 ,2,*, 张洁夫,1,*, 傅廷栋2
,2,*, 张洁夫,1,*, 傅廷栋2Genome-wide association study of seed number per silique in rapeseed (Brassica napus L.)
SUN Cheng-Ming1,2, CHEN Feng1, CHEN Song1, PENG Qi1, ZHANG Wei1, YI Bin,2,*, ZHANG Jie-Fu,1,*, FU Ting-Dong2通讯作者:
收稿日期:2019-04-15接受日期:2019-08-9网络出版日期:2019-09-11
| 基金资助: |
Received:2019-04-15Accepted:2019-08-9Online:2019-09-11
| Fund supported: |
作者简介 About authors
E-mail:suncm8331537@gmail.com。

摘要
关键词:
Abstract
Keywords:
PDF (3259KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
孙程明, 陈锋, 陈松, 彭琦, 张维, 易斌, 张洁夫, 傅廷栋. 甘蓝型油菜每角粒数的全基因组关联分析[J]. 作物学报, 2020, 46(1): 147-153. doi:10.3724/SP.J.1006.2020.94060
SUN Cheng-Ming, CHEN Feng, CHEN Song, PENG Qi, ZHANG Wei, YI Bin, ZHANG Jie-Fu, FU Ting-Dong.
油菜是我国重要的油料作物, 种植面积约690万公顷, 产油量占国产植物油总量的55%以上, 是我国最大的食用植物油来源[1]。作为食用油消费大国, 我国食用油60%以上却依赖进口, 油菜的产量水平直接影响我国食用油的供给, 因此, 提高产量成为油菜育种最迫切的目标。每角粒数是油菜重要的产量构成因子, 解析其遗传基础对提高油菜产量具有重要意义。
油菜种子的形成起始于胚珠原基, 而胚珠原基需要经历珠被、珠心和胚囊的形成, 以及授粉、受精、种子发育等过程才能最终形成种子, 这一系列过程受到复杂的遗传网络调控。李永鹏等[2]研究表明, 部分胚珠原基在发育过程中败育而最终不能形成成熟的种子, 正常发育胚珠百分比是反映品种间每角粒数差异的重要因素。Yang等[3]进一步研究表明, 油菜每角粒数的自然变异由每子房胚珠数(30.7%)、可育胚珠比(18.2%)、可育胚珠授精率(7.1%)和授精胚珠发育成种子的比率(43.9%)的差异共同决定。
多位研究者采用连锁分析定位了每角粒数QTL, Shi等[4]利用TN DH及F2群体在13条染色体检测到69个每角粒数QTL, 解释的表型变异范围是3.2%~15.5%; Luo等[5]利用60K SNP芯片再次对TN DH群体进行QTL定位, 共检测到64个每角粒数QTL, 其中贡献率最大的2个QTL qSN.A1-3和qSN.A1-5均位于A01染色体, 分别解释20.4%和24.1%的表型变异; 漆丽萍[6]在A01、A06~A08、C01、C03和C05染色体检测到15个QTL, 其中贡献率最大的2个QTL qSN.A1.1和qSN.A1.2均位于A01染色体, 分别解释26.3%和26.6%的表型变异; Cai等[7]在A01~A04、A06、A08~A10、C01、C03、C05、C06和C08染色体检测到17个QTL, 可解释5.6%~26.4%的表型变异; Yang等[8]利用一个RIL群体在A02、A06、C01、C04和C06染色体检测到5个QTL, 可解释5.3%~24.8%的表型变异, 其中位于A06的QTL qSN.A6在多个环境中被重复检测到, 解释20.1%的表型变异。
近年来, 随着高密度SNP基因分型芯片和全基因组测序等技术的发展, 全基因组关联分析(genome-wide association study, GWAS)已广泛应用于油菜复杂性状的遗传结构解析[9,10,11,12]。本研究以496份具有代表性的油菜资源为关联群体, 利用60K SNP芯片对群体进行基因型分析, 并对每角粒数进行全基因组关联分析, 旨在挖掘显著的SNP位点, 分析其候选基因, 为油菜每角粒数的遗传改良奠定基础。
1 材料与方法
1.1 试验材料
用于关联分析的496份甘蓝型油菜包括国内外的地方品种、育成品种及高世代育种材料(附表1)。其中国内资源444份, 主要来自湖北、重庆、江苏、湖南、四川、陕西等油菜主产省市; 国外资源52份, 主要来自德国、瑞典、朝鲜、加拿大等国家。所有材料均由华中农业大学国家油菜工程技术研究中心提供。1.2 田间试验与表型调查
于2015年和2016年在江苏泰州(15TZ和16TZ)种植自然群体。采用完全随机区组设计, 设置2个重复, 单个材料每重复种2行, 每行15株, 行宽1.5 m, 行距40 cm。每年10月上旬大田直播, 田间管理按当地常规方式进行。在油菜成熟期, 取每小区8株长势一致的植株, 选主花序中部10个角果测量每角粒数。利用R软件对各环境的每角粒数表型进行统计分析和相关性分析, 并计算广义遗传力和最佳线性无偏预测值(best linear unbiased prediction, BLUP)[13,14]。1.3 基因型检测与分析
利用Illumina 60K SNP芯片对496份甘蓝型油菜资源进行基因型分析。在Genome Studio软件中, 剔除Call Freq参数 < 0.75, 稀有等位基因频率 < 0.05, AA、BB频率 < 0.03及GenTrain值 < 0.50的SNP, 基因型杂合的SNP按缺失值处理。将过滤后的33,218个SNP序列比对至油菜Darmor-bzh基因组, e-value阈值设为10-12, 保留19,167个单拷贝、位置清楚的SNP标记用于后续分析。1.4 全基因组关联分析
