 ,1,2,*
,1,2,*Revealing the genetic diversity of wheat varieties (lines) in China based on SNP markers
LIU Yi-Ke1, ZHU Zhan-Wang1, CHEN Ling1, ZOU Juan1,2, TONG Han-Wen1, ZHU Guang1, HE Wei-Jie1, ZHANG Yu-Qing1, GAO Chun-Bao,1,2,*通讯作者:
收稿日期:2019-06-3接受日期:2019-08-9网络出版日期:2019-09-11
| 基金资助: |
Received:2019-06-3Accepted:2019-08-9Online:2019-09-11
| Fund supported: |
作者简介 About authors
E-mail:hbliuyk@foxmail.com。

摘要
关键词:
Abstract
Keywords:
PDF (1629KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
刘易科, 朱展望, 陈泠, 邹娟, 佟汉文, 朱光, 何伟杰, 张宇庆, 高春保. 基于SNP标记揭示我国小麦品种(系)的遗传多样性[J]. 作物学报, 2020, 46(2): 307-314. doi:10.3724/SP.J.1006.2020.91039
LIU Yi-Ke, ZHU Zhan-Wang, CHEN Ling, ZOU Juan, TONG Han-Wen, ZHU Guang, HE Wei-Jie, ZHANG Yu-Qing, GAO Chun-Bao.
遗传多样性一般指种内个体之间或一个群体内不同个体的遗传变异总和。丰富的植物遗传资源多样性为育种工作者改良品种和培育理想特性新品种提供了种质基础, 理想特性包括农民偏好的产量潜力大、大粒等性状和育种家偏好的抗病虫和光敏性等性状[1]。20世纪60年代中期, 在绿色革命的引领下, 培育出对高产和肥料有严格要求的矮秆品种, 这些品种大面积种植满足了人类对粮食不断增加的需求, 然而也导致绝大多数具有某些优良性状的地方品种被淘汰, 原始和广适性基因灭绝, 遗传多样性急剧下降[1]。
我国是世界上最大的小麦生产国和消费国, 其安全生产对我国粮食安全具有重要意义[2]。在小麦遗传改良中, 突破性遗传资源鉴定和利用与小麦品种选育取得突破性进展密切相关。20世纪60年代矮秆基因的利用, 使小麦在降低株高的同时, 单产也大幅度提高; 与抗病性相关小麦1B/1R易位系种质的应用, 培育出许多抗病高产的小麦品种[3]; 在我国, 优秀骨干亲本如碧蚂4号、南大2419和小偃6号等的利用, 使我国小麦单产得到大幅度提高[4], 促成了我国主要麦区小麦品种数次更新换代。然而, 与其他作物一样, 小麦的遗传多样性随着驯化和现代育种选择压力不断下降, 导致遗传差异较大、适应当地环境的地方品种数量减少和遗传变异性降低[5,6]。
研究小麦种质资源遗传多样性, 可为小麦育种的亲本选配提供理论支撑, 有利于突破性种质和新品种的选育。SNP (Single Nucleotide Polymorphism)标记能较真实反映小麦品种间的亲缘关系[7,8,9], 可用于小麦品种遗传多样性研究[10]。Würschum等[9]利用小麦9K SNP芯片和91对SSR标记对172份欧洲优异冬小麦品系进行遗传多样性和群体结构研究表明, 小麦SNP可以作为基因组学研究的有效方法, 并可作为小麦改良的重要工具。曹廷杰等[10]利用小麦高密度90K芯片对2000—2013年间河南省审品种和部分大面积种植的国审定品种进行遗传多样性分析, 表明参试品种间遗传相似度较高, 而且品种间遗传多样性有逐渐降低的趋势。Baloch等[11]利用39,568个DArTseq和20,661个SNP标记对91个硬粒小麦品种的遗传特性研究表明, 无意识的农民选择和商业品种的缺乏可能导致了遗传物质的交换, 这在土耳其和叙利亚的硬粒小麦的遗传结构中表现得尤为明显。