Identification of gene co-expression modules of maize plant height and ear height by WGCNA
MA Juan, CAO Yan-Yong, WANG Li-Feng, LI Jing-Jing, WANG Hao, FAN Yan-Ping, LI Hui-Yong,*Institute of Cereal Crops, Henan Academy of Agricultural Sciences, Zhengzhou 450002, Henan, China
This study was supported by the National Key Research and Development Program of China.2016YFD100103 the Science and Technology Project of Henan province.192102110008
Abstract Plant height (PH) and ear height (EH) are important factors for maize plant type and grain yield. Weighted gene co-expression network analysis (WGCNA) is an important method to explain the relationships between gene network and complicated traits and identify the PH and EH associated genes. In this study, we used Zheng 58, Ye 478, Chang 7-2, Huangzaosi and its combinations Zhengdan 958, Anyu 5, Zheng 58/Huangzaosi, and Ye 478/Huangzaosi as materials and utilized transcriptome data under the planting densities of 45,000 plants hm -2and 67,500 plants hm -2 to construct a co-expression network by WGCNA, getting 24 and 21 co-expression modules, respectively. Among them, a total of 15 co-expression modules were significantly correlated with PH and EH, with the absolute correlation coefficients higher than 0.50. Six modules were overlapped between PH and EH. By gene function analysis, these overlapped modules were significantly enriched in development, photosynthesis, response to light stimulus, plant hormone, and carbohydrate biosynthesis/metabolism related activities. According to connectivity of genes in modules, AP2-EREBP transcription factor EREB14, thiaminase TENA2, phosphoglyceric kinase PGK, glutathione transferase GST2, and succinate dehydrogenase SUDH7 were considered as hub genes. From gene networks, EREB14 was connected with three known PH genes D8, DWF1, ZmGRF10, and C3H35 (C3H transcription factor), GATA4 (C2C2-GATA transcription factor), and ethylene homology ETR40. Reported PH genes An1 and GA20ox3 were also found in our co-expression modules. From the networks of the five known PH genes, ARF-transcription factor 7 (ARFTF7), ARFTF26, GST39, photosystem II oxygen evolving polypeptide PspB2, and photosystem I N subunit PasN1 had connections with these known PH genes. The identification of 15 co-expression modules and their hub genes, and analysis of their gene function and gene networks of key genes will be helpful for revealing the genetic basis of PH and EH. Keywords:maize;weighted gene co-expression network;transcriptome;plant height;ear height