用Structure 2.3.3进行群体结构Q矩阵的计算[15], 用SPAGeDi计算材料间的亲缘关系K矩阵[16]。将基因型、表型、Q矩阵和K矩阵导入Tassel 4.0后, 以Q矩阵和Q+K矩阵作协变量, 分别进行基于一般线性模型(General Linear Model, GLM)和混合线性模型(Mixed Linear Model, MLM)的全基因组关联分析[17]。参考前人文献[9,10,11,12], 关联分析的显著性阈值 (Bonferroni threshold) 定为1/总标记数, 即-lg (P) = 4.28。当1 Mb区间内存在多个显著SNP时, 如果两两间的r2≥0.1, 则将这些SNP归为一个关联位点, 以最小P值的SNP作为代表。用R软件包qqman绘制曼哈顿图和QQ (Quantile- Quantile)图[18]。1.5 候选基因挖掘
以r2 = 0.1作为衰减阈值, 用Tassel计算显著关联位点的LD衰减距离作为其置信区间, 提取置信区间内的基因CDS序列, 与拟南芥的基因CDS进行BLAST比对, 设e-value阈值为10-10, 以相似度最高的拟南芥基因信息来注释油菜基因。将落在置信区间的油菜每角粒数基因和拟南芥每角粒数基因的同源基因作为候选基因。2 结果与分析
2.1 每角粒数表型分析
496份油菜资源的每角粒数在2个环境中具有广泛的变异, 单个环境的每角粒数极差范围是15.24~18.90。单个环境表型平均值范围是(20.56±1.99)~(21.45±2.36), 变异系数范围是0.10~0.11 (表1和图1)。2个环境间的相关系数为0.55 (P ≤ 0.001), 达到极显著水平, 这表明本研究考察的表型数据具有较高的可靠性和重复性。每角粒数在2个环境的广义遗传力为57.7%。Table 1
表1
表1关联群体每角粒数性状的统计分析
Table 1
| 环境 Environment | 最小值 Min. | 最大值 Max. | 平均值±标准差 Mean ± SD | 变异系数 CV |
|---|---|---|---|---|
| 2014/2015 Taizhou | 8.83 | 27.73 | 21.45±2.36 | 0.11 |
| 2015/2016 Taizhou | 11.00 | 26.24 | 20.56±1.99 | 0.10 |
新窗口打开|下载CSV
图1
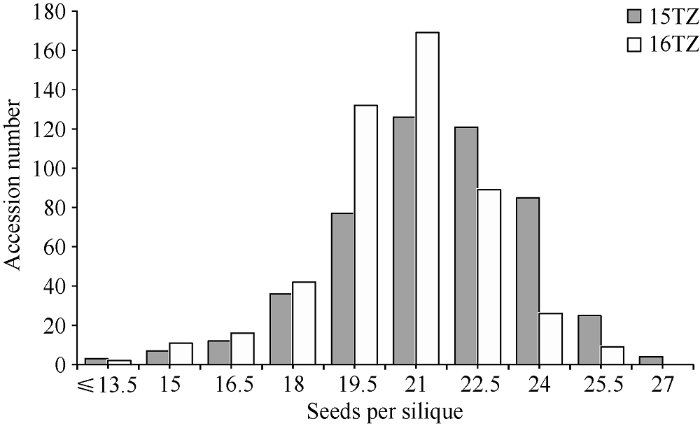 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1关联群体在2个环境的每角粒数分布
Fig. 1Distribution of seed number per silique of the association panel in two environments
2.2 每角粒数全基因组关联分析
利用MLM模型对各环境的每角粒数表型和BLUP值进行全基因组关联分析, 当阈值为-lg (P) = 4.28时, 在15TZ、16TZ和基于BLUP分别检测到6、2和2个显著位点(表2和图2)。位于C02的位点Bn-scaff_15712_6- p1336179在15TZ和16TZ两环境中被重复检测到。合并重叠的位点后得到9个显著位点, 分布在A01、A04、A07、A08、C01~C04染色体, 可解释3.26%~4.61%的表型变异。Table 2
表2
表2MLM每角粒数显著关联位点
Table 2
| 标记 Marker | 染色体 Chr. | 位置 Position | -lg (P) | 表型变异 R2 (%) | 环境 Environment | 已报道QTL Reported QTL |
|---|---|---|---|---|---|---|
| Bn-A01-p3904495 | A01 | 3,530,446 | 5.01 | 0.03832 | 16TZ | [5] |
| Bn-A04-p10393460 | A04 | 11,537,744 | 5.15 | 0.03946 | 15TZ | |
| Bn-A07-p22251229 | A07 | 23,650,401 | 4.78 | 0.03896 | 15TZ | |
| Bn-A08-p14749618 | A08 | 12,312,967 | 4.37 | 0.03265 | 15TZ | [7] |
| Bn-scaff_15936_1-p270915 | C01 | 36,405,870 | 4.66 | 0.03521 | BLUP | [5-6] |
| Bn-scaff_15712_6-p1336179 | C02 | 38,045,422 | 4.33 | 0.04469 | 15TZ, 16TZ | |
| Bn-scaff_23954_1-p220801 | C03 | 11,576,750 | 4.49 | 0.03371 | 15TZ | |
| Bn-scaff_16027_1-p367097 | C04 | 1,254,637 | 4.52 | 0.03698 | 15TZ | |
| Bn-scaff_23907_1-p3780 | C04 | 7,268,977 | 5.09 | 0.04605 | BLUP |
新窗口打开|下载CSV
图2
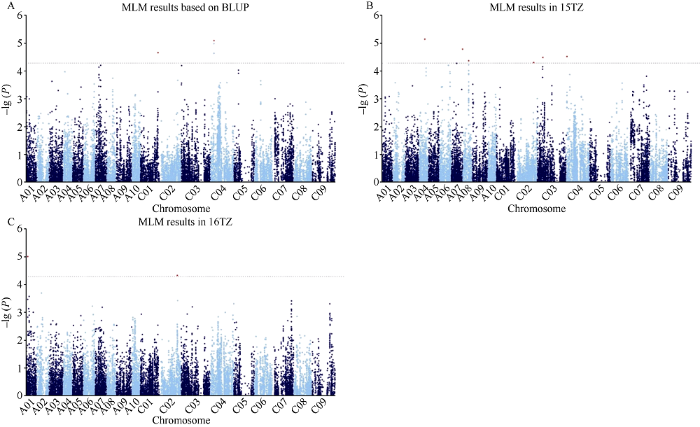 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2油菜每角粒数全基因组关联分析(MLM)
A: 每角粒数BLUP值MLM曼哈顿图; B: 15TZ每角粒数MLM曼哈顿图; C: 16TZ每角粒数MLM曼哈顿图。水平线代表Bonferroni阈值。
Fig. 2Genome-wide association study of rapeseed seed number per silique (MLM)
A: Manhattan plot of MLM based on BLUP value; B: Manhattan plot of MLM in15TZ; C: Manhattan plot of MLM in 16TZ. The dashed horizontal line depicts the Bonferroni significance threshold.