宋晓朋等[12]利用小麦90K SNP芯片对黄淮麦区144个小麦品种遗传多样性分析表明黄淮麦区小麦品种可以分为5个类群, 品种的分类与其生态种植区域和亲缘关系存在一定程度的相关。Joukhadar等[13]利用小麦90K SNP芯片对澳大利亚1840—2011年间育成的482份小麦品种进行遗传多样性、种群结构和祖先起源分析阐明了澳大利亚小麦育种的进化历史, 并为小麦基因组如何适应当地的生长条件做出了解释。Turuspekov等[14]利用小麦高密度90K芯片对哈萨克斯坦已育成或有潜力的90份小麦品种进行遗产多样性分析, 表明小麦品种的分类与其地理位置有显著关联。Müller等[15]利用小麦高密度15K芯片对瑞士502份栽培品种、地方品种和育种品系的遗产多样性分析, 表明现代小麦品种遗传上与地方品种不同, 可能与早期育种中地方品种的组配丢失有关。
目前在我国小麦育种过程中, 越来越多的杂交组合选配围绕部分大面积推广应用的高产品种[2]进行, 我国幅员辽阔、生态环境多样, 不同麦区育成的品种众多, 而利用高通量SNP芯片对我国主要麦区小麦品种的系统性遗传多样性研究尚未见报道。本研究选取我国主要小麦产区育成的部分主要小麦品种(系)和少量国外品种, 利用小麦高通量SNP芯片技术揭示我国小麦品种(系)的遗传多样性和亲缘关系, 以期为小麦新品种选育的亲本选配、遗传研究以及种质资源创制提供理论依据。
1 材料与方法
1.1 参试品种
供试品种(系)有240个(附表1), 包含我国小麦主产区育成的一些主要小麦品种(系), 其中超过50个小麦品种在2000—2016年间的最大年种植面积超过10万公顷, 包括骨干亲本济麦22号、宁麦9号、小偃6号、良星99、矮抗58和周麦18等。按育成区域划分, 包括西南麦区17个(代表品种为川麦和绵麦系列品种等)、长江中下游麦区50个(代表品种为扬麦、宁麦和鄂麦系列品种等)、黄淮南片94个(代表品种为郑麦、周麦和西农系列品种等)、黄淮北片47个(代表品种为济麦、山农和衡观系列品种等)、北部麦区品种(系) 21个(代表品种为中麦和兰天系列品种等)和国外品种11个(代表品种为CIMMIT高代品系等)。所有材料由粮食作物种质创新与遗传改良湖北省重点实验室保存。Supplementary table 1
附表1
附表1用于SNP分析的小麦品种(系)
Supplementary table 1
| 序号 | 品种 | 麦区 | 序号 | 品种 | 麦区 | 序号 | 品种 | 麦区 |
|---|---|---|---|---|---|---|---|---|
| 4 | 川麦42 | 西南麦区 | 37 | 周麦23 | 黄淮南片 | 239 | 淮麦18 | 黄淮南片 |
| 16 | 内麦8号 | 西南麦区 | 42 | 众麦1号 | 黄淮南片 | 25 | 济麦20 | 黄淮北片 |
| 18 | 绵阳99-7 | 西南麦区 | 44 | 洛麦21 | 黄淮南片 | 32 | 烟农22 | 黄淮北片 |
| 23 | 绵麦42 | 西南麦区 | 45 | 豫麦49-168 | 黄淮南片 | 33 | 青丰1号 | 黄淮北片 |
| 38 | 绵麦1403 | 西南麦区 | 47 | 项麦99 | 黄淮南片 | 43 | 科农9204 | 黄淮北片 |
| 39 | 西科麦4号 | 西南麦区 | 48 | 皖麦52 | 黄淮南片 | 53 | 山东664 | 黄淮北片 |
| 40 | XK0106-1-0806 | 西南麦区 | 49 | 周麦22 | 黄淮南片 | 54 | 山农189 | 黄淮北片 |
| 56 | 川麦50 | 西南麦区 | 51 | 周麦16 | 黄淮南片 | 60 | 烟农24 | 黄淮北片 |
| 61 | 川麦52 | 西南麦区 | 52 | 豫农202 | 黄淮南片 | 64 | 泰山21 | 黄淮北片 |
| 62 | 西科麦2号 | 西南麦区 | 55 | 豫麦52 | 黄淮南片 | 67 | 临汾138 | 黄淮北片 |
| 83 | 川麦51 | 西南麦区 | 57 | 新麦18 | 黄淮南片 | 68 | 邯3475 | 黄淮北片 |
