulent strains of Pseudomonas syringae, and peaks within 4 h after pathogen infection. In response to dexamethasone (DEX) treatment, plants expressing the CRK13 gene from a DEX-inducible promoter exhibited all tested features of pathogen defense activation, including rapid tissue collapse, accumulation of high levels of several defense-related gene transcripts including PR1, PR5 and ICS1, and accumulation of salicylic acid (SA). In addition, these plants suppressed growth of virulent pathogens by about 20-fold compared with the wild-type Col-0. CRK13-conferred pathogen resistance is salicylic acid-dependent. Gene expression analysis using custom cDNA microarrays revealed a remarkable overlap between the expression profiles of the plants overexpressing CRK13 and the plants treated with Pst DC3000 (avrRpm1). Our studies suggest that upregulation of CRK13 leads to hypersensitive response-associated cell death, and induces defense against pathogens by causing increased accumulation of salicylic acid.
DOI:10.1023/B:PLAN.0000009265.72567.58URL [本文引用: 1]
During the search for potential target genes of WRKY DNA-binding transcription factors, we have previously identified four pathogen-induced Arabidopsis genes (CRK5, CRK6, CRK10 and CRK11) encoding receptor-like protein kinases (RLKs) containing novel cysteine-rich repeats in their extracellular domains. In the present study, we transformed Arabidopsis plants with the RLK genes under control of the constitutive CaMV 35S promoter or a steroid-inducible Gal4 promoter. Expression of CRK5, but not the three other RLK genes, resulted in significant alterations in defense responses and leaf growth in transgenic plants. In transgenic plants harboring the 35S::CRK5 construct, significantly elevated and constitutive expression of CRK5 correlated with enhanced leaf growth and increased resistance to the bacterial pathogen Pseudomonas syringae. The enhanced disease resistance in the transgenic plants was associated with more rapidly induced expression of the PR1 gene after pathogen infection. In transgenic plants transformed with CRK5 under control of the steroid-inducible promoter, expression of the transgene was induced at relatively high levels after the steroid application and this induced expression of CRK5 triggered hypersensitive response-like cell death. Induced CRK5 expression also activated cell death in the npr1, ndr1 and eds1 mutants and in the transgenic nahG plants that fail to accumulate salicylic acid. Thus, the novel RLK is capable of activating multiple distinct defense responses depending on the manner and/or the levels of its over-expression in transgenic plants.
DOI:10.1016/j.jplph.2011.05.018URL [本文引用: 1]
In plants, the cysteine-rich repeat kinases (CRKs) are a sub-family of receptor-like protein kinases that contain the DUF26 motif in their extracellular domains. It has been shown that in Arabidopsis thaliana, CRK20 is transcriptionally induced by pathogens, salicylic acid and ozone (O(3)). However, its role in responses to biotic and abiotic stress remains to be elucidated. To determine the function of CRK20 in such responses, two CRK20 loss-of-function mutants, crk20-1 and crk20-2, were isolated from public collections of Arabidopsis T-DNA tagged lines and examined for responses to O(3) and Pseudomonas syringae pv. tomato (Pst) DC3000. crk20-1 and crk20-2 showed similar O(3) sensitivities and no differences in the expression of defense genes when compared with the wild-type. However, pathogen growth was significantly reduced, while there were no differences in the induction of salicylic acid related defense genes or salicylic acid accumulation. Furthermore, correlation analysis of CRK20 gene expression suggests that it has a role in the control of H(2)O and/or nutrient transport. We therefore propose that CRK20 promotes conditions that are favorable for Pst DC3000 growth in Arabidopsis, possibly through the regulation of apoplastic homeostasis, and consequently, of the environment of this biotrophic pathogen. (C) 2011 Elsevier GmbH.