利用GLM模型对各环境的每角粒数表型和BLUP值进行全基因组关联分析, 当阈值为-lg (P) = 4.28时, 在15TZ、16TZ和基于BLUP分别检测到10、6和5个显著位点。位于C04的位点Bn-scaff_23907_1-p3780基于BLUP和在15TZ均被重复检测到, 合并重叠的位点后得到20个显著位点, 分布在A01、A04、A07、A08、C01~C07和C09染色体, 可解释2.90%~4.83%的表型变异(表3和图3)。比较2个关联模型的结果, 所有MLM位点均得到GLM结果的验证, 合并2个模型的结果共得到20个显著位点。
Table 3
表3
表3GLM每角粒数显著关联位点
Table 3
| 标记 Marker | 染色体 Chr. | 位置 Position | -lg (P) | 表型变异 R2 (%) | 环境 Environment | 已报道QTL Reported QTL |
|---|---|---|---|---|---|---|
| Bn-A01-p3904495 | A01 | 3,530,446 | 5.05 | 0.0357 | 16TZ | [5] |
| Bn-A04-p10393460 | A04 | 11,537,744 | 5.79 | 0.0408 | 15TZ | |
| Bn-A07-p9916502 | A07 | 11,196,091 | 4.50 | 0.0404 | BLUP | |
| Bn-A07-p22251229 | A07 | 23,650,401 | 4.41 | 0.0322 | 15TZ | |
| Bn-A08-p14749618 | A08 | 12,312,967 | 4.74 | 0.0327 | 15TZ | [7] |
| Bn-scaff_15838_1-p1554155 | C01 | 1,926,096 | 4.41 | 0.0437 | 16TZ | [8] |
| Bn-scaff_15936_1-p270915 | C01 | 36,405,870 | 5.13 | 0.0353 | BLUP | [5-6] |
| Bn-scaff_15712_6-p1336179 | C02 | 38,045,422 | 4.66 | 0.0435 | 16TZ | |
| Bn-scaff_23954_1-p635109 | C03 | 11,205,203 | 4.78 | 0.0330 | 15TZ | |
| Bn-scaff_16027_1-p367097 | C04 | 1,254,637 | 4.62 | 0.0365 | 15TZ | |
| Bn-scaff_23907_1-p3780 | C04 | 7,268,977 | 5.59 | 0.0483 | 15TZ,BLUP | |
| Bn-scaff_19253_1-p524524 | C04 | 15,606,945 | 4.29 | 0.0290 | BLUP | |
| Bn-scaff_15936_1-p357665 | C05 | 9,508,115 | 4.78 | 0.0453 | BLUP | |
| Bn-scaff_16064_1-p1144443 | C06 | 24,511,029 | 4.74 | 0.0327 | 15TZ | |
| Bn-scaff_17484_1-p132976 | C07 | 5,857,563 | 4.60 | 0.0317 | 15TZ | |
| Bn-scaff_20084_1-p104549 | C07 | 9,638,283 | 5.36 | 0.0375 | 15TZ | |
| Bn-scaff_16069_1-p1651456 | C07 | 38,070,340 | 5.23 | 0.0365 | 15TZ | |
| Bn-scaff_16069_1-p3780494 | C07 | 40,184,749 | 4.35 | 0.0302 | 16TZ | |
| Bn-scaff_15808_1-p420800 | C09 | 37,129,614 | 5.15 | 0.0365 | 16TZ | [5] |
| Bn-scaff_15576_1-p74980 | C09 | 41,126,168 | 4.35 | 0.0303 | 16TZ | [5,19] |
新窗口打开|下载CSV
图3
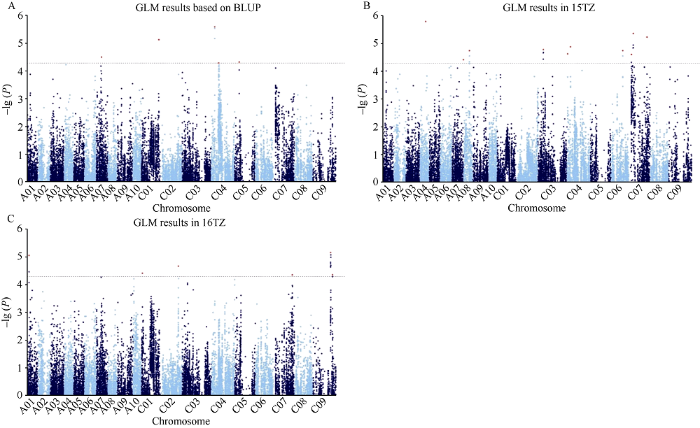 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3油菜每角粒数全基因组关联分析(GLM)
A: 每角粒数BLUP值GLM曼哈顿图; B: 15TZ每角粒数GLM曼哈顿图; C: 16TZ每角粒数GLM曼哈顿图。水平线代表Bonferroni阈值。
Fig. 3Genome-wide association study of rapeseed seed number per silique (GLM)
A: Manhattan plot of GLM based on BLUP value; B: Manhattan plot of GLM in 15TZ; C: Manhattan plot of GLM in 16TZ. The dashed horizontal line depicts the Bonferroni significance threshold.