| 138 | 西科麦5号 | 西南麦区 | 65 | 西农979 | 黄淮南片 | 69 | 邯6172 | 黄淮北片 |
| 199 | 川农42 | 西南麦区 | 71 | 荔垦2号 | 黄淮南片 | 70 | 冀麦30 | 黄淮北片 |
| 211 | 绵麦37 | 西南麦区 | 72 | 徐麦29 | 黄淮南片 | 75 | 烟5158 | 黄淮北片 |
| 212 | 川麦43 | 西南麦区 | 73 | 温麦6号 | 黄淮南片 | 76 | 临Y867 | 黄淮北片 |
| 215 | 绵麦185 | 西南麦区 | 74 | 新麦20 | 黄淮南片 | 78 | 济宁16号 | 黄淮北片 |
| 226 | 绵阳99-3 | 西南麦区 | 79 | 陕715 | 黄淮南片 | 85 | 科农199 | 黄淮北片 |
| 1 | 鄂麦596 | 长江中下游麦区 | 80 | 皖麦38 | 黄淮南片 | 87 | 良星99 | 黄淮北片 |
| 2 | 扬05-117 | 长江中下游麦区 | 81 | 温麦7号 | 黄淮南片 | 91 | 烟农19 | 黄淮北片 |
| 6 | 鄂麦27 | 长江中下游麦区 | 82 | 豫麦48 | 黄淮南片 | 92 | 长6359 | 黄淮北片 |
| 7 | 扬06-144 | 长江中下游麦区 | 84 | 新麦22 | 黄淮南片 | 98 | 临汾137 | 黄淮北片 |
| 13 | 荆辐麦1号 | 长江中下游麦区 | 86 | 连麦1号 | 黄淮南片 | 103 | 冀麦38 | 黄淮北片 |
| 14 | 扬麦12 | 长江中下游麦区 | 88 | 双抗7438 | 黄淮南片 | 107 | 石家庄8号 | 黄淮北片 |
| 21 | 鄂恩1号 | 长江中下游麦区 | 93 | 西农3517 | 黄淮南片 | 110 | 济麦21 | 黄淮北片 |
| 28 | 鄂麦580 | 长江中下游麦区 | 96 | 洛早7 | 黄淮南片 | 143 | 邯5316 | 黄淮北片 |
| 29 | CJ9306 | 长江中下游麦区 | 100 | 温麦18 | 黄淮南片 | 145 | 潍麦8号 | 黄淮北片 |
| 30 | 襄麦55 | 长江中下游麦区 | 101 | 温麦19 | 黄淮南片 | 149 | 泰农18 | 黄淮北片 |
| 36 | 鄂恩6号 | 长江中下游麦区 | 106 | 郑麦9023 | 黄淮南片 | 150 | 山农16 | 黄淮北片 |
| 41 | 扬06G5 | 长江中下游麦区 | 109 | 西农9871 | 黄淮南片 | 152 | 衡观115 | 黄淮北片 |
| 58 | 宁7840 | 长江中下游麦区 | 112 | 新麦11 | 黄淮南片 | 156 | 衡观4422 | 黄淮北片 |
| 59 | 扬麦158 | 长江中下游麦区 | 113 | 淮麦20 | 黄淮南片 | 164 | 荷麦13 | 黄淮北片 |
| 66 | 扬07-49 | 长江中下游麦区 | 115 | 漯6010 | 黄淮南片 | 165 | 泰农142 | 黄淮北片 |
| 77 | 鄂麦23 | 长江中下游麦区 | 116 | 新麦9817 | 黄淮南片 | 177 | 临Y7287 | 黄淮北片 |
| 89 | 镇麦6号 | 长江中下游麦区 | 117 | 新麦13 | 黄淮南片 | 179 | 济麦22 | 黄淮北片 |
| 95 | 宁麦16 | 长江中下游麦区 | 119 | 陕麦150 | 黄淮南片 | 180 | 山农15 | 黄淮北片 |
| 97 | 鄂恩5号 | 长江中下游麦区 | 120 | 周麦18 | 黄淮南片 | 184 | 临优2069 | 黄淮北片 |
| 102 | 镇麦5号 | 长江中下游麦区 | 122 | 豫农035 | 黄淮南片 | 191 | 烟5286 | 黄淮北片 |
| 105 | 鄂麦12 | 长江中下游麦区 | 126 | 小偃6号 | 黄淮南片 | 192 | 衡观35 | 黄淮北片 |