DOI:10.1371/journal.pgen.1006049URLPMID:27176732 [本文引用: 1]
Systemic acquired resistance, mediated by the Arabidopsis NPR1 gene and the rice NH1 gene, confers broad-spectrum immunity to diverse pathogens. NPR1 and NH1 interact with TGA transcription factors to activate downstream defense genes. Despite the importance of this defense response, the signaling components downstream of NPR1/NH1 and TGA proteins are poorly defined. Here we report the identification of a rice mutant, snim1, which suppresses NH1-mediated immunity and demonstrate that two genes encoding previously uncharacterized cysteine-rich-receptor-like kinases (CRK6 and CRK10), complement the snim1 mutant phenotype. Silencing of CRK6 and CRK10 genes individually in the parental genetic background recreates the snim1 phenotype. We identified a rice mutant in the Kitaake genetic background with a frameshift mutation in crk10; this mutant also displays a compromised immune response highlighting the important role of crk10. We also show that elevated levels of NH1 expression lead to enhanced CRK10 expression and that the rice TGA2.1 protein binds to the CRK10 promoter. These experiments demonstrate a requirement for CRKs in NH1-mediated immunity and establish a molecular link between NH1 and induction of CRK10 expression.
DOI:10.1111/j.1364-3703.2011.00736.xURLPMID:21819533 [本文引用: 1]
The receptor-like protein kinases (RLKs) constitute a large and diverse group of proteins controlling numerous plant physiological processes, including development, hormone perception and stress responses. The cysteine-rich RLKs (CRKs) represent a prominent subfamily of transmembrane-anchored RLKs. We have identified a putative barley (Hordeum vulgare) CRK gene family member, designated HvCRK1. The mature putative protein comprises 645 amino acids, and includes a putative receptor domain containing two characteristic 'domain 26 of unknown function' (duf26) domains in the N-terminal region, followed by a rather short 17-amino-acid transmembrane domain, which includes an AAA motif, two features characteristic of endoplasmic reticulum (ER)-targeted proteins and, finally, a characteristic putative protein kinase domain in the C-terminus. The HvCRK1 transcript was isolated from leaves inoculated with the biotrophic fungal pathogen Blumeria graminis f.sp. hordei (Bgh). HvCRK1 transcripts were observed to accumulate transiently following Bgh inoculation of susceptible barley. Transient silencing of HvCRK1 expression in bombarded epidermal cells led to enhanced resistance to Bgh, but did not affect R-gene-mediated resistance. Silencing of HvCRK1 phenocopied the effective penetration resistance found in mlo-resistant barley plants, and the possible link between HvCRK1 and MLO was substantiated by the fact that HvCRK1 induction on Bgh inoculation was dependent on Mlo. Finally, using both experimental and in silico approaches, we demonstrated that HvCRK1 localizes to the ER of barley cells. The negative effect on basal resistance against Bgh and the functional aspects of MLO- and ER-localized HvCRK1 signalling on Bgh inoculation are discussed.
URLPMID:15639879 [本文引用: 1]
MAPMAKER is one of the most widely used computer software package for constructing genetic linkage maps.However, the PC version, MAPMAKER 3.0 for PC, could not draw the genetic linkage maps that its Macintosh version, MAPMAKER 3.0 for Macintosh,was able to do. Especially in recent years, Macintosh computer is much less popular than PC. Most of the geneticists use PC to analyze their genetic linkage data. So a new computer software to draw the same genetic linkage maps on PC as the MAPMAKER for Macintosh to do on Macintosh has been crying for. Microsoft Excel,one component of Microsoft Office package, is one of the most popular software in laboratory data processing. Microsoft Visual Basic for Applications (VBA) is one of the most powerful functions of Microsoft Excel. Using this program language, we can take creative control of Excel, including genetic linkage map construction, automatic data processing and more. In this paper, a Microsoft Excel macro called MapDraw is constructed to draw genetic linkage maps on PC computer based on given genetic linkage data. Use this software,you can freely construct beautiful genetic linkage map in Excel and freely edit and copy it to Word or other application. This software is just an Excel format file. You can freely copy it from ftp://211.69.140.177 or ftp://brassica.hzau.edu.cn and the source code can be found in Excel's Visual Basic Editor.
