 ,1,2,*
,1,2,*Candidate genes screening for plant height and the first branch height based on QTL mapping and genome-wide association study in rapessed (Brassica napus L.)
HUO Qiang1,2,**, YANG Hong1,2,**, CHEN Zhi-You1,2, JIAN Hong-Ju1,2, QU Cun-Min1,2, LU Kun1,2, LI Jia-Na,1,2,*通讯作者:
第一联系人:
收稿日期:2019-04-28接受日期:2019-08-9网络出版日期:2019-09-02
| 基金资助: |
Received:2019-04-28Accepted:2019-08-9Online:2019-09-02
| Fund supported: |
作者简介 About authors
霍强,E-mail:354011524@qq.com。
杨鸿,E-mail:583791495@qq.com。

摘要
关键词:
Abstract
Keywords:
PDF (7210KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
霍强, 杨鸿, 陈志友, 荐红举, 曲存民, 卢坤, 李加纳. 基于QTL定位和全基因组关联分析筛选甘蓝型油菜株高和一次有效分枝高度的候选基因[J]. 作物学报, 2020, 46(2): 214-227. doi:10.3724/SP.J.1006.2020.94067
HUO Qiang, YANG Hong, CHEN Zhi-You, JIAN Hong-Ju, QU Cun-Min, LU Kun, LI Jia-Na.
甘蓝型油菜是四大油料作物之一, 且在饲料、绿肥、蔬菜、能源、旅游、蜜源等方面都有重要价值。作物的株型结构在作物从幼苗到成熟的生长期间以及作物对环境条件的响应中发生变化, 这种可塑性是由作物发育的灵活变化带来的[1], 同时作物株型的变化对作物的适应性和产量具有显著影响[2,3]。
株高是作物株型最突出的决定因素, 在作物中常被驯化或选择。其中矮秆基因已广泛应用于提高谷类作物的抗倒伏能力和收获指数[4]。第一次“绿色革命”, 将半矮化基因sd1引入水稻(Oryza sativa)和矮化基因Rht-B1b/Rht-D1b引入小麦(Triticum aestivum)来降低作物株高[5], 使2种作物增加其种植密度, 通过提高收获指数而大幅增产。拟南芥中, GIBBERELLIN INSENSITIVE (GAI)、REPRESSOR OF GIBBERELLIN (RGA)、RGA-LIKE (RGL1)、RGL2、RGL3是5个DELLA蛋白基因, 突变体gai和rga在DELLA蛋白结构域的序列缺失导致植株严重矮化[6,7]。GA20ox和GA3ox基因突变植株也表现出矮化现象[8]。作物的分枝同样是影响株型的重要因素。玉米的TB1基因可抑制侧生器官生长[9], 在小麦中过表达此基因可抑制小麦分蘖发育[10]。番茄LATERAL SUPPRESOR (LS)基因[11]、拟南芥LAS基因[12]和水稻MONOCULM1 (MOC1)基因[13]是同源基因, 以及番茄中BLIND (BL)基因[14], 它们的突变可显著减少营养分枝和生殖分枝的数量; 此外, 拟南芥MORE AXILLARY GROWTH (MAX)、豌豆RAMOUS (RMS)、矮牵牛DECREASED APICAL DOMINANCE (DAD)等基因也对分枝生长发育有着重要作用[15]。截至目前, 株高候选基因在模式植物中虽已经得到大量的克隆和鉴定, 但在甘蓝型油菜中仅有几例报道[16], 且一次有效分枝高度基因在甘蓝型油菜中尚无定位和克隆的报道。
利用QTL和GWAS相结合是定位数量性状候选基因的有效方法[17]。王嘉等[18]利用RIL (recombinant inbred lines)群体检测到分布于A01、A06、A07、A08、A10和C06染色体上的11个株高相关QTL, 单个QTL可解释的表型变异为5.00%~15.26%; 并检测到7个一次有效分枝高度相关QTL, 分别位于A06、C05和C06染色体上, 单个QTL解释5.04%~12.99%的表型变异。Zhao等[19]利用1个DH (doubled haploid)群体, 检测到18个位于A02、A03、A07、A10、C01、C03、C04、C06和C09染色体上的株高相关QTL和27个位于A01、A02、A03、A06、A07、A09、A10、C03、C05、C06和C09染色体上的一次有效分枝高度相关QTL。Cai等[2]利用DH群体, 检测20个株高相关QTL和16个一次有效分枝高度相关QTL。Luo等[20]对BnaTNDH (a doubled-haploid population)进行基因分型, 并对该群体之前的表型重新分析, 检测到80个株高相关QTL, 分布在除C01外的各个染色体上; 同时检测到35个与一次有效分枝高度相关QTL, 分布在A01、A02、A03、A05、A06、A09、A10、C05、C06、C07、C08、C09上, QTL的贡献率为3.19%~22.91%。贺亚军等[21]利用1个DH群体和1个永久性F2群体, 检测到9个株高相关QTL, 分布于A02、A09、C01、C02和C06连锁群上; 同时检测到11个一次有效分枝高度相关QTL, 分布于A01、A03、A09、C01和C03连锁群上, 单个QTL可解释4.01%~16.54%的表型变异。Shen等[22]利用DH群体检测到5个株高相关QTL和5个一次有效分枝高度相关QTL, 位于A02和A07染色体上。但以上报道均以单个方法筛选候选基因, 而使用QTL定位和全基因组关联分析(GWAS)筛选和鉴定候选基因具有重要意义。
虽然株高和一次有效分枝高度的相关基因挖掘已经有较多的报道[23,24,25], 但是本文利用不同的作图群体和具有物理位置的SNP标记, 筛选QTL和显著关联的SNP, 并利用其物理位置信息筛选候选基因, 结合qRT-PCR结果进一步验证筛选的候选基因, 这将为基因功能分析奠定基础。
1 材料与方法
1.1 试验材料
用于QTL定位的群体是以GH06为母本、中油821 (ZY821)为父本杂交获得F1, 自交的F2代通过“一粒传法”连续自交10代以上构建形成含186个株系的高世代重组自交系群体。该群体材料于2015—2016年度、2016—2017年度连续2年种植于重庆市北碚区歇马镇油菜基地(29oN, 106oE, 海拔238.6 m), 以育苗移栽方式, 随机区组设计, 2个重复, 每小区种植3行, 每行15株, 行距40 cm, 株距20 cm。按照常规生产方式进行田间管理。用于全基因组关联分析的自然群体是由全球油菜主要生产地征集的588份油菜品系组成[26], 其中大部分来自国内重庆、湖北、湖南、江苏、陕西等地, 部分来自加拿大、德国、瑞典、丹麦、澳大利亚等国家。该群体所有种植及田间管理同重组自交系群体。
1.2 性状考察
成熟期, 在每个小区中部选择5株长势一致的油菜植株统计调查表型性状, 分别测定其株高(plant height, PH)、一次有效分枝高度(the first branch height, BH)、结荚厚度(thickness of pod canopy, TPC)、经济产量(economic yield, EY)和收获指数(harvest index, HI)。株高指从子叶节到整株油菜主茎最高点的长度, 单位为cm; 一次有效分枝高度是指子叶节与主茎上最下部第一个有效分枝着生点之间的距离, 单位为cm; 结荚厚度是指整株油菜最高一个有效角果着生点与最低一个有效角果着生点的高度差, 单位为cm; 经济产量是指5株植株所有自然风干种子总重量的平均值, 以“g”为单位; 收获指数是指5株油菜单株经济产量之和与其生物产量之和的百分比。1.3 表型数据分析
利用Microsoft Excel 2007对高世代重组自交系群体和自然群体的表型数据进行整理和简单的统计分析; 利用IBM SPSS Statistics 19统计分析软件进行描述性统计分析并制作正态分布图、进行相关分析; 变异系数: CV = σ/μ, 其中σ为标准差, μ为平均值; 广义遗传力: h2= σ2G/(σ2G + σ2GE/e +σ2e/re), 其中σ2G表示基因型方差, σ2GE表示基因型与环境互作方差, σ2e为误差, e代表环境数目, r代表每个环境的重复数; 利用R软件对两年两群体的表型数据进行最佳线性无偏预测(best linear unbiased prediction, BLUP)。1.4 遗传图谱定位方法
取每个株系的5个幼嫩叶片混合, 提取DNA用于SNP标记分析。按照Illumina公司的Infinium HD Assay Ultra的说明进行DNA样品的预处理、与芯片杂交、洗脱、单碱基延伸、染色及包埋, 芯片准备好后利用Illumina HiSCAN扫描, 并利用GenomeStudio genotyping software v2011对扫描结果进行分析获得每个株系的基因型, SNP遗传图谱包括8575个SNP标记, 1201个bin, 覆盖甘蓝型油菜基因组6140.2 cM。采用WinQTL Cart2.5软件[27]的复合区间作图程序进行株高和一次有效分枝高度的QTL定位和效应检测。进行CIM分析时, 设置1 cM的步长(walking speed), 1000次回归, 显著水平0.01。遵照McCouch等[28]的方法命名QTL, 斜写的小写字母“q”加上性状的名字, 后面跟染色体, 最后一个数字表示QTL的序号。如q-2016PH-A01-1表示2016年株高在A01染色体上的第1个QTL。1.5 全基因组关联分析的方法
1.5.1 基因型数据测定与分析 对588份甘蓝型油菜进行重测序, 数据包含134.3亿个150 bp的双末端片段, 过滤后大约有530万个SNP。通过基因型分析, 最终获得385,692个可利用的SNP标记数据[26], 这些标记数据的最小基因型频率(minor allele frequency, MAF)大于0.05, 利用这些覆盖甘蓝型油菜全基因组的高密度标记, 对甘蓝型油菜自然群体进行群体结构(Q)、亲缘关系(K)和连锁不平衡(LD)分析, 并用这些标记结合重庆2年的株高和一次有效分枝高度的表型数据进行全基因组关联分析。1.5.2 群体结构、亲缘关系分析与连锁不平衡分析
利用Structure 2.3.4软件把每份甘蓝型油菜材料归类到特定的2个亚群里, 以此确定总群体的群体结构。运用TASSEL 5.0软件对自然群体中任意的2份材料进行亲缘关系的评估, 计算两特定材料间的遗传相似度与任意材料间的遗传相似度的相对值, 用0替代系数小于0的相对值。本群体中约2/3的材料间亲缘关系值小于0.05, 群体材料间亲缘关系越弱, 对进一步关联分析的影响越小[29]。运用TASSEL 5.0软件对群体内连锁不平衡(LD)进行分析计算。利用染色体上非等位基因位点间的r2值来估算甘蓝型油菜染色体中A和C染色体组LD的衰减, A和C染色体组的LD随着物理距离的增加而下降, 但A和C染色体组的衰减程度不同, 在同一物理位置A染色体组比C染色体组衰减快[26]。
1.5.3 全基因组关联分析 采用naive、Q、PCA、K、Q+K和PCA+K六种统计模型来评估群体结构、亲缘关系的影响进行表型和SNP关联分析(Q: 群体结构; PCA: 主成分; K: 亲缘关系)。运用TASSEL 5.0软件进行这6种模型的关联分析, 其中使用一般线性模型(GLM)分析naive、Q和PCA模型, 使用混合线性模型(MLM)分析K、Q+K和PCA+K模型。利用SAS软件对这6种模型的运算结果, 对其-lg (P)的观测值和-lg (P)期望值绘制Quantile-quantile散点图, 通过QQ图比较后确定最佳模型, 在最佳模型下进行株高和一次有效分枝高度性状的全基因组关联分析, 利用R软件作Manhattan图。本试验使用的SNP数据为385,692个, P值小于阈值(1/385,692 = 2.593E-06)的位点为显著关联位点。对于GWAS结果, 定位区间为显著关联SNP位点左右延伸500 kb[30]。
1.6 候选基因的筛选
根据甘蓝型油菜“Darmor-bzh”参考基因组[31], 提取株高和一次有效分枝高度性状定位的QTL置信区间内的基因; 根据分析得到的株高和一次有效分枝高度关联SNP标记, 提取各位点500 kb内的基因; 将需要功能分析的基因序列与拟南芥所有基因序列进行BLASTN比对, E-value阈值为1E-10, 并以同源性最高的拟南芥基因作为待分析基因的功能注释来筛选与株高和一次有效分枝高度相关的候选基因。1.7 候选基因的验证
对株高和一次有效分枝高度极端材料在蕾薹期取3个生物学重复的茎尖并提取RNA。根据前期所取材料的茎尖提取的RNA, 使用TaKaRa公司的PrimeScript RT reagent Kit with gDNA Eraser反转录合成cDNA, 将得到的cDNA产物稀释10倍后作为模板, 用于qRT-PCR验证。以油菜内参基因UBC21作为对照, 使用TaKaRa公司的SYBR Premix Ex TaqII试剂盒进行qRT-PCR验证候选基因的相对表达量。设置每个样品3次技术重复。在Bio-Rad定量PCR仪上进行qRT-PCR扩增。在网站http://biodb. swu.edu.cn/qprimerdb上查找设计候选基因的qRT-PCR引物(表1), 由上海生工生物工程有限公司合成。Table 1
表1
表1候选基因引物及其扩增产物
Table 1
| 基因名 Gene name | 引物序列 Primer sequence (5°-3°) |
|---|---|
| BnaA03g47940D | F: ATGGAGGCAATGAAGATGAAAC; R: GATAGACAAAGAAGCATCAGAG |
| BnaA07g28720D | F: CAGCTTCTTCGCTTACAAGATC; R: ATGCTCGATGGCTTATACTCAA |
| BnaA07g28820D | F: TTTTACTCGTCGTGCTTCTCTC; R: GACCTTCGAAAAACATGATGCT |
| BnaC06g30400D | F: ACAAGAATAATCGTTCTGCTGC; R: CCAACAGTTTCACTCTTCAAGG |
| BnaC06g30410D | F: TGGAAGCGTTACTGATTTGATG; R: TGAATATTGAGTGCAAGTAAGC |