| 111 | 扬06G86 | 长江中下游麦区 | 128 | 郑育麦9987 | 黄淮南片 | 194 | 衡观136 | 黄淮北片 |
| 118 | 扬06-164 | 长江中下游麦区 | 130 | 洛新998 | 黄淮南片 | 200 | 烟农21 | 黄淮北片 |
| 123 | 扬麦15 | 长江中下游麦区 | 132 | 泛麦5号 | 黄淮南片 | 203 | 济麦17 | 黄淮北片 |
| 124 | 华2533 | 长江中下游麦区 | 136 | 漯4-168 | 黄淮南片 | 209 | 鲁麦21号 | 黄淮北片 |
| 125 | 宁麦13 | 长江中下游麦区 | 139 | 新1817 | 黄淮南片 | 213 | 济南17 | 黄淮北片 |
| 127 | 扬07-129 | 长江中下游麦区 | 140 | 许科1号 | 黄淮南片 | 217 | 济麦19 | 黄淮北片 |
| 131 | 扬07-44 | 长江中下游麦区 | 142 | 郑麦004 | 黄淮南片 | 219 | 山农8355 | 黄淮北片 |
| 133 | 苏麦3号 | 长江中下游麦区 | 147 | 陕麦139 | 黄淮南片 | 221 | 烟2415 | 黄淮北片 |
| 134 | 华2566 | 长江中下游麦区 | 148 | 04中36 | 黄淮南片 | 227 | 鲁农116 | 黄淮北片 |
| 137 | 宁麦9号 | 长江中下游麦区 | 151 | 西农2000 | 黄淮南片 | 232 | 泰山23 | 黄淮北片 |
| 141 | 宁麦8号 | 长江中下游麦区 | 154 | 豫麦49-198 | 黄淮南片 | 8 | 兰天13 | 北部麦区 |
| 146 | 镇麦168 | 长江中下游麦区 | 155 | 豫麦69 | 黄淮南片 | 9 | 兰天17号 | 北部麦区 |
| 157 | 扬麦13 | 长江中下游麦区 | 161 | 郑麦98 | 黄淮南片 | 15 | 宁春47号 | 北部麦区 |
| 158 | 襄麦25 | 长江中下游麦区 | 162 | 新9817 | 黄淮南片 | 24 | 宁冬11号 | 北部麦区 |
| 160 | 扬07-15 | 长江中下游麦区 | 163 | 矮早781-99 | 黄淮南片 | 46 | 兰天23号 | 北部麦区 |
| 167 | 鄂麦11 | 长江中下游麦区 | 168 | 小偃166 | 黄淮南片 | 50 | 宁冬10号 | 北部麦区 |
| 170 | 扬麦14 | 长江中下游麦区 | 169 | 连麦2号 | 黄淮南片 | 94 | 兰天18号 | 北部麦区 |
| 171 | 鄂352 | 长江中下游麦区 | 172 | 郑麦366 | 黄淮南片 | 104 | 兰天15号 | 北部麦区 |
| 174 | 荆州66 | 长江中下游麦区 | 173 | 西农88 | 黄淮南片 | 108 | 京冬17 | 北部麦区 |
| 175 | 鄂麦18 | 长江中下游麦区 | 176 | 陕农78 | 黄淮南片 | 121 | 北京0045 | 北部麦区 |
| 181 | 扬辐麦2号 | 长江中下游麦区 | 182 | 陕农757 | 黄淮南片 | 129 | 中麦12号 | 北部麦区 |
| 183 | 扬麦16 | 长江中下游麦区 | 186 | 陕159 | 黄淮南片 | 144 | 中农2号 | 北部麦区 |
| 185 | 扬麦11 | 长江中下游麦区 | 188 | 郑农17 | 黄淮南片 | 178 | 轮选987 | 北部麦区 |
| 198 | 荆麦103 | 长江中下游麦区 | 190 | 豫麦70 | 黄淮南片 | 187 | 京冬8号 | 北部麦区 |
| 204 | 鄂07901 | 长江中下游麦区 | 193 | 开麦18 | 黄淮南片 | 214 | 兰天12 | 北部麦区 |
| 207 | 扬麦17 | 长江中下游麦区 | 195 | 豫麦10号 | 黄淮南片 | 216 | 兰天21号 | 北部麦区 |
| 231 | Wuhan 1 | 长江中下游麦区 | 196 | 洛早2 | 黄淮南片 | 218 | 兰天00-30 | 北部麦区 |