DOI:10.1093/nar/25.24.4876URLPMID:9396791 [本文引用: 1]
CLUSTAL X is a new windows interface for the widely-used progressive multiple sequence alignment program CLUSTAL W. The new system is easy to use, providing an integrated system for performing multiple sequence and profile alignments and analysing the results. CLUSTAL X displays the sequence alignment in a window on the screen. A versatile sequence colouring scheme allows the user to highlight conserved features in the alignment. Pull-down menus provide all the options required for traditional multiple sequence and profile alignment. New features include: the ability to cut-and-paste sequences to change the order of the alignment, selection of a subset of the sequences to be realigned, and selection of a sub-range of the alignment to be realigned and inserted back into the original alignment. Alignment quality analysis can be performed and low-scoring segments or exceptional residues can be highlighted. Quality analysis and realignment of selected residue ranges provide the user with a powerful tool to improve and refine difficult alignments and to trap errors in input sequences. CLUSTAL X has been compiled on SUN Solaris, IRIX5.3 on Silicon Graphics, Digital UNIX on DECstations, Microsoft Windows (32 bit) for PCs, Linux ELF for x86 PCs, and Macintosh PowerMac.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1006/meth.2001.1262URLPMID:11846609 [本文引用: 1]
The two most commonly used methods to analyze data from real-time, quantitative PCR experiments are absolute quantification and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control. The 2(-Delta Delta C(T)) method is a convenient way to analyze the relative changes in gene expression from real-time quantitative PCR experiments. The purpose of this report is to present the derivation, assumptions, and applications of the 2(-Delta Delta C(T)) method. In addition, we present the derivation and applications of two variations of the 2(-Delta Delta C(T)) method that may be useful in the analysis of real-time, quantitative PCR data.
DOI:10.1002/prot.22494URLPMID:19603485 [本文引用: 1]
DOI:10.1073/pnas.1109047109URLPMID:22232673 [本文引用: 1]
Gene duplication plays key roles in organismal evolution. Duplicate genes, if they survive, tend to diverge in regulatory and coding regions. Divergences in coding regions, especially those that can change the function of the gene, can be caused by amino acid-altering substitutions and/or alterations in exon-intron structure. Much has been learned about the mode, tempo, and consequences of nucleotide substitutions, yet relatively little is known about structural divergences. In this study, by analyzing 612 pairs of sibling paralogs from seven representative gene families and 300 pairs of one-to-one orthologs from different species, we investigated the occurrence and relative importance of structural divergences during the evolution of duplicate and nonduplicate genes. We found that structural divergences have been very prevalent in duplicate genes and, in many cases, have led to the generation of functionally distinct paralogs. Comparisons of the genomic sequences of these genes further indicated that the differences in exon-intron structure were actually accomplished by three main types of mechanisms (exon/intron gain/loss, exonization/pseudoexonization, and insertion/deletion), each of which contributed differently to structural divergence. Like nucleotide substitutions, insertion/deletion and exonization/pseudoexonization occurred more or less randomly, with the number of observable mutational events per gene pair being largely proportional to evolutionary time. Notably, however, compared with paralogs with similar evolutionary times, orthologs have accumulated significantly fewer structural changes, whereas the amounts of amino acid replacements accumulated did not show clear differences. This finding suggests that structural divergences have played a more important role during the evolution of duplicate than nonduplicate genes.