| BnaA01g29630D | F: ATAAAGACTCTATCGCGATCGG; R: ATCCAAGGAGCGATATTAGTGG |
| BnaA01g30830D | F: ACTCTTTCGGTTCTTTTGTTGG; R: GTTTTGTTACGTCCGAAGAACA |
| BnaA02g31980D | F: GACGATGTCAAGCTTATAAGCG; R: TAACCCCTAAACAAACCTCTCC |
| BnaA02g37010D | F: TTCTTGAATTGGCAGAACGATC; R: CTTCCGTGACAACAACCTTTAG |
| BnaA09g48360D | F: CAGTCAAAGCCATCCATGAAAA; R: CGAAGCTAATAGTGTGTTGGTG |
| BnaA09g49220D | F: TTGCTTCTTTTGTCAACCTAGC; R: CAGAAACAGTGCATACAGCTAC |
| BnaC02g14570D | F: GTGTTCTCAGGGAGATATCTCG; R: CACGAGGATCTTCAAATCCAAC |
新窗口打开|下载CSV
2 结果与分析
2.1 重组自交系群体与自然群体的表型变异
重组自交系群体中, 亲本GH06和ZY821的株高分别为198.1 cm和186.6 cm, 一次有效分枝高度分别为90.7 cm和67.8 cm, t检验株高在亲本间未达到显著差异(P>0.05), 但一次有效分枝高度达到显著差异(P<0.01)。株高在2016年和2017年的变异系数分别为5.24%和5.75%, 一次有效分枝高度的变异系数为18.58%和17.73%; 自然群体中, 株高在2016年和2017年的变异系数分别为8.57%和10.16%, 一次有效分枝高度的变异系数分别为26.65%和27.86% (表2)。2年间两性状的变异系数较为稳定, 株高的变异系数相对于一次有效分枝高度较小。株高在重组自交系和自然群体中的广义遗传力分别为75.45%和87.42%, 一次有效分枝高度分别为68.77%和82.21%; 在同一群体中, 株高较一次有效分枝高度的遗传力高。自然群体中, 测量株高和一次有效分枝高度极端材料YC4、YC15、YC11的表型数据, YC4和YC15株高分别为213.9 cm和185.7 cm; YC11和YC4的一次有效分枝高度分别为100.8 cm和70.3 cm, 2年极端材料的株高及一次有效分枝高度都达到显著差异(P<0.01)。Table 2
表2
表2两群体株高和一次有效分枝高度的表型数据
Table 2
| 性状 Trait | 群体 Population | 年份 Year | 均值±标准差 Mean ± SD | 范围 Range | 变异系数 CV (%) | 广义遗传力 h2 (%) |
|---|---|---|---|---|---|---|
| ZY821父本株高 | RIL | 2016 | 190.80±6.85 | |||
| ZY821♂ PH | 2017 | 184.33±11.74 | ||||
| GH06母本株高 | RIL | 2016 | 217.40±5.61 | |||
| GH06♀ PH | 2017 | 187.33±10.8 | ||||
| ZY821父本一次有效分枝高度 | RIL | 2016 | 86.25±5.35 | |||
| ZY821♂ BH | 2017 | 68.63±11.54 | ||||
| GH06母本一次有效分枝高度 | RIL | 2016 | 83.00±6.70 | |||
| GH06♀ BH | 2017 | 95.14±15.67 | ||||
| 株高PH | RIL | 2016 | 213.82±11.20 | 187.0-241.6 | 5.24 | 75.45 |
| 2017 | 207.26±11.91 | 163.0-238.4 | 5.75 | |||
| N | 2016 | 211.84±18.15 | 162.2-259.0 | 8.57 | 87.42 | |
| 2017 | 204.39±20.76 | 127.4-262.4 | 10.16 | |||
| 一次有效分枝高度BH | RIL | 2016 | 85.26±15.84 | 49.6-118.0 | 18.58 | 68.77 |
| 2017 | 82.85±14.69 | 50.1-123.6 | 17.73 | |||
| N | 2016 | 84.17±22.43 | 19.0-142.8 | 26.65 | 82.21 | |
| 2017 | 82.90±23.09 | 11.0-141.1 | 27.86 |
新窗口打开|下载CSV
重组自交系群体和自然群体2年株高和一次有效分枝高度均表现出连续正态分布(图1), 说明这2个性状都符合由多基因控制的数量性状特点, 满足QTL作图的基本要求, 适合全基因组关联分析。
图1
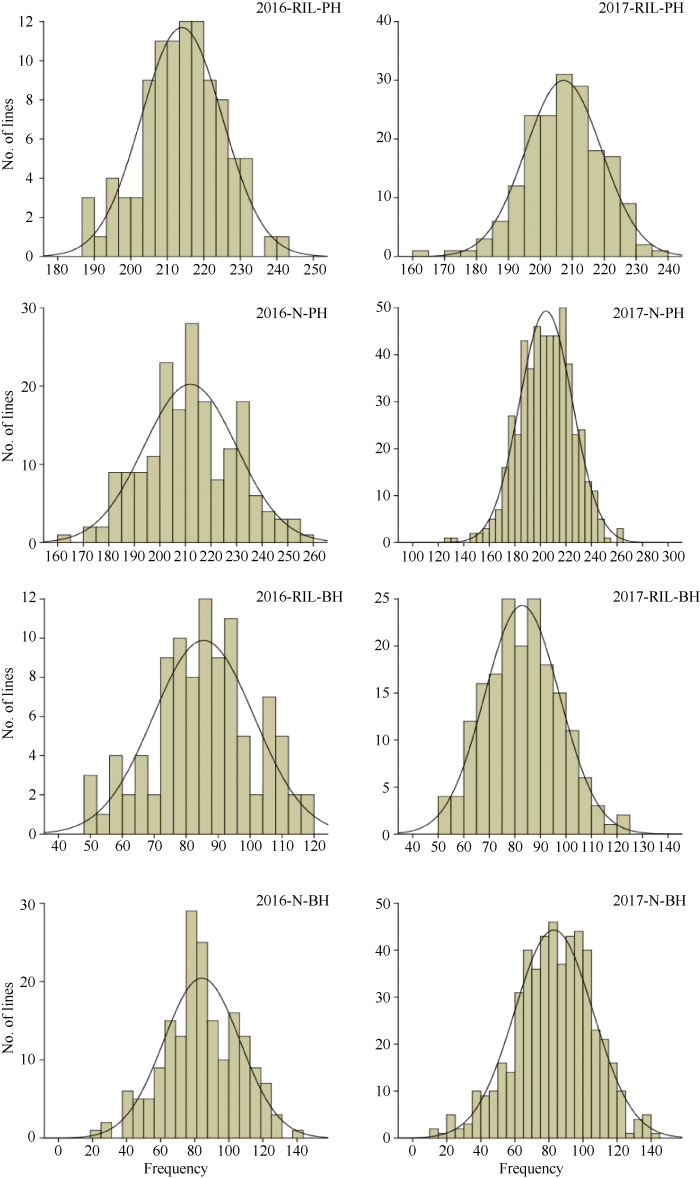 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1两群体株高和一次有效分枝高度的频次分布
PH: 株高; BH: 一次有效分枝高度; RIL: 重组自交系; N: 自然群体。
Fig. 1Frequency distribution of plant height and the first branch height in two populations
PH: plant height; BH: the first branch height; RIL: recombinant inbred lines; N: natural population.
2.2 重组自交系群体QTL定位分析
利用WinQTL2.5对株高和一次有效分枝高度共检测到19个QTL, 分布在8条染色体上, 其中在A09染色体上检测到的QTL数量最多(图2)。位于A01和A09上的QTL加性效应为负值, 表明这些QTL的增效基因来自父本, 其余的QTL则来自母本(表3)。图2
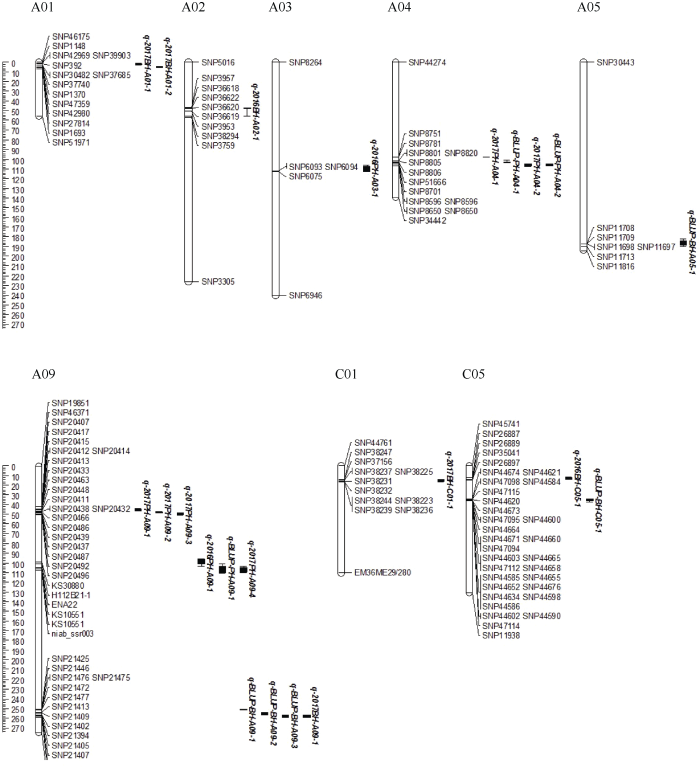 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2株高和一次有效分枝高度QTL在SNP连锁群上分布情况
Fig. 2Putative QTL locations of plant height and the first branch height on the SNP genetic map
Table 3
表3
表3在两年中检测到的株高和一次有效分枝高度QTL
Table 3
| 位点 QTL | 染色体 Chr. | 阈值 LOD score | 加性效应 Additive effect | 贡献率 R2 (%) | 标记区间 SNP interval | 物理区间 Physical position (bp) | 环境 Environment |
|---|---|---|---|---|---|---|---|
| 株高 PH | |||||||
| q-2016PH-A03-1 | A03 | 2.87 | 4.27 | 13.29 | SNP6093-SNP6075 | 18421721-18543828 | 2016 |
| q-2016PH-A09-1 | A09 | 3.51 | -3.81 | 11.28 | KS30880-H112B21-1 | 2016 | |
| q-2017PH-A04-1 | A04 | 2.79 | 2.71 | 4.92 | SNP8751-SNP8781 | 14085289-14244618 | 2017 |
| q-2017PH-A04-2 | A04 | 3.26 | 2.93 | 5.75 | SNP8596-SNP8650 | 13373664-13531885 | 2017 |
| q-2017PH-A09-1 | A09 | 5.20 | -5.58 | 9.63 | SNP46371-SNP20466 | 22590345-23259990 | 2017 |
| q-2017PH-A09-2 | A09 | 4.16 | -5.81 | 7.55 | SNP20486-SNP20487 | 23517031-23521212 | 2017 |
| q-2017PH-A09-3 | A09 | 4.72 | -5.40 | 8.51 | SNP20492-SNP20496 | 23603951-23642701 | 2017 |
| q-2017PH-A09-4 | A09 | 4.31 | -4.55 | 7.86 | KS10551-KS10551 | 2017 | |
| q-BLUP-PH-A04-1 | A04 | 4.25 | 1.43 | 7.60 | SNP8701-SNP8801 | 13798050-14401987 | BLUP |
| q-BLUP-PH-A04-2 | A04 | 3.45 | 1.31 | 6.27 | SNP8596-SNP8650 | 13373664-13531885 | BLUP |
| q-BLUP-PH-A09-1 | A09 | 2.60 | -1.36 | 4.60 | ENA22-niab_ssr003 | BLUP | |
| 一次有效分枝高度 BH | |||||||
| q-2016BH-A02-1 | A02 | 4.14 | 6.24 | 13.56 | SNP3957-SNP3759 | 22556597-23778808 | 2016 |
| q-2016BH-C05-1 | C05 | 5.26 | 7.49 | 19.10 | SNP26887-SNP26897 | 4224409-4258940 | 2016 |
| q-2017BH-A01-1 | A01 | 4.78 | -5.53 | 9.15 | SNP1148-SNP1370 | 17881769-19687948 | 2017 |
| q-2017BH-A01-2 | A01 | 3.08 | -4.64 | 6.03 | SNP47359-SNP1693 | 20189127-21190484 | 2017 |
| q-2017BH-A09-1 | A09 | 2.69 | -3.46 | 5.36 | SNP21394-SNP46209 | 32375997-32618214 | 2017 |
| q-2017BH-C01-1 | C01 | 6.16 | 6.22 | 12.17 | SNP38247-SNP38236 | 11120369-11162114 | 2017 |
| q-BLUP-BH-A05-1 | A05 | 2.76 | 1.25 | 5.12 | SNP11708-SNP11713 | 20293807-20374178 | BLUP |
| q-BLUP-BH-A09-1 | A09 | 4.60 | -1.66 | 9.32 | SNP21425-SNP21477 | 32728408-33029776 | BLUP |
| q-BLUP-BH-A09-2 | A09 | 6.52 | -1.95 | 13.09 | SNP21413-SNP21402 | 32435925-32608817 | BLUP |
| q-BLUP-BH-A09-3 | A09 | 4.62 | -1.64 | 9.48 | SNP21405-SNP21415 | 32514368-32619386 | BLUP |
| q-BLUP-BH-C05-1 | C05 | 2.77 | 1.23 | 5.51 | SNP44674-SNP47114 | 11206177-11417643 | BLUP |
新窗口打开|下载CSV
对于株高, 2016年检测到2个QTL, 2017年检测到6个QTL, 基于2年数据的BLUP值检测到3个QTL, 其中q-2017PH-A04-2 (A04: 13,373,664-13,531,885)和q-BLUP-PH-A04-2 (A04: 13,373,664-13,531,885)重合, q-2017PH-A09-1 (22,590,345-23,259,990)、q-2017PH- A09-2 (23,517,031-23,521,212)、q-2017PH-A09-3 (23,603,951- 23,642,701)有交叉重叠区段, 因此共检测到8个QTL。这些QTL主要分布于A03、A04、A09染色体, LOD值介于2.60~5.20, 表型变异贡献率为4.60%~13.29%。
对于一次有效分枝高度, 在2016年和2017年分别检测到2个和4个QTL, 基于2年数据的BLUP值检测到5个QTL, 其中q-2017BH-A09-1 (32,375,997- 32,618,214)、q-BLUP-BH-A09-2 (32,435,925-32,608,817)、q-BLUP-BH-A09-3 (32,514,368-32,619,386)有交叉重叠的区段, 因此共检测到9个QTL。这些QTL主要分布于A01、A02、A05、A09、C01和C05染色体, LOD值介于2.76~6.52, 表型变异贡献率为5.12%~19.10%。
2.3 自然群体的全基因组关联分析
对株高和一次有效分枝高度关联分析6种模型下的结果绘制Quantile-Quantile散点图(QQ plot), 将各模型下的曲线与期望曲线拟合度最高的模型作为最佳模型。由图3可知, 各性状的最佳模型如下, 株高(PH)都为P+K模型, 一次有效分枝高度(BH)除2016年为K模型外, 其余也为P+K模型。在最佳模型下, 利用385,692个SNP, 以P值小于阈值(1/385,692 = 2.593E-06)确定显著关联SNP位点(附表1), 并绘制Manhattan图(图3)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图32年株高和一次有效分枝高度在各模型下的QQ图及最佳模型下的Manhattan图
PH:株高; BH: - -次有效分枝高度; BLUP:最佳线性无偏预测。
Fig.3Quantile-quantile plot for six models and Manhattan plots of association analysis using the optimal model for plant height and the first branch height in two years
PH: plant height; BH: the first branch height; BLUP: best linear unbiased prediction.