| 233 | 宁麦11 | 长江中下游麦区 | 197 | 平安3号 | 黄淮南片 | 225 | 宁春4号 | 北部麦区 |
| 240 | 扬07-81 | 长江中下游麦区 | 201 | 百农160 | 黄淮南片 | 230 | 宁春43号 | 北部麦区 |
| 3 | 偃展4110 | 黄淮南片 | 202 | 陕627 | 黄淮南片 | 234 | 兰天22号 | 北部麦区 |
| 5 | 陕农138 | 黄淮南片 | 205 | 小偃107 | 黄淮南片 | 237 | 中麦9号 | 北部麦区 |
| 11 | 洛早6 | 黄淮南片 | 206 | 豫麦38 | 黄淮南片 | 10 | SYN1 | 国外 |
| 12 | 小偃22 | 黄淮南片 | 208 | 新麦19 | 黄淮南片 | 63 | 14FHBSN6404 | 国外 |
| 17 | 新麦16 | 黄淮南片 | 210 | 淮麦17 | 黄淮南片 | 90 | 14FHBSN6418 | 国外 |
| 19 | 平安6号 | 黄淮南片 | 220 | 中育10号 | 黄淮南片 | 99 | 14FHBSN6405 | 国外 |
| 20 | 金丰3号 | 黄淮南片 | 222 | 陕253 | 黄淮南片 | 114 | 14FHBSN6402 | 国外 |
| 22 | 豫麦70-36 | 黄淮南片 | 223 | 陕麦175 | 黄淮南片 | 135 | GAMENYA | 国外 |
| 26 | 郑麦9094 | 黄淮南片 | 224 | 新麦208 | 黄淮南片 | 153 | 14FHBSN6409 | 国外 |
| 27 | 郑育麦958 | 黄淮南片 | 228 | 皖麦50 | 黄淮南片 | 159 | Ocoroni | 国外 |
| 31 | 科大9612 | 黄淮南片 | 229 | 矮抗58 | 黄淮南片 | 166 | 14FHBSN6411 | 国外 |
| 34 | 徐麦27 | 黄淮南片 | 236 | 徐麦216 | 黄淮南片 | 189 | 14FHBSN6408 | 国外 |
| 35 | 周麦17 | 黄淮南片 | 238 | 濮麦10号 | 黄淮南片 | 235 | Mayoor | 国外 |
新窗口打开|下载CSV
1.2 SNP芯片分型
选取小麦幼苗叶片, 按Saghai-Maroof等[16]的CTAB法提取基因组DNA。利用Illumina 90K iSelect SNP标记对240个小麦品种进行全基因组扫描[17], 由美国农业部农业研究服务小谷物基因分型实验室(美国北达科他州立大学)采用Illumina’s GenomeStudio Software (https://www.illumina.com/ techniques/microarrays/array-data-analysis-experimentaldesign/ genomestudio.html)进行样本的原始SNP分型。1.3 SNP位点的多态性信息含量分析
利用多态性信息含量(polymorphism information content, PIC)对SNP位点的遗传多样性进行评价。按照Powermarker v3.25软件的格式要求对芯片分型数据进行整理并导入计算, 单个SNP位点的PIC值按照公式PIC = 1-∑Pij2计算, Pij表示第i个标记第j个等位变异出现的频率。1.4 品种(系)的遗传多样性及聚类分析
将SNP分型数据转换为0、1二元数据格式, 建立原始矩阵。用NTSYS-PC2.1统计软件计算品种(系)间遗传相似系数。采用非加权配对算术平均法(UPGMA)构建遗传关系树状图, 结合iTOL (https://itol.embl.de/tree/)网站进行聚类图的修饰和亚群区分。1.5 主成分分析
利用TASSEL5.2.51对供试品种进行主成分分析(principle component analysis, PCA), 利用ggplot2工具绘制主成分分析图。2 结果与分析
2.1 多态性SNP位点的分布