PDF (4874KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 马娟, 曹言勇, 王利锋, 李晶晶, 王浩, 范艳萍, 李会勇. 利用WGCNA鉴定玉米株高和穗位高基因共表达模块[J]. 作物学报, 2020, 46(3): 385-394. doi:10.3724/SP.J.1006.2020.93021 MA Juan, CAO Yan-Yong, WANG Li-Feng, LI Jing-Jing, WANG Hao, FAN Yan-Ping, LI Hui-Yong. Identification of gene co-expression modules of maize plant height and ear height by WGCNA[J]. Acta Crops Sinica, 2020, 46(3): 385-394. doi:10.3724/SP.J.1006.2020.93021
玉米自交系郑58、掖478、昌7-2、黄早四及其组配的杂交种郑单958 (郑58/昌7-2)、安玉5号(掖478/昌7-2)、郑58/黄早四和掖478/黄早四于2016年种植在河南省农业科学院新乡原阳试验田。采用随机区组试验设计, 3次重复。种植密度为45,000株 hm-2和67,500株 hm-2。授粉后15 d, 采集8个材料的穗位叶提取RNA, 用于转录组测序。采用Illumina X Ten对2种密度条件8个材料3个生物学重复进行双端测序。48个样品的转录组测序数据见NCBI (National Center for Biotechnology Information)的Sequence Read Archive数据库(登录号为 SRP136913)。利用Tophat v2.0.10将clean reads比对到玉米B73参考基因组(ftp://ftp.ensemblgenomes.org/pub/release-29/plants/fasta/zea_mays/dna/Zea_mays.AGPv3.29.dna.toplevel.fa.gz)。利用Cufflinks v.2.1.1计算基因在两种密度条件下8个材料3个生物学重复转录本的表达量。利用每千碱基外显子百万片段数(fragments per kilobase of exon per million fragments mapped, FPKM)值衡量基因的表达水平。在两种密度条件下, 分别选取24个样品FPKM值均大于1的基因, 用于构建基因共表达网络分析。授粉后45 d, 每个材料每个重复测量5个植株的株高和穗位高。利用R语言计算方差分析和Duncan’s多重比较。
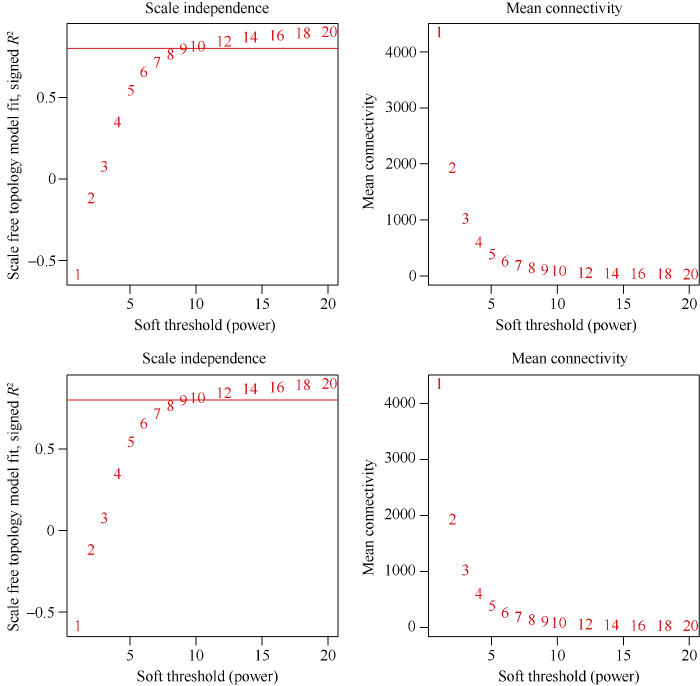
A和B: 左图纵坐标是无尺度网络模型指数; 右图纵坐标每一个软阈值对应的平均连接度; 横坐标均代表软阈值β。 Fig. 1Determination of soft threshold β at 45,000 plants hm-2
(A) and 67,500 plants hm-2 (B) A and B: The ordinate represents the index of scale free network model in left figure. The ordinate represents the average link degree of each soft threshold in right figure. The abscissa represents the soft threshold β.
图中数字表示均值±标准误。柱中字母为Duncan’s多重比较结果, 不同字母表示材料间在P < 0.05水平差异显著。*表示0.05显著水平。 Fig. 2Plant height and ear height of eight materials at 45,000 plants hm-2 and 67,500 plants hm-2
Number represents mean±SE. Bars with different letters are significantly different at P < 0.05 as determined by Duncan’s multiple comparison. * denotes significant at the 0.05 probability level.