Supplementary table 1
附表1
附表1最佳模型下株高和一次有效分枝高度显著位点表
Supplementary table 1
| 性状 Trait | 环境 Env. | 模型 Model | 位点 SNP | 染色体 Chr. | 位置 Position | 域值 -lg (P) | 贡献率 R2 (%) |
|---|---|---|---|---|---|---|---|
| PH | 2016 | K+PCA | S3_24415510 | A03 | 24415510 | 5.69 | 20.05 |
| 2017 | K+PCA | S2_4664084 | A02 | 4664084 | 5.84 | 6.72 | |
| S7_19604035 | A07 | 19604035 | 6.24 | 7.41 | |||
| S7_19609237 | A07 | 19609237 | 6.01 | 6.81 | |||
| S7_19665408 | A07 | 19665408 | 5.85 | 7.06 | |||
| S7_19781536 | A07 | 19781536 | 5.87 | 7.36 | |||
| S7_19889261 | A07 | 19889261 | 5.64 | 7.20 | |||
| S7_19890772 | A07 | 19890772 | 5.66 | 6.94 | |||
| S7_19902486 | A07 | 19902486 | 5.77 | 7.52 | |||
| S7_19993228 | A07 | 19993228 | 5.63 | 6.59 | |||
| S7_19993448 | A07 | 19993448 | 6.07 | 6.88 | |||
| S7_19993458 | A07 | 19993458 | 5.69 | 6.42 | |||
| S7_19994131 | A07 | 19994131 | 6.45 | 7.69 | |||
| S7_19997901 | A07 | 19997901 | 5.71 | 6.67 | |||
| S7_19998470 | A07 | 19998470 | 6.37 | 7.52 | |||
| S7_19999615 | A07 | 19999615 | 6.07 | 7.05 | |||
| S7_19999649 | A07 | 19999649 | 5.63 | 7.52 | |||
| S7_20000927 | A07 | 20000927 | 6.31 | 7.47 | |||
| S7_20003694 | A07 | 20003694 | 5.79 | 6.83 | |||
| S7_20005472 | A07 | 20005472 | 6.67 | 7.80 | |||
| S7_20006012 | A07 | 20006012 | 6.50 | 7.73 | |||
| S7_20006528 | A07 | 20006528 | 5.87 | 7.09 | |||
| S7_20009397 | A07 | 20009397 | 5.86 | 6.67 | |||
| S7_20009417 | A07 | 20009417 | 6.19 | 7.04 | |||
| S7_20033861 | A07 | 20033861 | 6.11 | 6.88 | |||
| S7_20050137 | A07 | 20050137 | 5.96 | 8.10 | |||
| S7_20050232 | A07 | 20050232 | 6.22 | 7.74 | |||
| S7_20053742 | A07 | 20053742 | 6.72 | 7.72 | |||
| S7_20056543 | A07 | 20056543 | 5.65 | 6.53 | |||
| S7_20079034 | A07 | 20079034 | 5.76 | 7.36 | |||
| S7_20107214 | A07 | 20107214 | 7.69 | 9.22 | |||
| S7_20107455 | A07 | 20107455 | 6.40 | 7.44 | |||
| S7_20107531 | A07 | 20107531 | 6.31 | 8.24 | |||
| S7_20108117 | A07 | 20108117 | 5.75 | 6.74 | |||
| S7_20125834 | A07 | 20125834 | 6.37 | 7.28 | |||
| S7_20144336 | A07 | 20144336 | 6.16 | 7.20 | |||
| S7_20154451 | A07 | 20154451 | 6.42 | 8.22 | |||
| S7_20160199 | A07 | 20160199 | 5.84 | 7.20 | |||
| S7_20171415 | A07 | 20171415 | 5.87 | 6.65 | |||
| S7_20171839 | A07 | 20171839 | 6.46 | 7.34 | |||
| S7_20178594 | A07 | 20178594 | 6.55 | 7.85 | |||
| S7_20178844 | A07 | 20178844 | 6.23 | 7.54 | |||
| S7_20178851 | A07 | 20178851 | 5.93 | 7.18 | |||
| S7_20186093 | A07 | 20186093 | 5.79 | 6.56 | |||
| S7_20188262 | A07 | 20188262 | 5.83 | 6.87 | |||
| S7_20207662 | A07 | 20207662 | 5.72 | 6.70 | |||
| S7_20547520 | A07 | 20547520 | 5.71 | 6.83 | |||
| S16_30960716 | C06 | 30960716 | 6.60 | 7.81 | |||
| S16_30960741 | C06 | 30960741 | 7.02 | 8.31 | |||
| S16_31108747 | C06 | 31108747 | 5.89 | 6.78 | |||
| BLUP | K+PCA | S2_4664084 | A02 | 4664084 | 6.36 | 7.07 | |
| S2_5390778 | A02 | 5390778 | 5.90 | 7.03 | |||
| S7_19604035 | A07 | 19604035 | 6.64 | 7.71 | |||
| S7_19607230 | A07 | 19607230 | 6.02 | 6.75 | |||
| S7_19609237 | A07 | 19609237 | 5.96 | 6.47 | |||
| S7_19665408 | A07 | 19665408 | 6.05 | 6.92 | |||
| S7_19749218 | A07 | 19749218 | 6.29 | 7.29 | |||
| S7_19781536 | A07 | 19781536 | 6.10 | 7.37 | |||
| S7_19856864 | A07 | 19856864 | 5.73 | 6.34 | |||
| S7_19889261 | A07 | 19889261 | 5.75 | 7.08 | |||
| S7_19902486 | A07 | 19902486 | 5.92 | 7.19 | |||
| S7_19902495 | A07 | 19902495 | 5.69 | 7.19 | |||
| S7_19902618 | A07 | 19902618 | 5.59 | 6.50 | |||
| S7_19911177 | A07 | 19911177 | 5.83 | 6.72 | |||
| S7_19923829 | A07 | 19923829 | 5.68 | 6.33 | |||
| S7_19923850 | A07 | 19923850 | 5.84 | 6.42 | |||
| S7_19989673 | A07 | 19989673 | 5.61 | 6.35 | |||
| S7_19993448 | A07 | 19993448 | 6.07 | 6.54 | |||
| S7_19993458 | A07 | 19993458 | 5.61 | 6.01 | |||
| S7_19994131 | A07 | 19994131 | 6.02 | 6.74 | |||
| S7_19997901 | A07 | 19997901 | 6.04 | 6.71 | |||
| S7_19998042 | A07 | 19998042 | 5.80 | 6.38 | |||
| S7_19998470 | A07 | 19998470 | 5.88 | 6.50 | |||
| S7_19999615 | A07 | 19999615 | 6.09 | 6.66 | |||
| S7_20000927 | A07 | 20000927 | 6.31 | 7.10 | |||
| S7_20003694 | A07 | 20003694 | 6.14 | 6.84 | |||
| S7_20005472 | A07 | 20005472 | 6.76 | 7.50 | |||
| S7_20006012 | A07 | 20006012 | 6.97 | 7.94 | |||
| S7_20006186 | A07 | 20006186 | 5.73 | 6.36 | |||
| S7_20006210 | A07 | 20006210 | 5.77 | 6.38 | |||
| S7_20006234 | A07 | 20006234 | 5.81 | 6.73 | |||
| S7_20006528 | A07 | 20006528 | 6.10 | 7.05 | |||
| S7_20008610 | A07 | 20008610 | 5.71 | 6.89 | |||
| S7_20009397 | A07 | 20009397 | 6.23 | 6.79 | |||
| S7_20009417 | A07 | 20009417 | 6.59 | 7.15 | |||
| S7_20030774 | A07 | 20030774 | 5.75 | 6.16 | |||
| S7_20050137 | A07 | 20050137 | 6.12 | 7.78 | |||
| S7_20050232 | A07 | 20050232 | 6.84 | 8.25 | |||
| S7_20050979 | A07 | 20050979 | 5.61 | 6.24 | |||
| S7_20053742 | A07 | 20053742 | 6.52 | 7.07 | |||
| S7_20057981 | A07 | 20057981 | 5.77 | 7.00 | |||
| S7_20082865 | A07 | 20082865 | 5.76 | 7.54 | |||
| S7_20107214 | A07 | 20107214 | 7.94 | 8.87 | |||
| S7_20107455 | A07 | 20107455 | 6.83 | 7.64 | |||
| S7_20107531 | A07 | 20107531 | 7.26 | 9.02 | |||
| S7_20108117 | A07 | 20108117 | 5.95 | 6.64 | |||
| S7_20125834 | A07 | 20125834 | 6.23 | 6.77 | |||
| S7_20144336 | A07 | 20144336 | 6.62 | 7.31 | |||
| S7_20154451 | A07 | 20154451 | 6.55 | 7.99 | |||
| S7_20160199 | A07 | 20160199 | 6.36 | 7.53 | |||
| S7_20171415 | A07 | 20171415 | 6.24 | 6.68 | |||
| S7_20171839 | A07 | 20171839 | 6.30 | 6.80 | |||
| S7_20178594 | A07 | 20178594 | 6.59 | 7.55 | |||
| S7_20178844 | A07 | 20178844 | 5.93 | 6.86 | |||
| S7_20186093 | A07 | 20186093 | 6.22 | 6.69 | |||
| S7_20188262 | A07 | 20188262 | 6.11 | 6.84 | |||
| S7_20198799 | A07 | 20198799 | 5.65 | 6.49 | |||
| S7_20207662 | A07 | 20207662 | 5.79 | 6.43 | |||
| S7_20429083 | A07 | 20429083 | 5.63 | 6.38 | |||
| S7_20547520 | A07 | 20547520 | 6.69 | 7.75 | |||
| S7_20567053 | A07 | 20567053 | 5.67 | 6.06 | |||
| S12_38247607 | C02 | 38247607 | 5.70 | 6.43 | |||
| S16_30166343 | C06 | 30166343 | 5.96 | 6.92 | |||
| S16_30650628 | C06 | 30650628 | 5.62 | 6.27 | |||
| S16_30960716 | C06 | 30960716 | 6.66 | 7.47 | |||
| S16_30960741 | C06 | 30960741 | 7.24 | 8.08 | |||
| S16_31108747 | C06 | 31108747 | 6.36 | 6.99 | |||
| BH | 2016 | K | S13_3400444 | C03 | 3400444 | 5.67 | 16.87 |
| S13_12620221 | C03 | 12620221 | 5.70 | 16.89 | |||
| S14_36535069 | C04 | 36535069 | 5.76 | 18.36 | |||
| S19_41983822 | C09 | 41983822 | 5.75 | 17.28 | |||
| 2017 | K+PCA | S2_1549097 | A02 | 1549097 | 5.80 | 7.63 | |
| S2_1553600 | A02 | 1553600 | 6.39 | 7.73 | |||
| S2_5424292 | A02 | 5424292 | 6.14 | 7.01 | |||
| S2_5503092 | A02 | 5503092 | 6.19 | 7.32 | |||
| S2_5827516 | A02 | 5827516 | 6.66 | 7.95 | |||
| S2_6130942 | A02 | 6130942 | 5.76 | 7.02 | |||
| S12_10249838 | C02 | 10249838 | 6.06 | 6.23 | |||
| S15_7059739 | C05 | 7059739 | 5.92 | 6.80 | |||
| BLUP | K+PCA | S2_1553600 | A02 | 1553600 | 5.81 | 6.55 | |
| S2_5503092 | A02 | 5503092 | 6.09 | 6.83 | |||
| S2_5827516 | A02 | 5827516 | 6.81 | 7.69 | |||
| S2_9351215 | A02 | 9351215 | 5.98 | 6.95 | |||
| S2_9351221 | A02 | 9351221 | 5.89 | 6.82 | |||
| S12_10249838 | C02 | 10249838 | 5.62 | 6.32 | |||
| S15_7059739 | C05 | 7059739 | 6.00 | 6.49 |
新窗口打开|下载CSV
对于株高, 2016年在A03染色体上检测到1个SNP; 2017年检测到49个SNP, 分别位于A02 (1个)、A07 (45个)和C06 (3个)染色体; 2年BLUP也检测到67个SNP, 分别位于A02 (2个)、A07 (59个)、C02 (1个)和C06 (5个)染色体。在这些位点中, 有42个SNP在2017年和基于BIUP都被检测到, 因此对于株高共检测到75个SNP, 它们的贡献率为6.01%~20.05%, 其中2016年A03染色体上的S3_24415510位点贡献率最大。
对于一次有效分枝高度, 2016年在C03(2个)、C04(1个)和C09(1个)染色体上检测到4个SNP; 2017年检测到8个SNP, 分别位于A02(6个)、C02(1个)和C05(1个)染色体; 基于2年BLUP检测到7个SNP, 分别位于A02(5个)、C02(1个)和C05(1个)染色体。其中有5个SNP在2017年和BIUP中都被检测到, 因此共检测到14个一次有效分枝高度SNP。这些SNP的贡献率为6.23%~18.36%, 其中2016年C04染色体上的S14_36535069位点贡献率最大。株高和一次有效分枝高度在GWAS和遗传连锁定位结果中均没有重合位点。
2.4 候选基因筛选
本文将所得的QTL置信区间内的基因与关联分析SNP位点上下游500 kb区间内的基因比对[30], 初步筛选出24个与油菜株高和一次有效分枝高度相关的基因。并将其基因序列与拟南芥基因序列进行BLAST比对, 结合前人已报道的拟南芥同源基因功能, 筛选可能在本研究中发挥作用的候选基因(表4)。这些基因在拟南芥同源基因中参与植物生长发育, 影响细胞增殖、细胞扩增, 调节细胞周期、细胞壁形成, 参与赤霉素、亚精胺等合成代谢途径、核糖体组成等。Table 4
表4
表4株高和一次有效分枝高度候选基因
Table 4