2.3 候选基因挖掘
基于油菜基因组注释信息, 在C09染色体Bn-scaff_ 15576_1-p74980位点下游82 kb找到已克隆的油菜每角粒数基因BnaC9.SMG7b, 该基因是拟南芥SMG7的同源基因(表4)。BnaC9.SMG7b参与调控油菜大孢子母细胞的减数分裂, 该基因的自然缺失导致部分雌配子体发育异常, 无法形成正常功能的胚珠, 最终导致油菜每角果粒数显著降低[19]。此外, 在A04染色体Bn-A04-p10393460位点上游522 kb找到候选基因BnaA04g13080, 该基因的拟南芥同源基因GRDP1编码一个短富含甘氨酸结构域的蛋白, 该基因突变导致植株角果长度缩短、每角粒数减少、粒重降低[20]。在C03染色体Bn-scaff_23954_1-p635109位点下游162 kb找到候选基因BnaC03g21140, 该基因的拟南芥同源基因DA2编码一个E3泛素连接酶, 其功能缺失突变体每角粒数增加、粒重增加[21]。此外, 本研究还找到其他候选基因, 包括拟南芥已知每角粒数调控基因GLE1[22]、SPATULA [23]、HVA22D [24]和MSI1[25]在油菜中的同源拷贝。Table 4
表4
表4每角粒数关联位点候选基因信息
Table 4
| 标记 Marker | 油菜基因 Rapeseed gene | 染色体 Chr. | 位置 Position | 拟南芥同源基因 Ar. homolog |
|---|---|---|---|---|
| Bn-A04-p10393460 | BnaA04g13080 | A04 | 11,015,882 | GRDP1 |
| Bn-A07-p9916502 | BnaA07g13170 | A07 | 11,744,966 | GLE1 |
| Bn-A08-p14749618 | BnaA08g15580 | A08 | 12,923,783 | SPATULA |
| Bn-scaff_23954_1-p635109 | BnaC03g21140 | C03 | 11,367,292 | DA2 |
| Bn-scaff_16069_1-p3780494 | BnaC07g39210 | C07 | 40,210,953 | HVA22D |
| Bn-scaff_15808_1-p420800 | BnaC09g33680 | C09 | 36,922,347 | MSI1 |
| Bn-scaff_15576_1-p74980 | BnaC09g38310 | C09 | 41,208,383 | SMG7b |
新窗口打开|下载CSV
3 讨论
每角粒数是油菜重要的产量构成因子, 增加每角粒数有利于提高单株籽粒产量, 从而提高群体的籽粒总量。本研究对496份油菜资源的每角粒数进行2个环境的考察, 分析表明本群体的每角粒数广义遗传力为57.7%。利用2个关联模型MLM和GLM, 检测到20个显著位点, 分布于12条染色体。本研究中每角粒数关联位点的效应较小(< 5%), 在2个环境间能被重复检测到的位点少, 表明该群体中每角粒数主要受微效多基因调控, 且基因效应受环境影响较大。本研究获得的每角粒数QTL有6个位点与前人结果一致(表2和表3), 其中位于C01染色体的位点Bn-scaff_15936_1-p270915, 物理位置36.41 Mb, 与Luo等[5]检测到的每角粒数QTL qSN.C1-2 (范围: 36.53~37.48 Mb)和漆丽萍[6]检测到的QTL qSN.C1b (范围: 35.69~36.81 Mb)重叠。位于C09染色体的位点Bn-scaff_15576_ 1-p74980物理位置是41.13 Mb, 与Li等[19]已克隆的每角粒数基因BnaC9.SMG7b (位置: 41.21 Mb)和Luo等[5]检测到的QTL qSN.C9-5 (范围: 41.28~41.67 Mb)重叠。这些结果证实了本研究结果的可靠性。此外, 本研究还检测到14个新位点, 如位于A04染色体的Bn-A04-p10393460和C04染色体的Bn-scaff_23907_1-p3780, 表型贡献率均相对较高; 位于C02染色体的位点Bn-scaff_15712_6- p1336179在2个环境中被重复检测到, 这些位点值得进一步验证和研究。
目前油菜中已报道的每角粒数基因较少, Li等[19]利用图位克隆的方法, 在C09染色体克隆了每角粒数基因BnaC9.SMG7b; Yang等[26]应用CRISPR/Cas9系统敲除CLV3在油菜A04和C04染色体的2个拷贝, 双突变植株叶片增多, 每角粒数和粒重均增加; Shah等[27]发现拟南芥AP1在油菜A02染色体的拷贝突变, 导致了油菜株高、分枝高度、分枝数、每角粒数和单株产量等性状的改变。本研究在Bn-scaff_15576_1-p74980位点下游82 kb找到BnaC9.SMG7b(表4)。此外, 还在另外6个位点附近找到了候选基因, 这些基因的拟南芥同源基因均参与每角粒数的调控, 部分基因还有一因多效的作用, 如GRDP1[20]和DA2[22]同时影响角果长、粒型和粒重等性状。后续研究可通过转基因或CRISPR/Cas9敲除, 进一步验证这些候选基因的功能。
附表 请见网络版: 1) 本刊网站http://zwxb.chinacrops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical-zuowxb.aspx。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]

本文在分析了我国食用植物油供给形势、油菜产业在食用植物油供给中的地位的基础上,对我国油菜产业的发展历史进行了回顾,提出在新的历史时期必须推动以“三高”(“高油、高产、高效”)为标志的我国油菜产业发展的第四次飞跃来应对新的挑战。
URL [本文引用: 1]
本文在分析了我国食用植物油供给形势、油菜产业在食用植物油供给中的地位的基础上,对我国油菜产业的发展历史进行了回顾,提出在新的历史时期必须推动以“三高”(“高油、高产、高效”)为标志的我国油菜产业发展的第四次飞跃来应对新的挑战。
[本文引用: 1]
[本文引用: 1]
URLPMID:29163611 [本文引用: 1]
Seed number is one of the key traits related to plant evolution/domestication and crop improvement/breeding. In rapeseed germplasm, the seed number per pod (SNPP) shows a very wide variation from several to nearly 30; however, the underlying causations/mechanisms for this variation are poorly known. In the current study, the genetic and cytological bases for the natural variation of SNPP in rapeseed was firstly and systematically investigated using the representative four high-SNPP and five low-SNPP lines. The results of self- or cross-pollination experiment between the high- and low-SNPP lines showed that the natural variation of SNPP was mainly controlled by maternal effect (mean = 0.79), followed by paternal effect (mean = 0.21). Analysis of the data using diploid seed embryo-cytoplasmic-maternal model further showed that the maternal genotype, embryo, and cytoplasm effects, respectively, explained 47.6, 35.2, and 7.5% of the genetic variance. In addition, the analysis of combining ability showed that for the SNPP of hybrid F1 was mainly determined by the general combining ability of parents (63.0%), followed by special combining ability of parental combination (37.0%). More importantly, the cytological observation showed that the SNPP difference between the high- and low-SNPP lines was attributable to the accumulative differences in its components. Of which, the number of ovules, the proportion of fertile ovules, the proportion of fertile ovules to be fertilized, and the proportion of fertilized ovules to develop into seeds accounted for 30.7, 18.2, 7.1, and 43.9%, respectively. The accordant results of both genetic and cytological analyses provide solid evidences and systematic insights to further understand the mechanisms underlying the natural variation of SNPP, which will facilitate the development of high-yield cultivars in rapeseed.
DOI:10.1534/genetics.109.101642URLPMID:19414564 [本文引用: 1]
Yield is the most important and complex trait for the genetic improvement of crops. Although much research into the genetic basis of yield and yield-associated traits has been reported, in each such experiment the genetic architecture and determinants of yield have remained ambiguous. One of the most intractable problems is the interaction between genes and the environment. We identified 85 quantitative trait loci (QTL) for seed yield along with 785 QTL for eight yield-associated traits, from 10 natural environments and two related populations of rapeseed. A trait-by-trait meta-analysis revealed 401 consensus QTL, of which 82.5% were clustered and integrated into 111 pleiotropic unique QTL by meta-analysis, 47 of which were relevant for seed yield. The complexity of the genetic architecture of yield was demonstrated, illustrating the pleiotropy, synthesis, variability, and plasticity of yield QTL. The idea of estimating indicator QTL for yield QTL and identifying potential candidate genes for yield provides an advance in methodology for complex traits.