Li QC, Li YX, Yang ZZ, LiuC, Liu ZZ, Li CH, PengB, ZhangY, WangD, Tan WW, Sun BC, Shi SY, Song CY, Zhang ZM, Pan GT, LiY, Wang TY . QTL mapping for plant height and ear height by using multiple related RIL populations in maize Acta Agron Sin, 2013,39:1521-1529 (in Chinese with English abstract). [本文引用: 1]
He KH, Chang LG, Cui TT, Qu JZ, Guo DW, Xu ST, Zhang XH, Zhang RH, Xue JQ, Liu JC . Mapping QTL for plant height and ear height in maize under multi-environments Sci Agric Sin, 2016,49:1443-1452 (in Chinese with English abstract). [本文引用: 1]
LiuK, Zhang XH, Sun GY, Yan PS, Guo HP, Chen SY, Xue YD, Guo ZY, Xie HL, Tang JH, Li WH . Genome-wide association studies of plant type traits in maize Sci Agric Sin, 2018,51:821-834 (in Chinese with English abstract). [本文引用: 1]
LiK, Zhang XX, Guan ZR, Shen YO, Pan GT . Genome-wide association analysis of plant height and ear height in maize J Maize Sci, 2017,25(6):1-7 (in Chinese with English abstract). [本文引用: 2]
LiX, ZhouZ, DingJ, WuY, ZhouB, WangR, MaJ, WangS, ZhangX, XiaZ, ChenJ, WuJ . Combined linkage and association mapping reveals QTL and candidate genes for plant and ear height in maize Front Plant Sci, 2016,7:833. [本文引用: 5]
WengJ, XieC, HaoZ, WangJ, LiuC, LiM, ZhangD, BaiL, ZhangS, LiX . Genome-wide association study identifies candidate genes that affect plant height in Chinese elite maize (Zea mays L.) inbred lines PLoS One, 2011,6:e29229. [本文引用: 1]
FujiokaS, YamaneH, Spray CR, GaskinP, MacmillanJ, Phinney BO, TakahashiN . Qualitative and quantitative analyses of gibberellins in vegetative shoots of normal, dwarf-1, dwarf-2, dwarf-3, and dwarf-5 seedlings of Zea mays L Plant Physiol, 1988,88:1367-1372. [本文引用: 1]
Winkler RG, HelentjarisT . The maize Dwarf3 gene encodes acytochrome P450-mediated early step in gibberellin biosynthesis Plant Cell, 1995,7:1307-1317. [本文引用: 2]
Thornsberry JM, Goodman MM, DoebleyJ, KresovichS, NielsenD, Buckler ES . Dwarf8 polymorphisms associate with variation in flowering time Nat Genet, 2001,28:286-289. [本文引用: 1]
Lawit SJ, Wych HM, XuD, KunduS, Tomes DT . Maize DELLA proteins dwarf plant8 and dwarf plant9 as modulators of plant development Plant Cell Physiol, 2010,51:1854-1868. [本文引用: 4]
Multani DS, Briggs SP, Chamberlin MA, Blakeslee JJ, Murphy AS, Johal GS . Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants Science, 2003,302:81-84. [本文引用: 2]
ZhangB, Horvath S. A general framework for weighted gene co-expression network analysis Stat Appl Genet Mol Biol, 2005, 4: Article 17. [本文引用: 1]