| 基因 Gene | 物理位置 Physical position | 拟南芥同源基因 Homologs in A. thaliana | 功能注释 Functional annotation |
|---|---|---|---|
| 株高PH | |||
| BnaA02g09200D | A02:4555438-4557323 | AT5G55180 | O-Glycosyl hydrolases family 17 protein (MCO15.13) |
| BnaA02g09270D | A02:4576434-4582446 | AT5G55120 | GDP-L-galactose phosphorylase VITAMIN C DEFECTIVE 5 (VTC5) |
| BnaA03g47940D | A03:24664280-24664459 | AT4G26320 | Arabinogalactan protein 13 (AGP13) |
| BnaA04g17050D | A04:13894646-13897338 | AT5G65940 | Beta-hydroxyisobutyryl-CoA hydrolase 1 (CHY1) |
| BnaA04g17120D | A04:13961721-13963012 | AT2G29350 | Senescence-associated gene 13 (SAG13) |
| BnaA04g17490D | A04:14227152-14227949 | AT2G30370 | Allergen-like protein (CHAL) |
| BnaA07g27280D | A07:19859720-19860312 | AT1G68590 | Ribosomal protein PSRP-3/Ycf65 |
| BnaA07g28720D | A07:20705370-20707220 | AT1G70210 | CYCD1;1/CYCLIN D1;1 |
| BnaA07g28820D | A07:20767232-20773649 | AT1G70710 | Glycosyl hydrolase 9B1 (GH9B1) |
| BnaC02g35320D | C02:38070292-38074778 | AT2G04030 | Chaperone protein htpG family protein (CR88) |
| BnaC06g30260D | C06:31058017-31061313 | AT1G68580 | Agenet domain-containing protein / bromo-adjacent homology (BAH) domain-containing protein (F24J5.18) |
| BnaC06g30400D | C06:31172511-31173663 | AT1G69220 | Protein kinase superfamily protein (SIK1) |
| BnaC06g30410D | C06:31173893-31177895 | AT1G69220 | Protein kinase superfamily protein (SIK1) |
| 一次有效分枝高度BH | |||
| BnaA01g25960D | A01:18111689-18112090 | AT3G47370 | Ribosomal protein S10p/S20e family protein (RPS20B) |
| BnaA01g27550D | A01:19232447-19236961 | AT3G16840 | P-loop containing nucleoside triphosphate hydrolases superfamily protein (RH13) |
| BnaA01g28120D | A01:19629034-19630394 | AT3G16240 | Delta tonoplast integral protein (DELTA-TIP) |
| BnaA01g29630D | A01:20485424-20487751 | AT3G14067 | Subtilase family protein (SASP) |
| BnaA01g30830D | A01:21042542-21043677 | AT3G12145 | Leucine-rich repeat (LRR) family protein (FLOR1) |
| BnaA02g37010D | A02:1536206-1537938 | AT5G61780 | TUDOR-SN protein 2 (Tudor2) |
| BnaA02g31980D | A02:23032463-23034656 | AT5G25460 | Transmembrane protein, putative (Protein of unknown function, DUF642) (DGR2) |
| BnaA09g48360D | A09:32409788-32410789 | AT1G09580 | Emp24/gp25L/p24 family/GOLD family protein (F14J9.28) |
| BnaA09g49220D | A09:32795595-32796344 | AT1G07830 | Ribosomal protein L29 family protein (F24B9.7) |
| BnaC02g14570D | C02:10095432-10098417 | AT5G53120 | Spermidine synthase 3 (SPDS3) |
| BnaC02g14600D | C02:10119161-10122317 | AT5G53090 | NAD(P)-binding Rossmann-fold superfamily protein (MFH8.1) |
新窗口打开|下载CSV
2.5 候选基因的验证
挑选了其中5个株高候选基因进行验证(图4), 其中BnaC07g28720D、BnaC07g28820D基因的相对表达量在株高较高的材料(YC4)中较高, 在株高较低的材料(YC15)中较低; BnaA03g47940D、BnaC06g30400D、BnaC06g30410D基因的相对表达量在株高较低的材料中较高, 在株高较高的材料中较低。图4
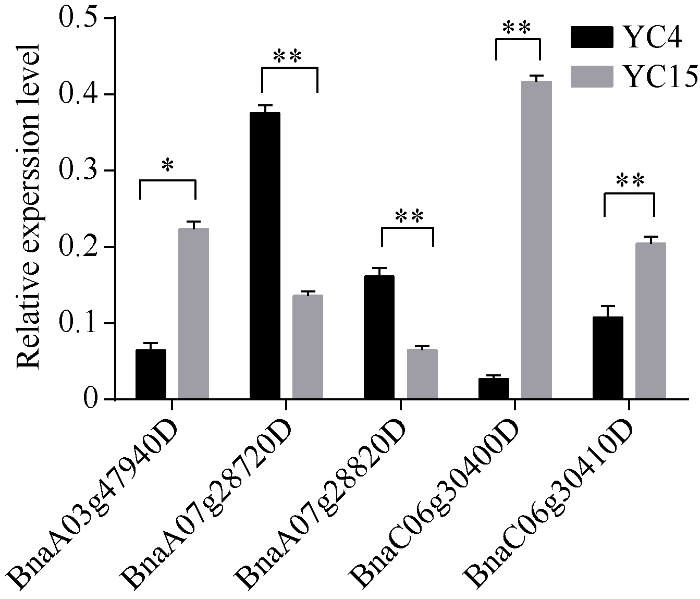 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4株高候选基因在茎尖中的相对表达量
误差线表示3次生物学重复的标准差。*和**分别表示基因在不同材料茎尖中的表达水平有显著(P < 0.05)和极显著(P < 0.01)差异。
Fig. 4Relative expression of the candidate genes of plant height in stem apex
The error bar shows the standard deviation of three biological replicates. * and ** indicate significantly different expression in stem apex to the control at P < 0.05 and P < 0.01, respectively.
从一次有效分枝高度的候选基因中, 挑选了7个进行验证(图5), 其中BnaA01g30830D、BnaA0 2g31980D、BnaA02g37010D、BnaA09g49220D、BnaC02g14570D基因的相对表达量在一次有效分枝高度较高的材料(YC11)中较高, 在一次有效分枝高度较低的材料(YC4)中较低; BnaA01g29630D、BnaA09g48360D基因的相对表达量在一次有效分枝高度较低的材料中较高, 在一次有效分枝高度较高的材料中较低。
图5
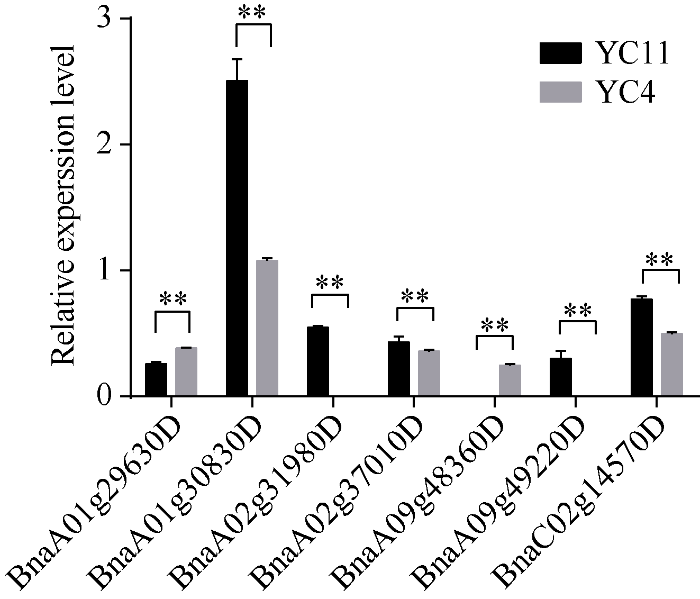 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5一次有效分枝高度候选基因在茎尖中的相对表达量
误差线表示3次生物学重复的标准差。*和**分别表示基因在不同材料茎尖中的表达水平有显著(P < 0.05)和极显著(P < 0.01)差异。
Fig. 5Relative expression of the candidate genes of the first branch height in stem apex
The error bar shows the standard deviation of three biological replicates. * and ** indicate significantly different expression in stem apex to the control at P < 0.05 and P < 0.01, respectively.
3 讨论
株高和一次有效分枝高度是甘蓝型油菜的2个重要株型性状。株高是作物根颈部到顶部之间的距离, 也是影响作物倒伏的重要的因素。一次有效分枝高度是油菜下部第一个有效分枝生长着生点与子叶节之间的距离, 而分枝则作为角果生长的载体。适宜的株高和一次有效分枝高度, 有利于增强植株的抗倒伏性和提高机械化利用率, 进而提高产量并促进油菜产业发展。结荚厚度包含了所有产量构成因素, 经济产量和收获指数又是衡量产量的2个重要指标。在试验过程中, 对株高和一次有效分枝高度的2016年表型值、2017年表型值和2年的BLUP值都进行了QTL定位和GWAS分析。BLUP能较为有效地利用亲缘表型信息、考虑不同世代的遗传差异并矫正环境效应, 但由于实际中数据的不完整、模型的真实度及其方差组分等的真值未知等问题, 通过BLUP得到的校正值并不一定是最佳的无偏预测, 因此本试验同时将3个值都进行了分析。两性状在2016年和2017年都未检测到重复位点, 利用BLUP值检测到的位点中, 发现与2017年有重复或交叉重叠, 没有与2016年重叠。一方面可能是性状本身受环境的影响较大, 另一方面可能是2016年材料缺失较为严重。
在重组自交系群体中检测到的10个株高性状QTL分别位于A03、A04和A09染色体上, 其中在2016年、2017年和BLIP值中检测到的位于A09染色体上的QTL加性效应均为负值, 说明A09染色体上的QTL增效作用均来自父本的等位基因位点, 且A09上的QTL数量最多, 各位点的贡献率相对较大; 另外, 在2017年和基于BLUP值检测中, A04染色体上有一个重复QTL (q-BLUP-PH-A04-2)。本研究中的q-2016PH-A03-1、q-2017PH-A09-1、q-2017PH- A09-2、q-2017PH-A09-3所在的区段在其他研究中也被重复检测到[21,32-33]。在重组自交系群体中检测到9个一次有效分枝高度性状QTL, 分别位于A01、A02、A05、A09、C01和C05染色体, 其中q-BLUP- BH-C05-1所在区段与王嘉等[18]研究中被检测到的区段有重叠区域。
在自然群体中, 株高性状检测到的A02染色体上的S2_4664084位点和A07染色体上的S7_20033861、S7_20050137等位点与Sun等[24]研究中的位点相近, 最近距离只有7.5 kb; A02染色体上有3个重复SNP, C02和C05染色体上各有1个重复SNP; 一次有效分枝高度性状检测到的A02染色体上的S2_6130942位点与Zheng等[23]研究中的位点相近。
在株高候选基因的拟南芥同源基因中, 检测到的与细胞增殖、细胞扩增、细胞周期和细胞壁相关的基因有AGP13、GH9B1、SIK1、CYCD1。AGP13编码的阿拉伯半乳聚糖蛋白是参与植物生长发育的细胞外蛋白多糖[34]。AGP13与植物细胞壁形成和重建相关, 在拟南芥的茎生长速率最快阶段, AGP13表达增加[35], 由此推测其可能在控制油菜株高性状过程中起关键作用。另外, AGP13是一个IAA特异调控的基因, 它在一定浓度IAA处理后表现下调[36,37,38]; 在一个细胞大小和数量严重减少的矮小gds1拟南芥突变体中, AGP13下调[39]。GH9B1/CEL1是内切-1,4-β-葡聚糖酶(EGase)基因, 参与细胞伸长, 且与细胞壁的重组和生长有关[40], 由此推测在油菜主茎伸长过程中细胞伸长发挥作用。CEL1蛋白存在于正在发育伸长的幼嫩组织细胞壁中, 特别是在增厚的细胞壁中。CEL1在细胞扩增及增强内切-1,4-β-葡聚糖酶活性与纤维素生物合成方面具有重要作用[41]。SIK1编码一个假定的丝氨酸/苏氨酸激酶, 类似Hippo/STE20激酶, SIK1与MOB1相互作用, 调控拟南芥细胞生长和增值, 在突变体sik1-4中, 细胞数量、细胞大小呈倍性水平降低[42]。CYCD1编码与CDC2A发生物理作用的d型细胞周期蛋白在种子萌发早期有关键作用并影响细胞分裂。CYCD1受AUX1和SNLs调控, 在生长中发挥重要作用[43]; 其中在细胞分裂素受体突变体(wol/cre1)的全基因组分析中发现CYCD1下调表达[44], 因此该基因可能通过调控植物激素转导途径来控制油菜株高。
在一次有效分枝高度候选基因的拟南芥同源基因中, SPDS3编码一种新的亚精胺合成酶, 它是亚精胺合酶SPDS1和SPDS2的旁系同源物。SPDS1和SPDS2转录水平在根系中高于其他器官, 但SPDS3在茎节间和胚芽以及根系中的表达都高; 另外发现SPDS3可能参与ABA介导的胁迫反应[45]。Tudor2参与了拟南芥GA的生物合成和种子萌发, 在AtTudor2 T-DNA插入突变体和AtTudor2 RNAi转基因株系中, 用于GA生物合成的关键酶AtGA20ox3的表达明显下调[46], 由此推测在油菜GA生物合成过程中起调控作用进而影响一次有效分枝高度。DGR2对L-半乳糖-1,4-内酯(L-GalL)表现特异性应答, 其dgr2突变体表现根短且莲座小, DGR2也对细胞壁的合成和延伸有影响[47]。SASP编码衰老相关的枯草蛋白酶, sasp1突变体的花序分枝和角果数量增多。SASP可能参与细胞外膜或质膜的靶向定位或释放质外体信号, 并且在生殖发育和衰老的进程中抑制花序分枝和角果的产生, 从而最大限度地减少分枝和果实间对资源的竞争[48], 同时, 在生长后期的拟南芥茎中它被鉴定出是一种细胞壁蛋白, 而且是蛋白酶抑制剂[49]。FLOR1编码一种富含亮氨酸的重复蛋白, 该蛋白直接与MADS结构域转录因子相互作用[50]。FLOR1在诱导开花时, 茎尖分生组织转化
为花序分生组织, 其表达增加并调控开花时间[51]; 从诱导花到心皮和雄蕊成熟的过程中, FLOR1也在特定的细胞中发挥重要作用, 它可能成为联合MADS结构域转录因子在生长发育调控和信号传导中发挥重要作用[52]。本研究探究了株高和一次有效分枝高度对产量的影响并且挖掘出可能与油菜株高和一次有效分枝高度相关的候选基因, 为油菜株型改良以及后续基因的研究提供理论依据, 为下一步的功能验证奠定基础。
4 结论
结合遗传连锁定位和全基因组关联分析, 在A02、A03、A04、A07、C02、C06鉴定出与株高相关的位点, 并筛选出BnaC07g28720D、BnaC07 g28820D、BnaA03g47940D、BnaC06g30400D、BnaC06g30410D等候选基因; 在A01、A02、A09、C02找到与一次有效分枝高度相关的位点, 筛选出BnaA01g30830D、BnaA02g31980D、BnaA02g37010D、BnaA09g49220D、BnaC02g14570D、BnaA01g29630D、BnaA09g48360D等候选基因, 并验证了部分基因在极端材料中的表达情况。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1146/annurev-arplant-042817-040422URLPMID:29553800 [本文引用: 1]

Shoot architecture is determined by the organization and activities of apical, axillary, intercalary, secondary, and inflorescence meristems and by the subsequent development of stems, leaves, shoot branches, and inflorescences. In this review, we discuss the unifying principles of hormonal and genetic control of shoot architecture including advances in our understanding of lateral branch outgrowth; control of stem elongation, thickness, and angle; and regulation of inflorescence development. We focus on recent progress made mainly in Arabidopsis thaliana, rice, pea, maize, and tomato, including the identification of new genes and mechanisms controlling shoot architecture. Key advances include elucidation of mechanisms by which strigolactones, auxins, and genes such as IDEAL PLANT ARCHITECTURE1 and TEOSINTE BRANCHED1 control shoot architecture. Knowledge now available provides a foundation for rational approaches to crop breeding and the generation of ideotypes with defined architectural features to improve performance and productivity.