DOI:10.1007/s00122-017-2911-7URLPMID:28455767 [本文引用: 3]
A comprehensive linkage atlas for seed yield in rapeseed. Most agronomic traits of interest for crop improvement (including seed yield) are highly complex quantitative traits controlled by numerous genetic loci, which brings challenges for comprehensively capturing associated markers/genes. We propose that multiple trait interactions underlie complex traits such as seed yield, and that considering these component traits and their interactions can dissect individual quantitative trait loci (QTL) effects more effectively and improve yield predictions. Using a segregating rapeseed (Brassica napus) population, we analyzed a large set of trait data generated in 19 independent experiments to investigate correlations between seed yield and other complex traits, and further identified QTL in this population with a SNP-based genetic bin map. A total of 1904 consensus QTL accounting for 22 traits, including 80 QTL directly affecting seed yield, were anchored to the B. napus reference sequence. Through trait association analysis and QTL meta-analysis, we identified a total of 525 indivisible QTL that either directly or indirectly contributed to seed yield, of which 295 QTL were detected across multiple environments. A majority (81.5%) of the 525 QTL were pleiotropic. By considering associations between traits, we identified 25 yield-related QTL previously ignored due to contrasting genetic effects, as well as 31 QTL with minor complementary effects. Implementation of the 525 QTL in genomic prediction models improved seed yield prediction accuracy. Dissecting the genetic and phenotypic interrelationships underlying complex quantitative traits using this method will provide valuable insights for genomics-based crop improvement.
[本文引用: 2]
[本文引用: 2]
URLPMID:26880301 [本文引用: 1]
An optimized plant architecture (PA) is fundamental for high-yield breeding but the genetic control of the important trait is largely unknown in rapeseed. Here plant architecture factors (PAFs) were proposed to consist of main inflorescence length proportion (MILP), branch height proportion (BHP), and branch segment proportion (BSP). Comparison of different genotypes in a DH population grown in diverse environments showed that an optimized PAF performance with MILP and BHP between 0.3-0.4 was important for high yield potential. In total, 163 unique quantitative trait loci (QTLs) for PA- and plant yield (PY)-related traits were mapped onto a high-density genetic map. Furthermore, 190 PA-related candidate genes for 91 unique PA QTLs and 2350 PY epistatic interaction loci-pairs were identified, which explain 2.8-51.8% and 5.2-23.6% of phenotypic variation, respectively. Three gene categories, transcription factor, auxin/IAA, and gibberellin, comprise the largest proportions of candidate genes for PA-related QTLs. The effectiveness of QTL candidate genes prediction was demonstrated by cloning of three candidate genes, Bna.A02.CLV2, Bna.A09.SLY2, and Bna.C07.AHK4. The study thus outlines a gene network for control of PA-related traits and provides novel information for understanding the establishment of ideal PA and for developing effective breeding strategies for yield improvement in rapeseed and other crops.
URLPMID:27067010 [本文引用: 1]
Seed number per pod (SNPP) is one of the major yield components and breeding targets in rapeseed that shows great variation and is invaluable for genetic improvement. To elucidate the genetic architecture and uncover the mechanism of SNPP, we identified five quantitative trait loci (QTLs) using the BnaZNRIL population, which were integrated with those of previous studies by physical map to demonstrate a complex and relatively complete genetic architecture of SNPP. A major QTL, qSN.A6, was successfully fine-mapped from 1910 to 267?kb using near-isogenic line (NIL). In addition, qSN.A6 exhibited an antagonistic pleiotropy on seed weight (SW), which is caused by a physiological interaction in which SNPP acts &quot;upstream&quot; of SW. Because the negative effect of qSN.A6 on SW cannot fully counteract its positive effect on SNPP, it also enhanced the final yield (17.4%), indicating its great potential for utilization in breeding. The following genetic and cytological experiments further confirmed that the different rate of ovule abortion was responsible for the ~5 seed difference between Zhongshuang11 and NIL-qSN.A6. This systematic approach to dissecting the comprehensive genetic architecture of SNPP and characterizing the underlying mechanism has advanced the understanding of SNPP and will facilitate the development of high-yield cultivars.
URLPMID:27512396 [本文引用: 2]
Plant height is a key morphological trait of rapeseed. In this study, we measured plant height of a rapeseed population across six environments. This population contains 476 inbred lines representing the major Chinese rapeseed genepool and 44 lines from other countries. The 60K Brassica Infinium? SNP array was utilized to genotype the association panel. A genome-wide association study (GWAS) was performed via three methods, including a robust, novel, nonparametric Anderson-Darling (A-D) test. Consequently, 68 loci were identified as significantly associated with plant height (P &lt; 5.22 × 10(-5)), and more than 70% of the loci (48) overlapped the confidence intervals of reported QTLs from nine mapping populations. Moreover, 24 GWAS loci were detected with selective sweep signals, which reflected the signatures of historical semi-dwarf breeding. In the linkage disequilibrium (LD) decay range up-and downstream of 65 loci (r (2) &gt; 0.1), we found plausible candidates orthologous to the documented Arabidopsis genes involved in height regulation. One significant association found by GWAS colocalized with the established height locus BnRGA in rapeseed. Our results provide insights into the genetic basis of plant height in rapeseed and may facilitate marker-based breeding.
URLPMID:30858362 [本文引用: 2]
Brassica napus (2n?=?4x?=?38, AACC) is an important allopolyploid crop derived from interspecific crosses between Brassica rapa (2n?=?2x?=?20, AA) and Brassica oleracea (2n?=?2x?=?18, CC). However, no truly wild B. napus populations are known; its origin and improvement processes remain unclear. Here, we resequence 588 B. napus accessions. We uncover that the A subgenome may evolve from the ancestor of European turnip and the C subgenome may evolve from the common ancestor of kohlrabi, cauliflower, broccoli, and Chinese kale. Additionally, winter oilseed may be the original form of B. napus. Subgenome-specific selection of defense-response genes has contributed to environmental adaptation after formation of the species, whereas asymmetrical subgenomic selection has led to ecotype change. By integrating genome-wide association studies, selection signals, and transcriptome analyses, we identify genes associated with improved stress tolerance, oil content, seed quality, and ecotype improvement. They are candidates for further functional characterization and genetic improvement of B. napus.
.