ZhangX, Hirsch CN, Sekhon RS, De LeonN, Kaeppler SM . Evidence for maternal control of seed size in maize from phenotypic and transcriptional analysis J Exp Bot, 2016,67:1907-1917. [本文引用: 1]
MaJ, ZhangD, CaoY, WangL, LiJ, LübberstedtT, WangT, LiY, LiH . Heterosis-related genes under different planting densities in maize (Zea mays L.) J Exp Bot, 2018,69:5077-5087. [本文引用: 1]
Yang YX, Sang ZQ, XuC, Dai WS, ZouC . Identification of maize flowering gene co-expression modules by WGCNA Acta Agron Sin, 2019,45:161-174 (in Chinese with English abstract). [本文引用: 1]
PengH, HeX, GaoJ, MaH, ZhangZ, ShenY, PanG, LinH . Transcriptomic changes during maize roots development responsive to Cadmium (Cd) pollution using comparative RNA seq- based approach Biochem Biophys Res Commun, 2015,464:1040-1047. [本文引用: 1]
ThirunavukkarasuN, HossainF, MohanS, ShirigaK, MittalS, SharmaR, Singh RK, Gupta HS . Genome-wide expression of transcriptomes and their co-expression pattern in subtropical maize (Zea mays L.) under waterlogging stress PLoS One, 2013,8:e70433. [本文引用: 1]
LyuY, LiangZ, GeM, QiW, ZhangT, LinF, PengZ, ZhaoH . Genome-wide identification and functional prediction of nitrogen-responsive intergenic and intronic long non-coding RNAs in maize ( Zea mays L.) BMC Genomics, 2016,17:350. [本文引用: 1]
ZhangS, YangW, ZhaoQ, ZhouX, JiangL, MaS, LiuX, LiYe, ZhangC, FanY, ChenR . Analysis of weighted co-regulatory networks in maize provides insights into new genes and regulatory mechanisms related to inositol phosphate metabolism BMC Genomics, 2016,17:129-146. [本文引用: 1]
TaoY, ZhengJ, XuZ, ZhangX, ZhangK, WangG . Functional analysis of ZmDWF1, a maize homolog of the Arabidopsis brassinosteroids biosynthetic DWF1/DIM gene Plant Sci, 2004,167:741-751. [本文引用: 2]
WuL, ZhangD, XueM, QianJ, HeY, WangS . Overexpression of the maize GRF10, an endogenous truncated growth regulating factor protein, leads to reduction in leaf size and plant height J Integr Plant Biol, 2014,56:1053-1063. [本文引用: 2]
HartwigT, Chuck GS, FujiokaS, KlempienA, WeizbauerR, Potluri DP, ChoeS, Johal GS, SchulzB . Brassinosteroid control of sex determination in maize Proc Natl Acad Sci USA, 2011,108:19814-19819. [本文引用: 2]