DOI:10.1038/srep21625URLPMID:26880301 [本文引用: 2]
An optimized plant architecture (PA) is fundamental for high-yield breeding but the genetic control of the important trait is largely unknown in rapeseed. Here plant architecture factors (PAFs) were proposed to consist of main inflorescence length proportion (MILP), branch height proportion (BHP), and branch segment proportion (BSP). Comparison of different genotypes in a DH population grown in diverse environments showed that an optimized PAF performance with MILP and BHP between 0.3-0.4 was important for high yield potential. In total, 163 unique quantitative trait loci (QTLs) for PA- and plant yield (PY)-related traits were mapped onto a high-density genetic map. Furthermore, 190 PA-related candidate genes for 91 unique PA QTLs and 2350 PY epistatic interaction loci-pairs were identified, which explain 2.8-51.8% and 5.2-23.6% of phenotypic variation, respectively. Three gene categories, transcription factor, auxin/IAA, and gibberellin, comprise the largest proportions of candidate genes for PA-related QTLs. The effectiveness of QTL candidate genes prediction was demonstrated by cloning of three candidate genes, Bna.A02.CLV2, Bna.A09.SLY2, and Bna.C07.AHK4. The study thus outlines a gene network for control of PA-related traits and provides novel information for understanding the establishment of ideal PA and for developing effective breeding strategies for yield improvement in rapeseed and other crops.
DOI:10.1016/j.copbio.2006.02.004URLPMID:16504498 [本文引用: 1]
Plant architecture, referring here to the aerial part of a higher plant, is mainly determined by factors affecting shoot branching, plant height and inflorescence morphology. Significant progress has been made in isolating and characterizing genes that are directly involved in the formation of plant architecture, especially those controlling the initiation and outgrowth of axillary buds, elongation of stems and architecture of inflorescences. Most of these genes are conserved between dicotyledonous and monocotyledonous plants, indicating that these plants share similar regulatory pathways to establish their shape. The conservation of these genes makes them of great agronomical importance for improving crop yields.
DOI:10.1007/s00122-010-1306-9URL [本文引用: 1]
Although dwarf genes have been widely used to improve lodging resistance and enhance harvest index in cereal crops, lodging is still a serious problem in rapeseed (Brassica napus) production. A semi-dwarf B. napus mutant, ds-1, was identified through EMS mutagenesis of a microspore-cultured DH line. The mutant had a significant reduction in height due to a lower first branch position and shorter internodes when compared with wild-type cultivars. This dwarfism was inherited as a single semi-dominant gene, ds-1. DS-1 locus was mapped to chromosome A6, and co-segregated with a microsatellite marker BnEMS1125 derived from the gene BnRGA. BnRGA encodes a DELLA protein that functions as a GA signaling repressor. The expression of a mutant BnRGA allele from ds-1, Bnrga-ds, caused dwarf phenotypes in Arabidopsis. Comparative sequencing of RGA open-reading frames (ORFs) of ds-1 and wild-type cultivars revealed a single proline (P)-to-leucine (L) substitution that may lead to a gain-of-function mutation in GA signaling. The expression of the Arabidopsis homolog, Atrga-ds, bearing this site-directed mutation also rendered dwarf phenotypes in Arabidopsis, which demonstrated that the P-to-L mutation in the VHYNP motif of Bnrga-ds is responsible for the dwarfism. A yeast two-hybrid assay confirmed that this mutation inhibited the interaction between Bnrga-ds/Atrga-ds and the GA receptor, AtGID1A, in the presence of GA3, suggesting that the conserved proline residue in the VHYNP motif of DELLA protein directly participates in DELLA-GID1 interaction. Identification and characterization of the dwarf gene ds-1 will facilitate its utilization in improving lodging resistance in Brassica breeding.
DOI:10.1038/35093585URLPMID:11584298 [本文引用: 1]
The origin of agriculture led to the domestication of many plant species and to the exploitation of natural resources. It took almost 10,000 years for food grain production to reach 1 billion tons, in 1960, and only 40 years to reach 2 billion tons, in 2000. This unprecedented increase, which has been named the 'green revolution', resulted from the creation of genetically improved crop varieties, combined with the application of improved agronomic practices.
DOI:10.1073/pnas.251534098URLPMID:11717468 [本文引用: 1]
RGA and GAI are homologous genes that encode putative transcriptional regulators that repress gibberellin (GA) signaling in Arabidopsis. Previously we showed that the green fluorescent protein (GFP)-RGA fusion protein is localized to the nucleus in transgenic Arabidopsis, and expression of this fusion protein rescues the rga null mutation. The GA signal seems to derepress the GA response pathway by degrading the repressor protein RGA. The GA-insensitive, semidominant, semidwarf gai-1 mutant encodes a mutant protein with a 17-amino acid deletion within the DELLA domain of GAI. It was hypothesized that this mutation turns the gai protein into a constitutive repressor of GA signaling. Because the sequences missing in gai-1 are identical between GAI and RGA, we tested whether an identical mutation (rga-Delta 17) in the RGA gene would confer a phenotype similar to gai-1. We demonstrated that expression of rga-Delta 17 or GFP-(rga-Delta 17) under the control of the RGA promoter caused a GA-unresponsive severe dwarf phenotype in transgenic Arabidopsis. Analysis of the mRNA levels of a GA biosynthetic gene, GA4, showed that the feedback control of GA biosynthesis in these transgenic plants was less responsive to GA than that in wild type. Immunoblot and confocal microscopy analyses indicated that rga-Delta17 and GFP-(rga-Delta 17) proteins were resistant to degradation after GA application. Our results illustrate that the DELLA domain in RGA plays a regulatory role in GA-induced degradation of RGA. Deletion of this region stabilizes the rga-Delta 17 mutant protein, and regardless of the endogenous GA status rga-Delta 17 becomes a constitutively active repressor of GA signaling.
DOI:10.1101/gad.11.23.3194URLPMID:9389651 [本文引用: 1]
The Arabidopsis gai mutant allele confers a reduction in gibberellin (GA) responsiveness. Here we report the molecular cloning of GAI and a closely related gene GRS. The predicted GAI (wild-type) and gai (mutant) proteins differ only by the deletion of a 17-amino-acid segment from within the amino-terminal region. GAI and GRS contain nuclear localization signals, a region of homology to a putative transcription factor, and motifs characteristic of transcriptional coactivators. Genetic analysis indicates that GAI is a repressor of GA responses, that GA can release this repression, and that gai is a mutant repressor that is relatively resistant to the effects of GA. Mutations at SPY and GAR2 suppress the gai phenotype, indicating the involvement of GAI, SPY, and GAR2 in a signaling pathway that regulates GA responses negatively. The existence of this pathway suggests that GA modulates plant growth through derepression rather than through simple stimulation.
DOI:10.1111/j.1365-313X.2007.03356.xURLPMID:18069939 [本文引用: 1]
The activity of the gibberellin (GA) biosynthetic enzymes GA 20-oxidases (GA20ox) is of particular importance in determining GA concentration in many plant species. In Arabidopsis these enzymes are encoded by a family of five genes: AtGA20ox1-AtGA20ox5. Transcript analysis indicated that they have different expression patterns and may thus participate differentially in GA-regulated developmental processes. We have used reverse genetics to determine the physiological roles of AtGA20ox1 and AtGA20ox2, the most highly expressed GA20ox genes during vegetative and early reproductive development. AtGA20ox1 and AtGA20ox2 act redundantly to promote hypocotyl and internode elongation, flowering time, elongation of anther filaments, the number of seeds that develop per silique and elongation of siliques, with AtGA20ox1 making the greater contribution to internode and filament elongation, and AtGA20ox2 making the greater contribution to flowering time and silique length. Pollination of the double mutant with wild-type pollen indicated that the GA promoting silique elongation is of maternal origin. The ga20ox2 phenotype revealed that GA promotes the number of stem internodes that elongate upon bolting, and does so independently of its effect on internode elongation. Comparison of the phenotype of the double mutant with that of the highly GA-deficient ga1-3 mutant indicates that other GA20ox genes contribute to all the developmental processes examined, and, in some cases such as root growth and leaf expansion, make major contributions, as these processes were unaffected in the double mutant. In addition, the effects of the mutations are mitigated by the homeostatic mechanism that acts on expression of other GA dioxygenase and GID1 receptor genes.
DOI:10.1038/386485a0URLPMID:9087405 [本文引用: 1]
The domestication of crop plants has often involved an increase in apical dominance (the concentration of resources in the main stem of the plant and a corresponding suppression of axillary branches). A striking example of this phenomenon is seen in maize (Zea mays spp. mays), which exhibits a profound increase in apical dominance compared with its probable wild ancestor, teosinte (Zea mays ssp. parviglumis). Previous research has identified the teosinte branched1 (tb1) gene as a major contributor to this evolutionary change in maize. We have cloned tb1 by transposon tagging and show here that it encodes a protein with homology to the cycloidea gene of snapdragon. The pattern of tb1 expression and the morphology of tb1 mutant plants suggest that tb1 acts both to repress the growth of axillary organs and to enable the formation of female inflorescences. The maize allele of tb1 is expressed at twice the level of the teosinte allele, suggesting that gene regulatory changes underlie the evolutionary divergence of maize from teosinte.
DOI:10.1007/s00299-008-0543-8URL [本文引用: 1]
The number of viable shoots influences the overall architecture and productivity of wheat (Triticum aestivum L.). The development of lateral branches, or tillers, largely determines the resultant canopy. Tillers develop from the outgrowth of axillary buds, which form in leaf axils at the crown of the plant. Tiller number can be reduced if axillary buds are not formed or if the outgrowth of these buds is restricted. The teosinte branched1 (tb1) gene in maize, and homologs in rice and Arabidopsis, genetically regulate vegetative branching. In maize, increased expression of the tb1 gene restricts the outgrowth of axillary buds into lateral branches. In this study, the maize tb1 gene was introduced through transformation into the wheat cultivar “Bobwhite” to determine the effect of tb1 overexpression on wheat shoot architecture. Examination of multiple generations of plants reveals that tb1 overexpression in wheat results in reduced tiller and spike number. In addition, the number of spikelets on the spike and leaf number were significantly greater in tb1-expressing plants, and the height of these plants was also reduced. These data reveal that the function of the tb1 gene and genetic regulation of lateral branching via the tb1 mode of action is conserved between wheat, rice, maize and Arabidopsis. Thus, the tb1 gene can be used to alter plant architecture in agriculturally important crops like wheat.
DOI:10.1073/pnas.96.1.290URLPMID:9874811 [本文引用: 1]
The ability of the shoot apical meristem to multiply and distribute its meristematic potential through the formation of axillary meristems is essential for the diversity of forms and growth habits of higher plants. In the lateral suppressor mutant of tomato the initiation of axillary meristems is prevented, thus offering the unique opportunity to study the molecular mechanisms underlying this important function of the shoot apical meristem. We report here the isolation of the Lateral suppressor gene by positional cloning and show that the mutant phenotype is caused by a complete loss of function of a new member of the VHIID family of plant regulatory proteins.
DOI:10.1006/dbio.1999.9572URLPMID:10656774 [本文引用: 1]
Shoot development is reiterative: shoot apical meristems (SAMs) give rise to branches made of repeating leaf and stem units with new SAMs in turn formed in the axils of the leaves. Thus, new axes of growth are established on preexisting axes. Here we describe the formation of axillary meristems and floral meristems in Arabidopsis by monitoring the expression of the SHOOT MERISTEMLESS and AINTEGUMENTA genes. Expression of these genes is associated with SAMs and organ primordia, respectively. Four stages of axillary meristem development and previously undefined substages of floral meristem development are described. We find parallels between the development of axillary meristems and the development of floral meristems. Although Arabidopsis flowers develop in the apparent absence of a subtending leaf, the expression patterns of AINTEGUMENTA and SHOOT MERISTEMLESS RNAs during flower development suggest the presence of a highly reduced, &quot;cryptic&quot; leaf subtending the flower in Arabidopsis. We hypothesize that the STM-negative region that develops on the flanks of the inflorescence meristem is a bract primordium and that the floral meristem proper develops in the &quot;axil&quot; of this bract primordium. The bract primordium, although initially specified, becomes repressed in its growth.
DOI:10.1038/nature01518URLPMID:12687001 [本文引用: 1]
Tillering in rice (Oryza sativa L.) is an important agronomic trait for grain production, and also a model system for the study of branching in monocotyledonous plants. Rice tiller is a specialized grain-bearing branch that is formed on the unelongated basal internode and grows independently of the mother stem (culm) by means of its own adventitious roots. Rice tillering occurs in a two-stage process: the formation of an axillary bud at each leaf axil and its subsequent outgrowth. Although the morphology and histology and some mutants of rice tillering have been well described, the molecular mechanism of rice tillering remains to be elucidated. Here we report the isolation and characterization of MONOCULM 1 (MOC1), a gene that is important in the control of rice tillering. The moc1 mutant plants have only a main culm without any tillers owing to a defect in the formation of tiller buds. MOC1 encodes a putative GRAS family nuclear protein that is expressed mainly in the axillary buds and functions to initiate axillary buds and to promote their outgrowth.
DOI:10.1073/pnas.022516199URLPMID:11805344 [本文引用: 1]
The multitude of forms observed in flowering plants is largely because of their ability to establish new axes of growth during postembryonic development. This process is initiated by the formation of secondary meristems that develop into vegetative or reproductive branches. In the blind and torosa mutants of tomato, initiation of lateral meristems is blocked during shoot and inflorescence development, leading to a strong reduction in the number of lateral axes. In this study, it is shown that blind and torosa are allelic. The Blind gene has been isolated by positional cloning, and it was found that the mutant phenotype is caused by a loss of function of an R2R3 class Myb gene. RNA interference-induced blind phenocopies confirmed the identity of the isolated gene. Double mutant analysis shows that Blind acts in a novel pathway different from the one to which the previously identified Lateral suppressor gene belongs. The findings reported add a new class of transcription factors to the group of genes controlling lateral meristem initiation and reveal a previously uncharacterized function of R2R3 Myb genes.