URLPMID:29725340 [本文引用: 2]
Cadmium is a potentially toxic heavy metal to human health. Rapeseed (Brassica napus L.), a vegetable and oilseed crop, might also be a Cd hyperaccumulator, but there is little information on this trait in rapeseed. We evaluated Cd accumulation in different oilseed accessions and employed a genome-wide association study to identify quantitative trait loci (QTLs) related to Cd accumulation. A total of 419 B. napus accessions and inbred lines were genotyped with a 60K Illumina Infinium SNP array of Brassica. Wide genotypic variations in Cd concentration and translocation were found. Twenty-five QTLs integrated with 98 single-nucleotide polymorphisms (SNPs) located at 15 chromosomes were associated with Cd accumulation traits. These QTLs explained 3.49-7.57% of the phenotypic variation observed. Thirty-two candidate genes were identified in these genomic regions, and they were 0.33-497.97 kb away from the SNPs. We found orthologs of Arabidopsis thaliana located near the significant SNPs on the B. napus genome, including NRAMP6 (natural resistance-associated macrophage protein 6), IRT1 (iron-regulated transporter 1), CAD1 (cadmium-sensitive 1), and PCS2 (phytochelatin synthase 2). Of them, four candidate genes were verified by qRT-PCR, the expression levels of which were significantly higher after exposure to Cd than in the controls. Our results might facilitate the study of the genetic basis of Cd accumulation and the cloning of candidate Cd accumulation genes, which could be used to help reduce Cd levels in edible plant parts and/or create more efficient hyperaccumulators.
DOI:10.1093/dnares/dsv035URLPMID:26659471 [本文引用: 2]
Flowering time adaptation is a major breeding goal in the allopolyploid species Brassica napus. To investigate the genetic architecture of flowering time, a genome-wide association study (GWAS) of flowering time was conducted with a diversity panel comprising 523 B. napus cultivars and inbred lines grown in eight different environments. Genotyping was performed with a Brassica 60K Illumina Infinium SNP array. A total of 41 single-nucleotide polymorphisms (SNPs) distributed on 14 chromosomes were found to be associated with flowering time, and 12 SNPs located in the confidence intervals of quantitative trait loci (QTL) identified in previous researches based on linkage analyses. Twenty-five candidate genes were orthologous to Arabidopsis thaliana flowering genes. To further our understanding of the genetic factors influencing flowering time in different environments, GWAS was performed on two derived traits, environment sensitivity and temperature sensitivity. The most significant SNPs were found near Bn-scaff_16362_1-p380982, just 13 kb away from BnaC09g41990D, which is orthologous to A. thaliana CONSTANS (CO), an important gene in the photoperiod flowering pathway. These results provide new insights into the genetic control of flowering time in B. napus and indicate that GWAS is an effective method by which to reveal natural variations of complex traits in B. napus.
URL [本文引用: 1]
For many horticultural crops, selection is based on quality as well as yield. To investigate the distribution of trait variation and identify those attributes appropriate for developing selection indices, we collected and organized information related to fruit size, shape, color, soluble solids, acid, and yield traits for 143 processing tomato (Solanum lycopersicum L.) lines from North America. Evaluation of the germplasm panel was conducted in a multiyear, multilocation trial. Data were stored in a flat-file format and in a trait ontology database, providing a public archive. We estimated variance components and proportion of variance resulting from genetics for each trait. Genetic variance was low to moderate (range, 0.03-0.51) for most traits, indicating high environmental influence on trait expression and/or complex genetic architecture. Phenotypic values for each line were estimated across environments as best linear unbiased predictors (BLUPs). Principal components (PC) analysis using the trait BLUPs provided a means to assess which traits explained variation in the germplasm. The first two PCs explained 28.0% and 16.2% of the variance and were heavily weighted by measures of fruit shape and size. The third PC explained 12.9% of the phenotypic variance and was determined by fruit color and yield components. Trait BLUPs and the first three PCs were also used to explore the relationship between phenotypes and the origin of the accessions. We were able to differentiate germplasm for fruit size, fruit shape, yield, soluble solids, and color based on origin, indicating regional breeding programs provide a source of trait variation. These analyses suggest that multitrait selection indices could be established that encompass quality traits in addition to yield. However, such indices will need to balance trait correlations and be consistent with market valuation.
DOI:10.1002/rcm.8315URLPMID:30366355 [本文引用: 1]
High-resolution mass spectrometry (HRMS) with high sample throughput has become an important analytical tool for the analysis of highly complex samples and data processing has become a major challenge for the user community. Evaluating direct-infusion HRMS data without automated tools for batch processing can be a time-consuming step in the analytical pipeline. Therefore, we developed a new browser-based software tool for processing HRMS data.
DOI:10.1111/j.1365-294X.2005.02553.xURLPMID:15969739 [本文引用: 1]
The identification of genetically homogeneous groups of individuals is a long standing issue in population genetics. A recent Bayesian algorithm implemented in the software STRUCTURE allows the identification of such groups. However, the ability of this algorithm to detect the true number of clusters (K) in a sample of individuals when patterns of dispersal among populations are not homogeneous has not been tested. The goal of this study is to carry out such tests, using various dispersal scenarios from data generated with an individual-based model. We found that in most cases the estimated 'log probability of data' does not provide a correct estimation of the number of clusters, K. However, using an ad hoc statistic DeltaK based on the rate of change in the log probability of data between successive K values, we found that STRUCTURE accurately detects the uppermost hierarchical level of structure for the scenarios we tested. As might be expected, the results are sensitive to the type of genetic marker used (AFLP vs. microsatellite), the number of loci scored, the number of populations sampled, and the number of individuals typed in each sample.
DOI:10.1016/j.yebeh.2019.106687URLPMID:31816478 [本文引用: 1]
Irritability is a adverse effect of many antiseizure medications (ASMs), but there are no validated measures currently available to characterize this behavioral risk. We examined both child and parent/guardian versions of the Affective Reactivity Index (ARI), a validated measure developed for application in adolescent psychiatry, to determine its sensitivity to ASM-related irritability. We hypothesized irritability increases associated with levetiracetam (LEV) but not lamotrigine (LTG) or oxcarbazepine (OXC).
DOI:10.1093/bioinformatics/btm308URLPMID:17586829 [本文引用: 1]
Association analyses that exploit the natural diversity of a genome to map at very high resolutions are becoming increasingly important. In most studies, however, researchers must contend with the confounding effects of both population and family structure. TASSEL (Trait Analysis by aSSociation, Evolution and Linkage) implements general linear model and mixed linear model approaches for controlling population and family structure. For result interpretation, the program allows for linkage disequilibrium statistics to be calculated and visualized graphically. Database browsing and data importation is facilitated by integrated middleware. Other features include analyzing insertions/deletions, calculating diversity statistics, integration of phenotypic and genotypic data, imputing missing data and calculating principal components.