TamotsuH, Rod WK, Chris AH, MasajiK . The involvement of gibberellin 20-oxidase genes in phytochrome-regulated petiole elongation of Arabidopsis Plant Physiol, 2005,138:1106-1116. [本文引用: 2]
ZhaoW, LangfelderP, FullerT, DongJ, LiA, HovarthS . Weighted gene coexpression network analysis: state of the art J Biopharm Stat, 2010,20:281-300. [本文引用: 1]
WangH, GuL, ZhangX, LiuM, JiangH, CaiR, ZhaoY, ChengB . Global transcriptome and weighted gene co-expression network analyses reveal hybrid-specific modules and candidate genes related to plant height development in maize Plant Mol Biol, 2018,98:187-203. [本文引用: 3]
MizoiJ, ShinozakiK, Yamaguchi-ShinozakiK . AP2/ERF family transcription factors in plant abiotic stress responses BBA-Gene Regul Mech, 2012,1819:86-96. [本文引用: 1]
HinzM, Wilson IW, YangJ, BuerstenbinderK, LlewellynD, Dennis ES, SauterM, DolferusR . Arabidopsis RAP2: 2. An ethylene response transcription factor that is important for hypoxia survival Plant Physiol, 2010,153:757-772. [本文引用: 1]
LicausiF, Ohme TakagiM, PerataP . APETALA2/ethylene responsive factor (AP2/ERF) transcription factors: mediators of stress responses and developmental programs New Phytol, 2013,199:639-649. [本文引用: 1]
CassaniE, BertoliniE, Cerino BF, LandoniM, GavinaD, SirizzottiA, PiluR . Characterization of the first dominant dwarf maize mutant carrying a single amino acid insertion in the VHYNP domain of the dwarf8 gene Mol Breed, 2009,24:375-385. [本文引用: 1]
TengF, ZhaiL, LiuR, BaiW, WangL, HuoD, TaoY, ZhengY, ZhangZ . ZmGA3ox2, a candidate gene for a major QTL, qPH3.1, for plant height in maize Plant J, 2013,73:405-416. [本文引用: 2]
ZhengL, ZhouY, ZengX, DiH, Weng JF, Li XH, Wang ZH . QTL Mapping of plant height in maize Crops, 2016, ( 2):8-13 (in Chinese with English abstract). [本文引用: 1]
 ,*河南省农业科学院粮食作物研究所, 河南郑州 450002
,*河南省农业科学院粮食作物研究所, 河南郑州 450002
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT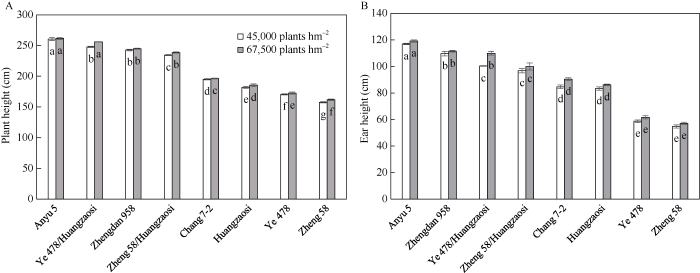 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT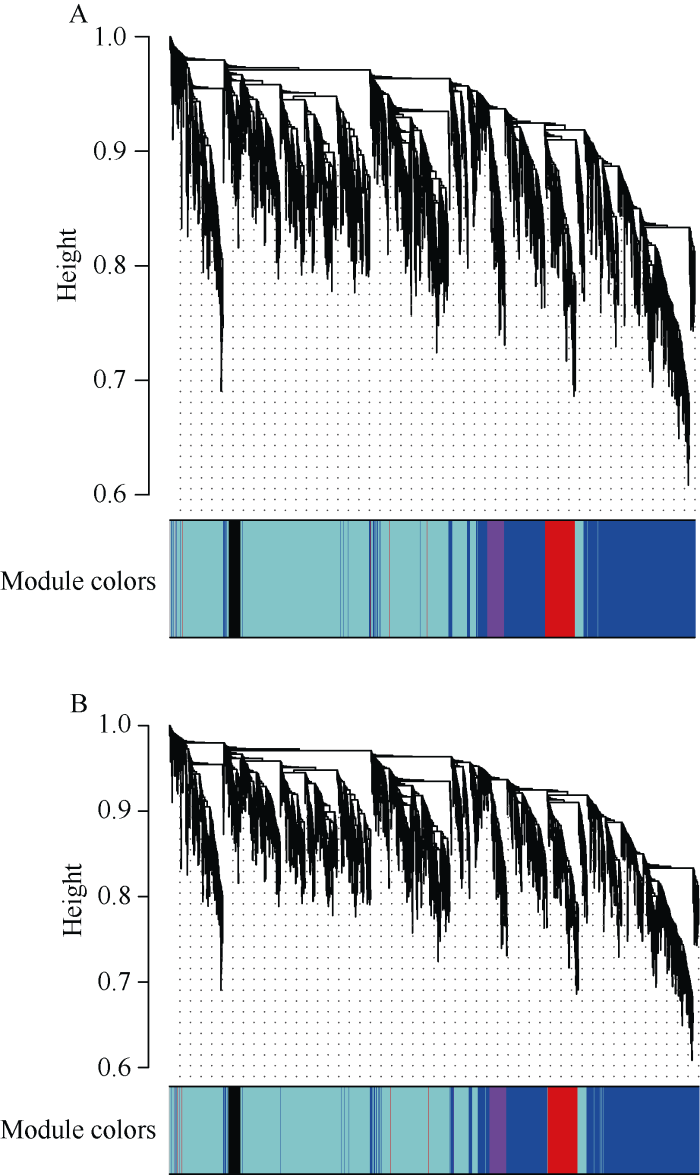 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT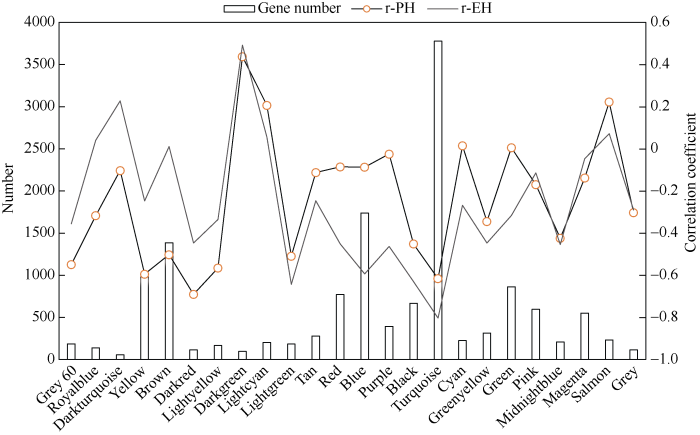 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT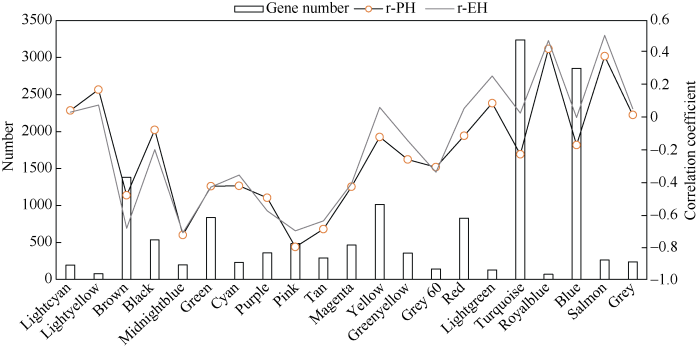 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT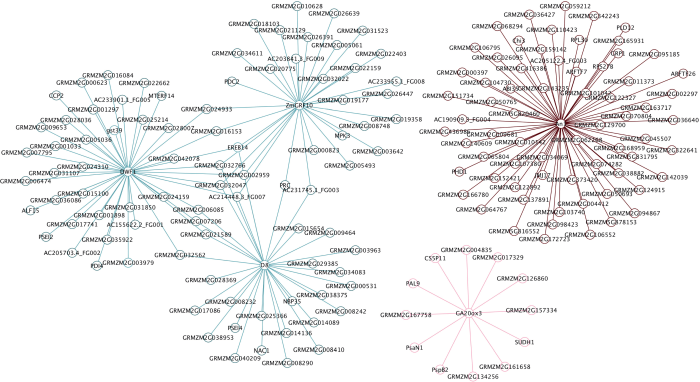 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}