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/nph.15632URLPMID:30536633 [本文引用: 1]
Plant architecture is the key factor affecting overall yield in many crops. The genetic basis underlying plant architecture in rapeseed (Brassica napus), a key global oil crop, is elusive. We characterized an ethyl methanesulfonate (EMS)-mutagenized rapeseed mutant, sca, which had multiple phenotypic alterations, including crinkled leaves, semi-dwarf stature, narrow branch angles and upward-standing siliques. We identified the underlying gene, which encodes an Aux/IAA protein (BnaA3.IAA7). A G-to-A mutation changed the glycine at the 84th position to glutamic acid (G84E), disrupting the conserved degron motif GWPPV and reducing the affinity between BnaA3.IAA7 and TIR1 (TRANSPORT INHIBITOR RESPONSE 1) in an auxin dosage-dependent manner. This change repressed the degradation of BnaA3.IAA7 and therefore repressed auxin signaling at low levels of auxin that reduced the length of internodes. The G84E mutation reduced branch angles by enhancing the gravitropic response. The heterozygote +/sca closely resembled a proposed ideal plant architecture, displaying strong yield heterosis through single-locus overdominance by improving multiple component traits. Our findings demonstrate that a weak gain-of-function mutation in BnaA3.IAA7 contributes to yield heterosis by improving plant architecture and would be valuable for breeding superior rapeseed hybrid cultivars and such a mutation may increase the yield in other Brassica crops.
DOI:10.1111/pbi.12894URLPMID:29406565 [本文引用: 1]
Capsaicinoids are unique compounds produced only in peppers (Capsicum spp.). Several studies using classical quantitative trait loci (QTLs) mapping and genomewide association studies (GWAS) have identified QTLs controlling capsaicinoid content in peppers; however, neither the QTLs common to each population nor the candidate genes underlying them have been identified due to the limitations of each approach used. Here, we performed QTL mapping and GWAS for capsaicinoid content in peppers using two recombinant inbred line (RIL) populations and one GWAS population. Whole-genome resequencing and genotyping by sequencing (GBS) were used to construct high-density single nucleotide polymorphism (SNP) maps. Five QTL regions on chromosomes 1, 2, 3, 4 and 10 were commonly identified in both RIL populations over multiple locations and years. Furthermore, a total of 109?610 SNPs derived from two GBS libraries were used to analyse the GWAS population consisting of 208 C.?annuum-clade accessions. A total of 69 QTL regions were identified from the GWAS, 10 of which were co-located with the QTLs identified from the two biparental populations. Within these regions, we were able to identify five candidate genes known to be involved in capsaicinoid biosynthesis. Our results demonstrate that QTL mapping and GBS-GWAS represent a powerful combined approach for the identification of loci controlling complex traits.
[本文引用: 2]
[本文引用: 2]
DOI:10.3389/fpls.2016.00017URLPMID:26858737 [本文引用: 1]
Seed yield (SY) is the most important trait in rapeseed, is determined by multiple seed yield-related traits (SYRTs) and is also easily subject to environmental influence. Many quantitative trait loci (QTLs) for SY and SYRTs have been reported in Brassica napus; however, no studies have focused on seven agronomic traits simultaneously affecting SY. Genome-wide QTL analysis for SY and seven SYRTs in eight environments was conducted in a doubled haploid population containing 348 lines. Totally, 18 and 208 QTLs for SY and SYRTs were observed, respectively, and then these QTLs were integrated into 144 consensus QTLs using a meta-analysis. Three major QTLs for SY were observed, including cqSY-C6-2 and cqSY-C6-3 that were expressed stably in winter cultivation area for 3 years and cqSY-A2-2 only expressed in spring rapeseed area. Trait-by-trait meta-analysis revealed that the 144 consensus QTLs were integrated into 72 pleiotropic unique QTLs. Among them, all the unique QTLs affected SY, except for uq.A6-1, including uq.A2-3, uq.C1-2, uq.C1-3, uq.C6-1, uq.C6-5, and uq.C6-6 could also affect more than two SYRTs. According to the constructed high-density consensus map and QTL comparison from literatures, 36 QTLs from five populations were co-localized with QTLs identified in this study. In addition, 13 orthologous genes were observed, including five each gene for SY and thousand seed weight, and one gene each for biomass yield, branch height, and plant height. The genomic information of these QTLs will be valuable in hybrid cultivar breeding and in analyzing QTL expression in different environments.
DOI:10.1007/s00122-017-2911-7URLPMID:28455767 [本文引用: 1]
A comprehensive linkage atlas for seed yield in rapeseed. Most agronomic traits of interest for crop improvement (including seed yield) are highly complex quantitative traits controlled by numerous genetic loci, which brings challenges for comprehensively capturing associated markers/genes. We propose that multiple trait interactions underlie complex traits such as seed yield, and that considering these component traits and their interactions can dissect individual quantitative trait loci (QTL) effects more effectively and improve yield predictions. Using a segregating rapeseed (Brassica napus) population, we analyzed a large set of trait data generated in 19 independent experiments to investigate correlations between seed yield and other complex traits, and further identified QTL in this population with a SNP-based genetic bin map. A total of 1904 consensus QTL accounting for 22 traits, including 80 QTL directly affecting seed yield, were anchored to the B. napus reference sequence. Through trait association analysis and QTL meta-analysis, we identified a total of 525 indivisible QTL that either directly or indirectly contributed to seed yield, of which 295 QTL were detected across multiple environments. A majority (81.5%) of the 525 QTL were pleiotropic. By considering associations between traits, we identified 25 yield-related QTL previously ignored due to contrasting genetic effects, as well as 31 QTL with minor complementary effects. Implementation of the 525 QTL in genomic prediction models improved seed yield prediction accuracy. Dissecting the genetic and phenotypic interrelationships underlying complex quantitative traits using this method will provide valuable insights for genomics-based crop improvement.
DOI:10.3724/SP.J.1006.2018.00533URL [本文引用: 2]
株高是油菜重要的农艺性状之一。以油菜品种Express、SWU07构建的包含261个株系的DH群体和由其构建的包含234个株系的IF2群体为材料, 分析2年环境下株高及其相关性状QTL表明, 在2个群体的各年份环境中总共检测到41个株高及其相关性状QTL, 分布于甘蓝型油菜的13条染色体上, 其中9个与株高相关的QTL, 分布于A02、A09、C01、C02和C06连锁群, 分别揭示了3.85%~13.34%的表型变异, 15个与主花序长度相关的QTL, 分布于A01、A02、A05、A08、A09、C01、C03和C05连锁群, 分别揭示了3.82%~9.52%的表型变异; 11个与第1分枝高度相关的QTL, 分布于A01、A03、A09、C01和C03连锁群, 分别揭示了4.01%~16.54%的表型变异; 4个与分枝区段长相关的QTL, 分布于甘蓝型油菜的A07、A09、C03和C04连锁群, 揭示了4.79%~8.10%的表型变异; 2个与平均节间长相关的QTL, 分布于A07和C05连锁群, 分别揭示了4.29%~6.04%的表型变异。其中5个QTL在不同年份环境或不同群体中被重复检测到。这些QTL为油菜株高的遗传改良提供了有用的信息。
DOI:10.3724/SP.J.1006.2018.00533URL [本文引用: 2]
株高是油菜重要的农艺性状之一。以油菜品种Express、SWU07构建的包含261个株系的DH群体和由其构建的包含234个株系的IF2群体为材料, 分析2年环境下株高及其相关性状QTL表明, 在2个群体的各年份环境中总共检测到41个株高及其相关性状QTL, 分布于甘蓝型油菜的13条染色体上, 其中9个与株高相关的QTL, 分布于A02、A09、C01、C02和C06连锁群, 分别揭示了3.85%~13.34%的表型变异, 15个与主花序长度相关的QTL, 分布于A01、A02、A05、A08、A09、C01、C03和C05连锁群, 分别揭示了3.82%~9.52%的表型变异; 11个与第1分枝高度相关的QTL, 分布于A01、A03、A09、C01和C03连锁群, 分别揭示了4.01%~16.54%的表型变异; 4个与分枝区段长相关的QTL, 分布于甘蓝型油菜的A07、A09、C03和C04连锁群, 揭示了4.79%~8.10%的表型变异; 2个与平均节间长相关的QTL, 分布于A07和C05连锁群, 分别揭示了4.29%~6.04%的表型变异。其中5个QTL在不同年份环境或不同群体中被重复检测到。这些QTL为油菜株高的遗传改良提供了有用的信息。
DOI:10.3389/fpls.2018.00390URLPMID:29643859 [本文引用: 1]
Plant height (PH), branch initiation height (BIH), and stem diameter (SD) are three stem-related traits that play crucial roles in plant architecture and lodging resistance. Herein, we show one doubled haploid (DH) population obtained from a cross between Y689 (one Capsella bursa-pastoris derived Brassica napus intertribal introgression) and Westar (B. napus cultivar) that these traits were significantly positively correlated with one another and with flowering time (FT). Based on a high-density SNP map, a total of 102 additive quantitative trait loci (QTL) were identified across six environments. Seventy-two consensus QTL and 49 unique QTL were identified using a two-round strategy of QTL meta-analysis. Notably, a total of 19 major QTL, including 11 novel ones, were detected for these traits, which comprised two QTL clusters on chromosomes A02 and A07. Conditional QTL mapping was performed to preliminarily evaluate the genetic basis (pleiotropy or tight linkage) of the co-localized QTL. In addition, QTL by environment interactions (QEI) mapping was performed to verify the additive QTL and estimate the QEI effect. In the genomic regions of all major QTL, orthologs of the genes involved in phytohormone biosynthesis, phytohormone signaling, flower development, and cell differentiation in Arabidopsis were proposed as candidate genes. Of these, BnaA02g02560, an ortholog of Arabidopsis GASA4, was suggested as a candidate gene for PH, SD, and FT; and BnaA02g08490, an ortholog of Arabidopsis GNL, was associated with PH, BIH and FT. These results provide useful information for further genetic studies on stem-related traits and plant growth adaptation.
DOI:10.3389/fpls.2017.01246URLPMID:28769955 [本文引用: 2]
Plant architecture is crucial for rapeseed yield and is determined by plant height (PH), branch initiation height (BIH), branch number (BN) and leaf and inflorescence morphology. In this study, we measured three major factors (PH, BIH, and BN) in a panel of 333 rapeseed accessions across 4 years. A genome-wide association study (GWAS) was performed via Q + K model and the panel was genotyped using the 60 k Brassica Infinium SNP array. We identified seven loci for PH, four for BIH, and five for BN. Subsequently, by determining linkage disequilibrium (LD) decay associated with 38 significant SNPs, we gained 31, 15, and 17 candidate genes for these traits, respectively. We also showed that PH is significantly correlated with BIH, while no other correlation was revealed. Notably, a GA signaling gene (BnRGA) and a flowering gene (BnFT) located on chromosome A02 were identified as the most likely candidate genes associated with PH regulation. Furthermore, a meristem initiation gene (BnLOF2) and a NAC domain transcriptional factor (BnCUC3) that may be associated with BN were identified on the chromosome A07. This study reveals novel insight into the genetic control of plant architecture and may facilitate marker-based breeding for rapeseed.
DOI:10.3389/fpls.2016.01102URLPMID:27512396 [本文引用: 2]
Plant height is a key morphological trait of rapeseed. In this study, we measured plant height of a rapeseed population across six environments. This population contains 476 inbred lines representing the major Chinese rapeseed genepool and 44 lines from other countries. The 60K Brassica Infinium? SNP array was utilized to genotype the association panel. A genome-wide association study (GWAS) was performed via three methods, including a robust, novel, nonparametric Anderson-Darling (A-D) test. Consequently, 68 loci were identified as significantly associated with plant height (P &lt; 5.22 × 10(-5)), and more than 70% of the loci (48) overlapped the confidence intervals of reported QTLs from nine mapping populations. Moreover, 24 GWAS loci were detected with selective sweep signals, which reflected the signatures of historical semi-dwarf breeding. In the linkage disequilibrium (LD) decay range up-and downstream of 65 loci (r (2) &gt; 0.1), we found plausible candidates orthologous to the documented Arabidopsis genes involved in height regulation. One significant association found by GWAS colocalized with the established height locus BnRGA in rapeseed. Our results provide insights into the genetic basis of plant height in rapeseed and may facilitate marker-based breeding.
DOI:10.1016/j.plantsci.2015.05.012URLPMID:26566834 [本文引用: 1]
Crop plant architecture plays a highly important role in its agronomic performance. Plant height (PH) and primary branch number (PB) are two major factors that affect the plant architecture of rapeseed (Brassica napus). Previous studies have shown that these two traits are controlled by multiple quantitative trait loci (QTL); however, QTLs have not been delimited to regions less than 10cM. Genome-wide association study (GWAS) is a highly efficient approach for identifying genetic loci controlling traits at relatively high resolution. In this study, variations in PH and PB of a panel of 472 rapeseed accessions that had previously been analyzed by a 60k SNP array were investigated for three consecutive years and studied by GWAS. Eight QTLs on chromosome A03, A05, A07 and C07 were identified for PH, and five QTLs on A01, A03, A07 and C07 were identified for PB. Although most QTLs have been detected in previous studies based on linkage analyses, the two QTLs of PH on A05 and the QTL of PB on C07 were novel. In the genomic regions close to the GWAS peaks, orthologs of the genes involved in flower development, phytohormone biosynthesis, metabolism and signaling in Arabidopsis were identified.
DOI:10.1038/s41467-019-09134-9URLPMID:30858362 [本文引用: 3]
Brassica napus (2n?=?4x?=?38, AACC) is an important allopolyploid crop derived from interspecific crosses between Brassica rapa (2n?=?2x?=?20, AA) and Brassica oleracea (2n?=?2x?=?18, CC). However, no truly wild B. napus populations are known; its origin and improvement processes remain unclear. Here, we resequence 588 B. napus accessions. We uncover that the A subgenome may evolve from the ancestor of European turnip and the C subgenome may evolve from the common ancestor of kohlrabi, cauliflower, broccoli, and Chinese kale. Additionally, winter oilseed may be the original form of B. napus. Subgenome-specific selection of defense-response genes has contributed to environmental adaptation after formation of the species, whereas asymmetrical subgenomic selection has led to ecotype change. By integrating genome-wide association studies, selection signals, and transcriptome analyses, we identify genes associated with improved stress tolerance, oil content, seed quality, and ecotype improvement. They are candidates for further functional characterization and genetic improvement of B. napus.
[本文引用: 1]
DOI:10.1007/s10142-013-0328-1URLPMID:23813016 [本文引用: 1]
Salinity is one of the major constraints adversely influencing crop productivity. Saltol QTL is a major QTL associated with Na?-K? ratio and seedling stage salinity tolerance in rice. With an aim to understand the contribution of individual genes localized within saltol towards salinity tolerance, we analysed the transcript abundance of a set of these genes in seedlings of contrasting genotypes of rice. We hypothesize that this approach may be helpful in identifying new 'candidate genes' for improving salinity tolerance in crops. For this purpose, seedlings of Oryza sativa cv. IR64 (sensitive) and the landrace Pokkali (tolerant) were subjected to short/long durations of salinity. qRT-PCR analysis clearly exhibited differential regulation of genes encoding signaling related protein (SRPs), where higher transcript abundance for most of them was observed in Pokkali than IR64 under non-stress conditions, thereby indicating towards well preparedness of the former to handle stress, in anticipation. Genes encoding proteins of unknown function (PUFs), though, constitute a considerable portion of plant genome, have so far been neglected in most studies. Time course analysis of these genes showed a continuous increase in their abundance in Pokkali, while in IR64, their abundance increased till 24 h followed by a clear decrease, thereby justifying their nomenclature as 'salinity induced factors' (SIFs). This is the first report showing possible involvement of SIFs localized within salinity related QTL towards salinity stress response. Based on the phenotypes of insertional mutants, it is proposed that these SIFs may have a putative function in vegetative growth (SIFVG), fertility (SIFF), viability (SIFV) or early flowering (SIFEF).