[本文引用: 1]
DOI:10.1104/pp.15.01040URLPMID:26494121 [本文引用: 3]
Number of seeds per silique (NSS) is an important determinant of seed yield potential in Brassicaceae crops, and it is controlled by naturally occurring quantitative trait loci. We previously mapped a major quantitative trait locus, qSS.C9, on the C9 chromosome that controls NSS in Brassica napus. To gain a better understanding of how qSS.C9 controls NSS in B. napus, we isolated this locus through a map-based cloning strategy. qSS.C9 encodes a predicted small protein with 119 amino acids, designated as BnaC9.SMG7b, that shows homology with the Ever ShorterTelomere1 tertratricopeptide repeats and Ever Shorter Telomere central domains of Arabidopsis (Arabidopsis thaliana) SUPPRESSOR WITH MORPHOGENETIC EFFECTS ON GENITALIA7 (SMG7). BnaC9.SMG7b plays a role in regulating the formation of functional female gametophyte, thus determining the formation of functional megaspores and then mature ovules. Natural loss or artificial knockdown of BnaC9.SMG7b significantly reduces the number of functional ovules per silique and thus, results in decreased seed number, indicating that qSS.C9 is a positive regulator of NSS in B. napus. Sequence and function analyses show that BnaC9.SMG7b experiences a subfunctionalization process that causes loss of function in nonsense-mediated mRNA decay, such as in Arabidopsis SMG7. Haplotype analysis in 84 accessions showed that the favorable BnaC9.SMG7b alleles are prevalent in modern B. napus germplasms, suggesting that this locus has been a major selection target of B. napus improvement. Our results represent the first step toward unraveling the molecular mechanism that controls the natural variation of NSS in B. napus.
DOI:10.1016/j.bbrc.2017.03.015URLPMID:28285133 [本文引用: 2]
Glycine Rich Proteins (GRPs) are induced at different developmental stages and in specific plant tissues. Recently, we described a novel Arabidopsis gene encoding a short glycine-rich domain protein (AtGRDP1). This gene is involved in abiotic stress responsiveness; the Atgrdp1-null mutant seeds were more sensitive to stress, while the opposite phenotype was achieved by AtGRDP1 overexpression. In this study, we analyzed the phenotype of the fruits produced by Arabidopsis Atgrdp1 mutants and 35S::AtGRDP1 overexpression lines. Our analyses revealed important changes in silique length, seed number, seed weight and morphology in the analyzed lines. In particular, Atgrdp1 mutant lines exhibited several defects including short siliques, a diminished number of seeds per silique, and a reduction in seed size and weight as compared to Col-0. The overexpression of the AtGRDP1 gene also generated phenotypes with alterations in size of silique, number of seeds per silique, and size and weight of the seed. In addition, the expression analysis of AtGRDP1 gene showed that it was expressed in floral and fruit organs, with the highest expression level in mature siliques. The alterations in the siliques and seeds traits in the Atgrdp1 mutant line, as well as the phenotypes observed in AtGRDP1 overexpression lines, suggest a role of the AtGRDP1 gene in the Arabidopsis fruit development.
DOI:10.1105/tpc.113.115063URL [本文引用: 1]
Seed size in higher plants is determined by the coordinated growth of the embryo, endosperm, and maternal tissue. Several factors that act maternally to regulate seed size have been identified, such as AUXIN RESPONSE FACTOR2, APETALA2, KLUH, and DA1, but the genetic and molecular mechanisms of these factors in seed size control are almost totally unknown. We previously demonstrated that the ubiquitin receptor DA1 acts synergistically with the E3 ubiquitin ligase ENHANCER1 OF DA1 (EOD1)/BIG BROTHER to regulate the final size of seeds in Arabidopsis thaliana. Here, we describe another RING-type protein with E3 ubiquitin ligase activity, encoded by DA2, which regulates seed size by restricting cell proliferation in the maternal integuments of developing seeds. The da2-1 mutant forms large seeds, while overexpression of DA2 decreases seed size of wild-type plants. Overexpression of rice (Oryza sativa) GRAIN WIDTH AND WEIGHT2, a homolog of DA2, restricts seed growth in Arabidopsis. Genetic analyses show that DA2 functions synergistically with DA1 to regulate seed size, but does so independently of EOD1. Further results reveal that DA2 interacts physically with DA1 in vitro and in vivo. Therefore, our findings define the genetic and molecular mechanisms of three ubiquitin-related proteins DA1, DA2, and EOD1 in seed size control and indicate that they are promising targets for crop improvement.
DOI:10.1104/pp.112.202192URLPMID:22898497 [本文引用: 2]
Early embryogenesis in Arabidopsis (Arabidopsis thaliana) is distinguished by a predictable pattern of cell divisions and is a good system for investigating mechanisms of developmental pattern formation. Here, we identified a gene called LONO1 (LNO1) in Arabidopsis in which mutations can abolish the first asymmetrical cell division of the zygote, alter planes and number of cell divisions in early embryogenesis, and eventually arrest embryo development. LNO1 is highly expressed in anthers of flower buds, stigma papilla of open flowers, and embryo and endosperm during early embryogenesis, which is correlated with its functions in reproductive development. The homozygous lno1-1 seed is not viable. LNO1, a homolog of the nucleoporin NUP214 in human (Homo sapiens) and Nup159 in yeast (Saccharomyces cerevisiae), encodes a nucleoporin protein containing phenylalanine-glycine repeats in Arabidopsis. We demonstrate that LNO1 can functionally complement the defect in the yeast temperature-sensitive nucleoporin mutant nup159. We show that LNO1 specifically interacts with the Arabidopsis DEAD-box helicase/ATPase LOS4 in the yeast two-hybrid assay. Furthermore, mutations in AtGLE1, an Arabidopsis homolog of the yeast Gle1 involved in the same poly(A) mRNA export pathway as Nup159, also result in seed abortion. Our results suggest that LNO1 is a component of the nuclear pore complex required for mature mRNA export from the nucleus to the cytoplasm, which makes LNO1 essential for embryogenesis and seed viability in Arabidopsis.