DOI:10.1007/s00425-009-1062-8URLPMID:19937257 [本文引用: 1]
Stilbene synthase is a plant-specific polyketide synthase, and plays important roles in diverse metabolic processes. The genomic stilbene synthase gene was cloned from accession &quot;Baihe-35-1&quot; of Chinese wild Vitis pseudoreticulata, and a stilbene synthase of V. pseudoreticulata (VpSTS) transcripts expressed in the grape-powdery mildew interaction were determined by semi-quantitative RT-PCR. To monitor VpSTS expression in plant, the promoter region flanking the 5' VpSTS coding region was isolated from the genomic DNA of Chinese wild V. pseudoreticulata accession Baihe-35-1. Alignment of the VpSTS promoter sequence showed a 56.4% identity to Vitis vinifera. To identify the upstream region of the VpSTS gene required for promoter activity, a series of VpSTS promoter deletion derivatives was constructed. Each deletion construct was analyzed by Agrobacterium-mediated transient transformation in grapevine and tobacco leaves after infection by Uncinula necator and Alternaria alternata. In transiently transformed grapevine leaves, GUS activity was also determined after treatment with salicylic acid (SA) and 4 degrees C cold. Analysis of a series of 5' deletions of the VpSTS promoter in grapevine leaves indicated that the proximal 162 bp from the transcription initiation site was proved to be necessary for establishing both the constitutive and induced pattern of expression.
URLPMID:8896462 [本文引用: 1]
PR1 is a pathogenesis-related protein encoded in the parsley genome by a family of three genes (PR1-1, PR1-2 and PR1-3). Loss- and gain-of-function experiments in a transient expression system demonstrated the presence of two fungal elicitor responsive elements in each of the PR1-1 and PR1-2 promoters. These elements, W1, W2 and W3, contain the sequence (T)TGAC(C) and mutations that disrupt this sequence abolish function. Gel shift experiments demonstrated that W1, W2 and W3 are bound specifically by similar nuclear proteins. Three cDNA clones encoding sequence-specific DNA-binding proteins were isolated by South-Western screening and these proteins, designated WRKY1, 2 and 3, also bind specifically to W1, W2 and W3. WRKY1, 2 and 3 are members of the family of sequence-specific DNA-binding proteins, which we call the WRKY family. Treatment of parsley cells with the specific oligopeptide elicitor Pep25 induced a transient and extremely rapid increase in mRNA levels of WRKY1 and 3. WRKY2 mRNA levels in contrast showed a concomitant transient decrease. These rapid changes in WRKY mRNA levels in response to a defined signal molecule suggest that WRKY1, 2 and 3 play a key role in a signal transduction pathway that leads from elicitor perception to PR1 gene activation.
DOI:10.1104/pp.101.3.1117URLPMID:8310051 [本文引用: 1]
DOI:10.1105/tpc.5.11.1529URLPMID:8312738 [本文引用: 1]
An Arabidopsis cDNA (Atmyb2) that contains a sequence that encodes a transcription factor, which is a homolog of MYB, was cloned from a cDNA library prepared from dehydrated Arabidopsis rosette plants. A gene (Atmyb2) corresponding to the Atmyb2 cDNA was also cloned and its nucleotide sequence was determined. RNA gel blot analysis showed that the Atmyb2 mRNA was induced by dehydration and disappeared upon rehydration. The Atmyb2 mRNA also accumulated upon salt stress and with the onset of treatment with abscisic acid. A beta-glucuronidase reporter gene driven by the Atmyb2 promoter was induced by dehydration and salt stress in transgenic Arabidopsis plants. These observations indicate that Atmyb2 is responsive to dehydration at the transcriptional level. The putative protein (ATMYB2) encoded by Atmyb2 has 274 amino acids, a molecular mass of 32 kD, and a putative DNA binding domain that shows considerable homology to plant MYB-related proteins, such as maize C1. A fusion protein that included ATMYB2 was expressed in Escherichia coli, and it bound specifically to oligonucleotides that contained a consensus MYB recognition sequence (TAACTG), such as is found in the simian virus 40 enhancer and the maize bronze-1 promoter. Binding was sequence specific, as indicated by a gel mobility shift experiment. These results suggest that a MYB-related transcription factor is involved in the regulation of genes that are responsive to water stress in Arabidopsis.
DOI:10.1104/pp.101.3.1119URLPMID:8310052 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}