DOI:10.1038/ng1702URLPMID:16380716 [本文引用: 1]
As population structure can result in spurious associations, it has constrained the use of association studies in human and plant genetics. Association mapping, however, holds great promise if true signals of functional association can be separated from the vast number of false signals generated by population structure. We have developed a unified mixed-model approach to account for multiple levels of relatedness simultaneously as detected by random genetic markers. We applied this new approach to two samples: a family-based sample of 14 human families, for quantitative gene expression dissection, and a sample of 277 diverse maize inbred lines with complex familial relationships and population structure, for quantitative trait dissection. Our method demonstrates improved control of both type I and type II error rates over other methods. As this new method crosses the boundary between family-based and structured association samples, it provides a powerful complement to currently available methods for association mapping.
DOI:10.1038/srep36452URLPMID:27811979 [本文引用: 2]
Harvest index (HI), the ratio of seed mass to total biomass of the aboveground plant parts, is an important trait for harvestable yield of crops. Unfortunately, HI of Brassica napus is lower than that of other economically important crops. To identify candidate genes associated with high HI, a genome-wide association study of HI and four HI-related traits was conducted with 520 B. napus accessions cultivated in both Yunnan and Chongqing. We detected 294 single nucleotide polymorphisms significantly associated with the abovementioned traits, including 79 SNPs that affected two or more traits. Differentially expressed genes between extremely high- and low-HI accessions were identified in 8 tissues at two cultivated regions. Combination of linkage disequilibrium and transcriptome analyses revealed 33 functional candidate genes located within the confidence intervals of significant SNPs associated with more than one trait, such as SHOOT GRAVITROPISM 5 (Bna.SGR5), ATP-CITRATE LYASE A-3 (Bna.ACLA-3) and CAROTENOID CLEAVAGE DIOXYGENASE 1 (Bna.CCD1), their orthologs in the Arabidopsis thaliana have been shown to play key roles in photosynthesis, inflorescence, and silique development. Our results provide insight into the molecular mechanisms underlying establishment of high-HI B. napus and lay a foundation for characterization of candidate genes aimed at developing high-HI B. napus varieties.
DOI:10.1126/science.1253435URLPMID:25146293 [本文引用: 1]
Oilseed rape (Brassica napus L.) was formed ~7500 years ago by hybridization between B. rapa and B. oleracea, followed by chromosome doubling, a process known as allopolyploidy. Together with more ancient polyploidizations, this conferred an aggregate 72× genome multiplication since the origin of angiosperms and high gene content. We examined the B. napus genome and the consequences of its recent duplication. The constituent An and Cn subgenomes are engaged in subtle structural, functional, and epigenetic cross-talk, with abundant homeologous exchanges. Incipient gene loss and expression divergence have begun. Selection in B. napus oilseed types has accelerated the loss of glucosinolate genes, while preserving expansion of oil biosynthesis genes. These processes provide insights into allopolyploid evolution and its relationship with crop domestication and improvement.
DOI:10.3389/fpls.2018.01127URLPMID:30116254 [本文引用: 1]
Worldwide consumption of oil is increasing with the growing population in need for edible oil and the expansion of industry using biofuels. Then, demand for high yielding varieties of oil crops is always increasing. Brassica napus (rapeseed) is one of the most important oil crop in the world, therefore, increasing rapeseed yield through breeding is inevitable in order to cater for the high demand of vegetable oil and high-quality protein for live stocks. Quantitative trait loci (QTL) analysis is a powerful tool to identify important loci and which is also valuable for molecular marker assisted breeding. Seed-yield (SY) is a complex trait that is controlled by multiple loci and is affected directly by seed weight, seeds per silique and silique number. Some yield-related traits, such as plant height, biomass yield, flowering time, and so on, also affect the SY indirectly. This study reports the assembly of QTLs identified for seed-yield and yield-related traits in rapeseed, in one unique map. A total of 972 QTLs for seed-yield and yield-related were aligned into the physical map of B. napus Darmor-bzh and 92 regions where 198 QTLs overlapped, could be discovered on 16 chromosomes. Also, 147 potential candidate genes were discovered in 65 regions where 131 QTLs overlapped, and might affect nine different traits. At the end, interaction network of candidate genes was studied, and showed nine genes that could highly interact with the other genes, and might have more influence on them. The present results would be helpful to develop molecular markers for yield associated traits and could be used for breeding improvement in B. napus.
DOI:10.1534/genetics.109.101642URLPMID:19414564 [本文引用: 1]
Yield is the most important and complex trait for the genetic improvement of crops. Although much research into the genetic basis of yield and yield-associated traits has been reported, in each such experiment the genetic architecture and determinants of yield have remained ambiguous. One of the most intractable problems is the interaction between genes and the environment. We identified 85 quantitative trait loci (QTL) for seed yield along with 785 QTL for eight yield-associated traits, from 10 natural environments and two related populations of rapeseed. A trait-by-trait meta-analysis revealed 401 consensus QTL, of which 82.5% were clustered and integrated into 111 pleiotropic unique QTL by meta-analysis, 47 of which were relevant for seed yield. The complexity of the genetic architecture of yield was demonstrated, illustrating the pleiotropy, synthesis, variability, and plasticity of yield QTL. The idea of estimating indicator QTL for yield QTL and identifying potential candidate genes for yield provides an advance in methodology for complex traits.
DOI:10.1105/tpc.12.9.1751URLPMID:11006345 [本文引用: 1]
Arabinogalactan proteins (AGPs) are extracellular proteoglycans implicated in plant growth and development. We searched for classical AGPs in Arabidopsis by identifying expressed sequence tags based on the conserved domain structure of the predicted protein backbone. To confirm that these genes encoded bona fide AGPs, we purified native AGPs and then deglycosylated and deblocked them for N-terminal protein sequencing. In total, we identified 15 genes encoding the protein backbones of classical AGPs, including genes for AG peptides-AGPs with very short backbones (10 to 13 amino acid residues). Seven of the AGPs were verified as AGPs by protein sequencing. A gene encoding a putative cell adhesion molecule with AGP-like domains was also identified. This work provides a firm foundation for beginning functional analysis by using a genetic approach.
DOI:10.3724/SP.J.1006.2012.01802URL [本文引用: 1]
利用新版SG遗传图谱和282个SG-DH株系在中国西安、杭州和德国哥廷根3个生长环境下8个发育时期测定的株高数据,运用WinQTLCart2.5复合区间作图法以及结合条件遗传分析方法对其进行静态和动态QTL分析。结果显示, 来自品种Gaoyou等位基因在PHA3和PHC6两个QTL上同时存在时,可降低株高约20 cm;而当植株整合来自冬性品种Sollux的PHA9、PHC1和来自半冬性品种Gaoyou的PHA1、PHA3、PHC6时,株高可相应下降40 cm;环境对株高QTL的作用机理影响不大,但不同QTL的基因表达模式不同,存在来自双亲之一的等位基因控制株高和双亲等位基因在不同生长时期交替控制株高两种情况;通常株高QTL在中后期才能被检测到,但基因多在生长最为旺盛的短时期内表达,符合基因表达在先,性状表现在后的规律。解析株高性状在不同发育时期基因的累加效应和特定时段内的净表达效应,对克隆油菜株高基因和指导生产实践都将提供富有价值的科学信息和理论依据。
DOI:10.3724/SP.J.1006.2012.01802URL [本文引用: 1]
利用新版SG遗传图谱和282个SG-DH株系在中国西安、杭州和德国哥廷根3个生长环境下8个发育时期测定的株高数据,运用WinQTLCart2.5复合区间作图法以及结合条件遗传分析方法对其进行静态和动态QTL分析。结果显示, 来自品种Gaoyou等位基因在PHA3和PHC6两个QTL上同时存在时,可降低株高约20 cm;而当植株整合来自冬性品种Sollux的PHA9、PHC1和来自半冬性品种Gaoyou的PHA1、PHA3、PHC6时,株高可相应下降40 cm;环境对株高QTL的作用机理影响不大,但不同QTL的基因表达模式不同,存在来自双亲之一的等位基因控制株高和双亲等位基因在不同生长时期交替控制株高两种情况;通常株高QTL在中后期才能被检测到,但基因多在生长最为旺盛的短时期内表达,符合基因表达在先,性状表现在后的规律。解析株高性状在不同发育时期基因的累加效应和特定时段内的净表达效应,对克隆油菜株高基因和指导生产实践都将提供富有价值的科学信息和理论依据。
DOI:10.1104/pp.103.034736URLPMID:15047898 [本文引用: 1]
Although numerous physiological studies have addressed the interactions between brassinosteroids and auxins, little is known about the underlying molecular mechanisms. Using an Affymetrix GeneChip representing approximately 8,300 Arabidopsis genes, we studied comprehensive transcript profiles over 24 h in response to indole-3-acetic acid (IAA) and brassinolide (BL). We identified 409 genes as BL inducible, 276 genes as IAA inducible, and 637 genes in total. These two hormones regulated only 48 genes in common, suggesting that most of the actions of each hormone are mediated by gene expression that is unique to each. IAA-up-regulated genes were enriched in genes regulated in common. They were induced quickly by IAA and more slowly by BL, suggesting divergent physiological roles. Many were early auxin-inducible genes and their homologs, namely SAUR, GH3, and IAA. The comprehensive comparison also identified IAA- and BL-specific genes, which should help to elucidate the specific actions of each hormone. The identified genes were classified using hierarchical clustering based on the similarity of their responses to the two hormones. Gene classification also allowed us to analyze the frequency of cis-elements. The TGTCTC element, a core element of the previously reported auxin response element, was not enriched in genes specifically regulated by IAA but was enriched in the 5'-flanking region of genes up-regulated by both IAA and BL. Such gene classification should be useful for predicting the functions of unknown genes, to understand the roles of these two hormones, and the promoter analysis should provide insight into the interaction of transcriptional regulation by the two hormones.
DOI:10.1093/jxb/erp230URLPMID:19654206 [本文引用: 1]
The AUXIN RESPONSE FACTORs (ARFs) and the Aux/IAA proteins regulate various auxin responses through auxin perception mediated by the F-box proteins TIR1/AFBs. ARFs are transcription factors that modulate expression of auxin response genes and are negatively regulated by the Aux/IAA proteins. To gain insight into the regulatory mechanisms of Aux/IAA-ARF action at the genome level, the transcriptome regulated downstream of iaa1, a stabilized IAA1 mutant protein, was identified using dexamethasone (DEX)-controlled nuclear translocation of iaa1 during the auxin response. The expression of the iaa1-regulated auxin-responsive genes selected from microarray data was analysed with RNA-gel blot analysis and it was shown that auxin-regulated expression of these genes was significantly inhibited by DEX treatment. While cycloheximide-inducible expression of a majority of these genes was also DEX-suppressible, expression of some genes could not be suppressed by treatment with DEX. Expression analysis in a variety of arf mutant backgrounds suggested that all iaa1-regulated auxin-response genes examined are controlled by ARFs to different extents and that the same ARF protein can regulate the expression of these genes in response to auxin in a positive or a negative manner. However, arf mutations did not affect auxin-mediated down-regulation, indicating that ARFs might not play a critical role in down-regulation. The decrease in auxin-responsive gene expression in arf7 arf19 mutants was more severe than that of tir1/afb quadruple mutants. These results show the diversity and complexity of mechanisms of Aux/IAA-ARF- and auxin-regulated gene expression. These data also provide the opportunity for functional analysis of genes mediating the auxin-response downstream of Aux/IAA-ARFs.
DOI:10.1111/j.1365-313X.2004.02061.xURLPMID:15086809 [本文引用: 1]
We describe the development of a high-density Arabidopsis'whole genome' oligonucleotide probe array for expression analysis (the Affymetrix ATH1 GeneChip probe array) that contains approximately 22 750 probe sets. Precedence on the array was given to genes for which either expression evidence or a credible database match existed. The remaining space was filled with 'hypothetical' genes. The new ATH1 array represents approximately 23 750 genes of which 60% were detected in RNA from cultured seedlings. Sensitivity of the array, determined using spiking controls, was approximately one transcript per cell. The array demonstrated high technical reproducibility and concordance with real-time PCR results. Indole-3 acetic acid (IAA)-induced changes in gene expression were used for biological validation of the array. A total of 222 genes were significantly upregulated and 103 significantly downregulated by exposure to IAA. Of the genes whose products could be functionally classified, the largest specific classes of upregulated genes were transcriptional regulators and protein kinases, many fewer of which were represented among the downregulated genes. Over one-third of the auxin-regulated genes have no known function, although many belong to gene families with members that have previously been shown to be auxin regulated. For the 6714 genes represented both on this and the earlier Arabidopsis Genome (AG) array, both signal intensities and gene expression ratios were very similar. Mapping of the oligonucleotides on the ATH1 array to the latest (version 4.0) annotation showed that over 95% of the probe sets (based on version 2.0 annotation) still fully represented their original target genes.
DOI:10.1016/j.plantsci.2016.06.010URLPMID:27457991 [本文引用: 1]
Eukaryotic C3H-type zinc finger proteins (Znfs) comprise a large family of regulatory proteins involved in many aspects of plant stress response, growth and development. However, compared to mammalian, only a few plant Znfs have been functionally characterized. Here, T-DNA inserted gds1 (growth, development and splicing 1) mutant, displayed abnormal growth throughout the lifecycle owing to the reduction of cell size and number. Inverse PCR analysis revealed that the abnormal growth was caused by the disruption of At3g47120, which encodes a C3H42 protein belonging to the C-X7-C-X5-C-X3-H class of the Znf family. GDS1 was ubiquitously transcribed, but shows high levels of expression in young seedling and unexpanded new leaves. In gds1, the transcripts of many growth- and development-related genes were down-regulated, and the auxin response was dramatically reduced. A fluorescence-based assay revealed that the GDS1 protein was localized to the nucleus, prominently in the speckle compartments. Its arginine/serine dipeptide-rich-like (RS-like) domain was essential for nuclear localization. In addition, the SR1, SRm102 and U1-70K components of the U1 spliceosome interacted with GDS1 in the nuclear speckle compartments. Taken together, these suggest that GDS1, a nuclear-speckle-associated Znf, might play a significant role in splicing during plant growth and development.