DOI:10.1111/j.1365-313X.2008.03469.xURLPMID:18315540 [本文引用: 1]
The SPATULA (SPT) gene is involved in generating the septum, style and stigma: specialized tissues that arise from carpel margins. By matching sequences within the extended bHLH region of AtSPT across species databases, twelve orthologues were identified in eudicots, rice and a gymnosperm. Two conserved structural domains were revealed in addition to the bHLH region: an amphipathic helix and an acidic domain. These are conserved in the tomato orthologue, which can restore carpel function to spt mutants of Arabidopsis. The acidic domain is essential for SPT carpel function, and the amphipathic helix supports it. A bipartite sequence overlapping the bHLH domain is required for nuclear localization, and a mutation in the conserved beta strand adjacent to the bHLH C terminus results in the loss of SPT function. SPT apparently acts as a transcriptional activator, as the addition of the SRDX repression domain phenocopies the spt mutant phenotype. Expression of an artificially activating 35S:SPT-VP16 construct can induce carpelloid properties in sepals, and new defects in the gynoecium. These disruptions are associated with ectopic expression of the STYLISH2 gene, although STYLISH2 expression does not require SPT function. Ectopic expression of unmodified SPT does not induce such changes, implying that SPT acts in association with essential coactivators present only in regions where SPT is normally active. Because the VP16 activation domain can compensate to some extent for the loss of the amphipathic helix and acidic domain, these domains may normally interact with such co-activators.
DOI:10.1104/pp.108.131490URLPMID:19151132 [本文引用: 1]
Autophagy is an intracellular process in which a portion of cytoplasm is transported into vacuoles for recycling. Physiological roles of autophagy in plants include recycling nutrients during senescence, sustaining life during starvation, and the formation of central digestive vacuoles. The regulation of autophagy and the formation of autophagosomes, spherical double membrane structures containing cytoplasm moving toward vacuoles, are poorly understood. HVA22 is a gene originally cloned from barley (Hordeum vulgare), which is highly induced by abscisic acid and environmental stress. Homologs of HVA22 include Yop1 in yeast, TB2/DP1 in human, and AtHVA22a to -e in Arabidopsis (Arabidopsis thaliana). Reverse genetics followed by a cell biology approach were employed to study the function of HVA22 homologs. The AtHVA22d RNA interference (RNAi) Arabidopsis plants produced small siliques with reduced seed yield. This phenotype cosegregated with the RNAi transgene. Causes of the reduced seed yield include short filaments, defective carpels, and dysfunctional pollen grains. Enhanced autophagy was observed in the filament cells. The number of autophagosomes in root tips of RNAi plants was also increased dramatically. The yop1 deletion mutant of Saccharomyces cerevisiae was used to verify our hypothesis that HVA22 homologs are suppressors of autophagy. Autophagy activity of this mutant during nitrogen starvation increased in 5 min and reached a plateau after 2 h, with about 80% of cells showing autophagy, while the wild-type cells exhibited low levels of autophagy following 8 h of nitrogen starvation. We conclude that HVA22 homologs function as suppressors of autophagy in both plants and yeast. Potential mechanisms of this suppression and the roles of abscisic acid-induced HVA22 expression in vegetative and reproductive tissues are discussed.
DOI:10.1242/dev.009027URLPMID:17855429 [本文引用: 1]
Polycomb group (PcG) proteins are evolutionary conserved proteins that stably maintain established transcriptional patterns over cell generations. The FERTILIZATION INDEPENDENT SEED (FIS) PcG complex from plants has a similar composition to the Polycomb repressive complex 2 from animals. Mutations in FIS genes cause parent-of-origin-dependent seed abortion. Every seed inheriting a mutant fis allele from the mother is destined to abort, regardless of the presence of a wild-type paternal allele. We tested in Arabidopsis whether the parent-of-origin-dependent seed abortion caused by lack of the FIS subunit MSI1 is caused by parental imprinting of the MSI1 gene. Our data show that MSI1 is not an imprinted gene and that early paternal MSI1 expression is not sufficient to rescue msi1 mutant seeds. By contrast, expression of MSI1 in msi1 female gametophytes is necessary to restore normal seed development, strongly arguing that the female gametophytic effect of fis mutants is caused by a functional requirement for an intact FIS complex in the female gametophyte. Thus, FIS-mediated expression patterns established in the female gametophyte can impact on seed development, establishing fis mutants as true female gametophytic maternal-effect mutants.
.
DOI:10.1111/pbi.12872URLPMID:29250878 [本文引用: 1]
Multilocular silique is a desirable agricultural trait with great potential for the development of high-yield varieties of Brassica. To date, no spontaneous or induced multilocular mutants have been reported in Brassica napus, which likely reflects its allotetraploid nature and the extremely low probability of the simultaneous random mutagenesis of multiple gene copies with functional redundancy. Here, we present evidence for the efficient knockout of rapeseed homologues of CLAVATA3 (CLV3) for a secreted peptide and its related receptors CLV1 and CLV2 in the CLV signalling pathway using the CRISPR/Cas9 system and achieved stable transmission of the mutations across three generations. Each BnCLV gene has two copies located in two subgenomes. The multilocular phenotype can be recovered only in knockout mutations of both copies of each BnCLV gene, illustrating that the simultaneous alteration of multiple gene copies by CRISPR/Cas9 mutagenesis has great potential in generating agronomically important mutations in rapeseed. The mutagenesis efficiency varied widely from 0% to 48.65% in T0 with different single-guide RNAs (sgRNAs), indicating that the appropriate selection of the sgRNA is important for effectively generating indels in rapeseed. The double mutation of BnCLV3 produced more leaves and multilocular siliques with a significantly higher number of seeds per silique and a higher seed weight than the wild-type and single mutant plants, potentially contributing to increased seed production. We also assessed the efficiency of the horizontal transfer of Cas9/gRNA cassettes by pollination. Our findings reveal the potential for plant breeding strategies to improve yield traits in currently cultivated rapeseed varieties.
URLPMID:30594150 [本文引用: 1]
Increasing the productivity of rapeseed as one of the widely cultivated oil crops in the world is of upmost importance. As flowering time and plant architecture play a key role in the regulation of rapeseed yield, understanding the genetic mechanism underlying these traits can boost the rapeseed breeding. Meristem identity genes are known to have pleiotropic effects on plant architecture and seed yield in various crops. To understand the function of one of the meristem identity genes, APETALA1 (AP1) in rapeseed, we performed phenotypic analysis of TILLING mutants under greenhouse conditions. Three stop codon mutant families carrying a mutation in Bna.AP1.A02 paralog were analyzed for different plant architecture and seed yield-related traits.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}