DOI:10.1105/tpc.106.044743URLPMID:17172353 [本文引用: 1]
Transformation of plant cells with T-DNA of virulent agrobacteria is one of the most extreme triggers of developmental changes in higher plants. For rapid growth and development of resulting tumors, specific changes in the gene expression profile and metabolic adaptations are required. Increased transport and metabolic fluxes are critical preconditions for growth and tumor development. A functional genomics approach, using the Affymetrix whole genome microarray (approximately 22,800 genes), was applied to measure changes in gene expression. The solute pattern of Arabidopsis thaliana tumors and uninfected plant tissues was compared with the respective gene expression profile. Increased levels of anions, sugars, and amino acids were correlated with changes in the gene expression of specific enzymes and solute transporters. The expression profile of genes pivotal for energy metabolism, such as those involved in photosynthesis, mitochondrial electron transport, and fermentation, suggested that tumors produce C and N compounds heterotrophically and gain energy mainly anaerobically. Thus, understanding of gene-to-metabolite networks in plant tumors promotes the identification of mechanisms that control tumor development.
DOI:10.1007/s00299-006-0167-9URL [本文引用: 1]
Arabidopsis thaliana CEL1 protein was detected in young expanding tissues. Immunostaining revealed that CEL1 accumulated mostly in xylem cells. The primary, as well as the secondary xylem showed considerable CEL1 staining. CEL1 was also observed in young epidermal cells, in which the thicker lateral and tangential walls stained more intensely than the inner walls. In newly formed cell walls, the lateral tangential walls were labeled more intensively than the inner walls. Cellulase activity was found to be significantly higher in growing tissue compared to mature parts of the plant. Cel1 expression concurrently with cellulase activity could be restored in detached matured leaves by sucrose treatment after 48h in the culture medium.
DOI:10.1093/jxb/erv538URLPMID:26685188 [本文引用: 1]
Multicellular organisms co-ordinate cell proliferation and cell expansion to maintain organ growth. In animals, the Hippo tumor suppressor pathway is a master regulator of organ size. Central to this pathway is a kinase cascade composed of Hippo and Warts, and their activating partners Salvador and Mob1/Mats. In plants, the Mob1/Mats homolog MOB1A has been characterized as a regulator of cell proliferation and sporogenesis. Nonetheless, no Hippo homologs have been identified. Here we show that the Arabidopsis serine/threonine kinase 1 (SIK1) is a Hippo homolog, and that it interacts with MOB1A to control organ size. SIK1 complements the function of yeast Ste20 in bud site selection and mitotic exit. The sik1 null mutant is dwarf with reduced cell numbers, endoreduplication, and cell expansion. A yeast two-hybrid screen identified Mob1/Mats homologs MOB1A and MOB1B as SIK1-interacting partners. The interaction between SIK1 and MOB1 was found to be mediated by an N-terminal domain of SIK1 and was further confirmed by bimolecular fluorescence complementation. Interestingly, sik1 mob1a is arrested at the seedling stage, and overexpression of neither SIK1 in mob1a nor MOB1A in sik1 can rescue the dwarf phenotypes, suggesting that SIK1 and MOB1 may be components of a larger protein complex. Our results pave the way for constructing a complete Hippo pathway that controls organ growth in higher plants.
DOI:10.1038/ncomms13412URLPMID:27834370 [本文引用: 1]
Histone acetylation is known to affect the speed of seed germination, but the molecular regulatory basis of this remains ambiguous. Here we report that loss of function of two histone deacetylase-binding factors, SWI-INDEPENDENT3 (SIN3)-LIKE1 (SNL1) and SNL2, results in accelerated radicle protrusion and growth during seed germination. AUXIN RESISTANT 1 (AUX1) is identified as a key factor in this process, enhancing germination speed downstream of SNL1 and SNL2. AUX1 expression and histone H3 acetylation at lysines 9 and 18 is regulated by SNL1 and SNL2. The D-type cyclins encoding genes CYCD1;1 and CYCD4;1 display increased expression in AUX1 over-expression lines and the snl1snl2 double mutant. Accordingly, knockout of CYCD4;1 reduces seed germination speed of AUX1 over-expression lines and snl1snl2 suggesting the importance of cell cycling for radicle protrusion during seed germination. Together, our work identifies AUX1 as a link between histone acetylation mediated by SNL1 and SNL2, and radicle growth promoted by CYCD1;1 and CYCD4;1 during seed germination.
DOI:10.1104/pp.103.021436URLPMID:12913156 [本文引用: 1]
The phytohormone cytokinin is an important regulator of plant growth and development; however, relatively few genes that mediate cytokinin responses have been identified. Genome-wide analyses of Arabidopsis seedlings using the approximately 8,300-element Affymetrix Arabidopsis GeneChips (Affymetrix, Santa Clara, CA) to examine cytokinin-responsive genes were conducted, revealing at least 30 genes whose steady-state level of mRNA was elevated and at least 40 that were down-regulated at multiple time points after application of cytokinin. The cytokinin up-regulated genes include the type-A Arabidopsis response regulators (ARRs), which had been shown previously to be cytokinin primary response genes, cytokinin oxidase, which encodes an enzyme that degrades cytokinins, and several transcription factors. Cytokinin down-regulated genes include several peroxidases and kinases and an E3 ubiquitin ligase. We identified a common sequence motif enriched in the upstream regions of the most consistently cytokinin up-regulated genes. This motif is highly similar to the optimal DNA-binding sites for ARR1/ARR2, type-B ARRs that have been implicated in the transcriptional elevation of the type-A ARRs. Additionally, genome-wide analyses of cytokinin receptor mutants (wol/cre1) revealed large-scale changes in gene expression, including down-regulation of the type-A ARRs and several meristem and cell cycle genes, such as CycD3. Mutations in CRE1 reduced but did not eliminate the effect of cytokinin on gene expression for a subset of cytokinin-responsive genes and had little or no effect on others, suggesting functional redundancy among the cytokinin receptors.
DOI:10.1016/s0014-5793(02)03217-9URLPMID:12220656 [本文引用: 1]
The Arabidopsis genome contains four genes that encode proteins similar to both spermidine synthase and spermine synthase of other organisms. Our previous study revealed that one of these genes, designated ACAULIS5 (ACL5), encodes spermine synthase and that its null mutation results in a severe defect in the elongation of stem internodes. Here we report the characterization of the other three genes, designated SPDS1, SPDS2 and SPDS3. Our results showed that SPDS1 and SPDS2 possess spermidine synthase activity in yeast spermidine synthase-deficient mutants, but the enzyme activity of SPDS3 remained to be determined. RNA gel blot analysis revealed that all of these genes are expressed in all plant organs but show different responses to exogenous plant hormones, suggesting that they are involved in different aspects of growth by modulating the contents of polyamines in plant cells.
DOI:10.1007/s00425-010-1167-0URL [本文引用: 1]
The 4SN-Tudor domain protein is an almost ubiquitous eukaryotic protein with four Staphylococcal nuclease domains at the N terminus and a Tudor domain towards the C terminus. It has been found that Tudor-SN protein has multiple roles in governing gene expression during cell growth and development in animals. In plant, although Tudor-SN orthologs have been found in rice, pea and Arabidopsis, and are associated with cytoskeleton, their roles in growth and development are poorly understood. In this study, we investigated the function of Arabidopsis Tudor-SN protein, AtTudor. Our results indicated that the expression of AtTudor2 in seeds was evidently higher than in other tissues. Furthermore, we found that the expression of a key enzyme for GA biosynthesis, AtGA20ox3, was downregulated obviously in AtTudor2 T-DNA insertion mutant and AtTudor1/AtTudor2 RNAi transgenic lines. Together, our results suggest that AtTudor2 is involved in GA biosynthesis and seed germination of Arabidopsis.
DOI:10.1093/pcp/pcs014URL [本文引用: 1]
Unraveling the role of genes annotated as protein of unknown function is of importance in progression of plant science. l-Galactono-1,4-lactone (l-GalL) is the terminal precursor for ascorbic acid (AsA) biosynthesis in Arabidopsis thaliana, and a previous study showed two DUF (domains of unknown function) 642 family genes (At1g80240 and At5g25460, designated as DGR1 and DGR2, respectively) to be sensitive to it. In this work, leaves from wild-type Arabidopsis were fed with d-glucose, l-galactose, l-GalL and AsA, and the expression level of the At1g80240 and At5g25460 genes showed a specific response to l-GalL, but not to the other supplements despite the increases of the tissue AsA contents. Analysis of promoter-beta-glucuronidase (GUS) transgenic plants showed the two genes to be complementarily expressed at the root apex and in the rest of the root excluding the apex, respectively, in both young and old seedlings, and to be expressed at the leaf primordia. The GUS activity under the control of the At5g25460 promoter was high in the cotyledon and leaf veins of young seedlings. These findings were consistent with the results of quantitative real-time PCR. Interestingly, the T-DNA insertion mutant of At5g25460 (SALK_125079) displayed shorter roots and smaller rosettes than Col-0; however, no phenotypic difference was observed between the T-DNA insertion mutant of At1g80240 and the wild type. This is the first report on the expression and functional analysis of these two DUF642 family genes, with the results revealing the contribution of DGR genes to the development of Arabidopsis.
DOI:10.1093/jxb/eru409URLPMID:25371504 [本文引用: 1]
Senescence involves increased expression of proteases, which may participate in nitrogen recycling or cellular signalling. 2D zymograms detected two protein species with increased proteolytic activity in senescing leaves of Arabidopsis thaliana. A proteomic analysis revealed that both protein species correspond to a subtilisin protease encoded by At3g14067, termed Senescence-Associated Subtilisin Protease (SASP). SASP mRNA levels and enzyme activity increase during leaf senescence in leaves senescing during both the vegetative or the reproductive phase of the plant life cycle, but this increase is more pronounced in reproductive plants. SASP is expressed in all above-ground organs, but not in roots. Putative AtSASP orthologues were identified in dicot and monocot crop species. A phylogenetic analysis shows AtSASP and its putative orthologues clustering in one discrete group of subtilisin proteases in which no other Arabidospsis subtilisin protease is present. Phenotypic analysis of two knockout lines for SASP showed that mutant plants develop more inflorescence branches during reproductive development. Both AtSASP and its putative rice orthologue (OsSASP) were constitutively expressed in sasp-1 to complement the mutant phenotype. At maturity, sasp-1 plants produced 25% more inflorescence branches and siliques than either the wild-type or the rescued lines. These differences were mostly due to an increased number of second and third order branches. The increased number of siliques was compensated for by a small decrease (5.0%) in seed size. SASP downregulates branching and silique production during monocarpic senescence, and its function is at least partially conserved between Arabidopsis and rice.
DOI:10.3389/fpls.2015.00315URLPMID:26029221 [本文引用: 1]
Identifying the cell wall-ionically bound glycoside hydrolases (GHs) in Arabidopsis stems is important for understanding the regulation of cell wall integrity. For cell wall proteomics studies, the preparation of clean cell wall fractions is a challenge since cell walls constitute an open compartment, which is more likely to contain a mixture of intracellular and extracellular proteins due to cell leakage at the late growth stage. Here, we utilize a CaCl2-extraction procedure to isolate non-structural proteins from Arabidopsis whole stems, followed by the in-solution and in-gel digestion methods coupled with Nano-LC-MS/MS, bioinformatics and literature analyses. This has led to the identification of 75 proteins identified using the in-solution method and 236 proteins identified by the in-gel method, among which about 10% of proteins predicted to be secreted. Together, eight cell wall proteins, namely AT1G75040, AT5G26000, AT3G57260, AT4G21650, AT3G52960, AT3G49120, AT5G49360, and AT3G14067, were identified by the in-solution method; among them, three were the GHs (AT5G26000, myrosinase 1, GH1; AT3G57260, β-1,3-glucanase 2, GH17; AT5G49360, bifunctional XYL 1/α-L-arabinofuranosidase, GH3). Moreover, four more GHs: AT4G30270 (xyloglucan endotransferase, GH16), AT1G68560 (bifunctional α-l-arabinofuranosidase/XYL, GH31), AT1G12240 (invertase, GH32) and AT2G28470 (β-galactosidase 8, GH35), were identified by the in-gel solution method only. Notably, more than half of above identified GHs are xylan- or hemicellulose-modifying enzymes, and will likely have an impact on cellulose accessibility, which is a critical factor for downstream enzymatic hydrolysis of plant tissues for biofuels production. The implications of these cell wall proteins identified at the late growth stage for the genetic engineering of bioenergy crops are discussed.
DOI:10.1006/bbrc.2001.5875URLPMID:11689012 [本文引用: 1]
We are interested in identifying potential protein interactors of MADS domain transcription factors during Arabidopsis thaliana flower development. We based our biochemical search on a conserved motif in the MADS domain that includes putative phosphatase and phosphorylation sites that may mediate protein interactions. An affinity column with this motif and a few surrounding hypervariable amino acids derived from the AGAMOUS sequence was prepared and used to isolate potential interactors from floral crude extracts. Only two proteins were specifically bound to the affinity column. The first corresponds to a carpel specific storage protein, VSP1, that presents acid phosphatase activity, and the second is a novel leucine-rich repeat protein that we have named FLOR1. Coimmunoprecipitation, two-hybrid yeast, and affinity column assays show that the FLOR1-VSP1 complex interacts with AGAMOUS and that this transcription factor directly interacts with FLOR1. This is the first assay to show an interaction between plant MADS domain factors and non-MADS proteins.
DOI:10.1105/tpc.111.092791URL [本文引用: 1]
Flowering of Arabidopsis thaliana is induced by exposure to long days (LDs). During this process, the shoot apical meristem is converted to an inflorescence meristem that forms flowers, and this transition is maintained even if plants are returned to short days (SDs). We show that exposure to five LDs is sufficient to commit the meristem of SD-grown plants to flower as if they were exposed to continuous LDs. The MADS box proteins SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) and FRUITFULL (FUL) play essential roles in this commitment process and in the induction of flowering downstream of the transmissible FLOWERING LOCUS T (FT) signal. We exploited laser microdissection and Solexa sequencing to identify 202 genes whose transcripts increase in the meristem during floral commitment. Expression of six of these transcripts was tested in different mutants, allowing them to be assigned to FT-dependent or FT-independent pathways. Most, but not all, of those dependent on FT and its paralog TWIN SISTER OF FT (TSF) also relied on SOC1 and FUL. However, this dependency on FT and TSF or SOC1 and FUL was often bypassed in the presence of the short vegetative phase mutation. FLOR1, which encodes a leucine-rich repeat protein, was induced in the early inflorescence meristem, and flor1 mutations delayed flowering. Our data contribute to the definition of LD-dependent pathways downstream and in parallel to FT.
DOI:10.1016/j.plantsci.2004.03.009URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}