芯片中分布于21条染色体的81,587个SNP位点中, 22,904个具备多态性, 多态性比率为28.07%, 其中18,741个被定位于明确的染色体上并用于后续分析。每条染色体分布76~2038个多态性位点(图1-A), 差异较大, 4D上差异位点最少, 而1B上差异位点最多。同时, 多态性位点在A、B、D基因组上分布不均, 以B基因组最多, 为9376个, 占50.03%, A基因组上多态性位点为7672个, 占40.94%, 6A较多, 4A最少; D基因组上多态性位点最少, 仅有1693个, 占9.03%。图1
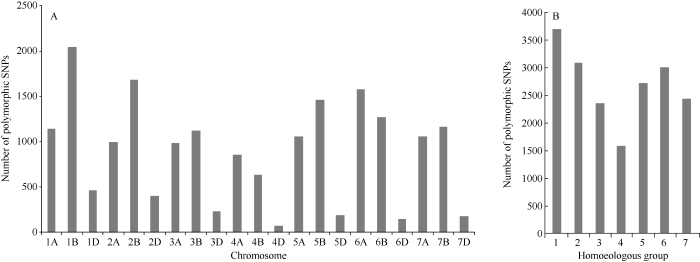 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1多态性SNP位点在染色体(A)和同源群(B)上的分布
Fig. 1Distribution of polymorphic SNPs on individual chromosome (A) and each homoeologous group (B)
多态性SNP位点在7个部分同源群中分布不均, 在第1和第2部分同源群上分布最多, 在第4部分同源群最少(图1-B)。
2.2 SNP位点的多态性信息含量分析
利用18,741个SNP分子标记, 在240份小麦材料中检测到37,482个等位变异, 每个位点均检测到2个等位变异。SNP分子标记多态性信息含量即PIC值的变幅为0.01~0.38, 平均值为0.26, 主要分布于0.10~0.38之间(图2)。图2
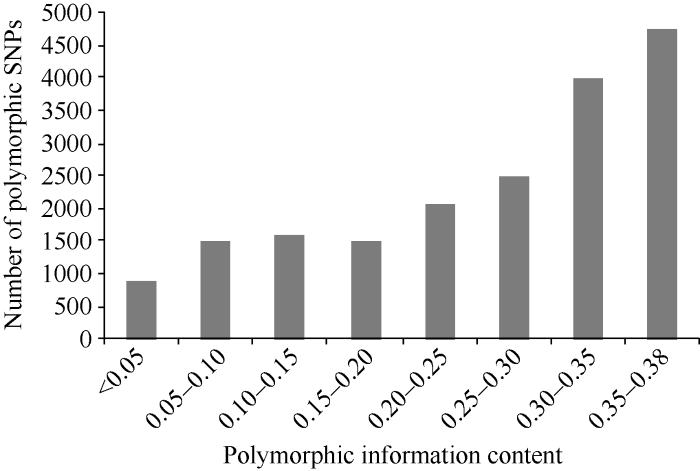 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2SNP标记在240份小麦材料中多态性信息含量(PIC)的分布
Fig. 2Distribution of polymorphic information content in 240 elite wheat lines based on SNP marker
2.3 小麦品种(系)间的遗传相似系数
利用NTSYS-PC2.1统计软件, 对240个品种(系)相互间的相似系数进行计算, 共获得28,680个遗传相似系数, 其平均变异系数为0.656, 变异范围为0.133~0.998 (表1)。品种(系)间的遗传系数主要集中在0.60~0.78之间, 有24,965个, 占87.05%, 而小于0.6的有2974个, 占10.37%, 而大于0.78的最少, 只有741个, 占2.58% (图3)。Table 1
表1
表1不同地区育成品种(系)的遗传相似系数
Table 1
| 地区 Region | 品种数 No. of varieties | 遗传相似系数 Genetic similarity | |
|---|---|---|---|
| 变幅 Range | 均值 Average | ||
| 西南麦区 Wheat region of southwest China | 17 | 0.609-0.983 | 0.718 |
| 长江中下游麦区Wheat region of the middle and lower reaches of Yangtze River | 50 | 0.161-0.998 | 0.712 |
| 黄淮南片 Wheat region of South Huang-Huai | 94 | 0.146-0.998 | 0.686 |
| 黄淮北片 Wheat region of North Huang-Huai | 47 | 0.559-0.991 | 0.680 |
| 北部麦区 Wheat region of Northern China | 21 | 0.538-0.989 | 0.660 |
| 国外 Abroad | 11 | 0.262-0.861 | 0.552 |
| 总计Total | 240 | 0.133-0.998 | 0.656 |
新窗口打开|下载CSV
图3
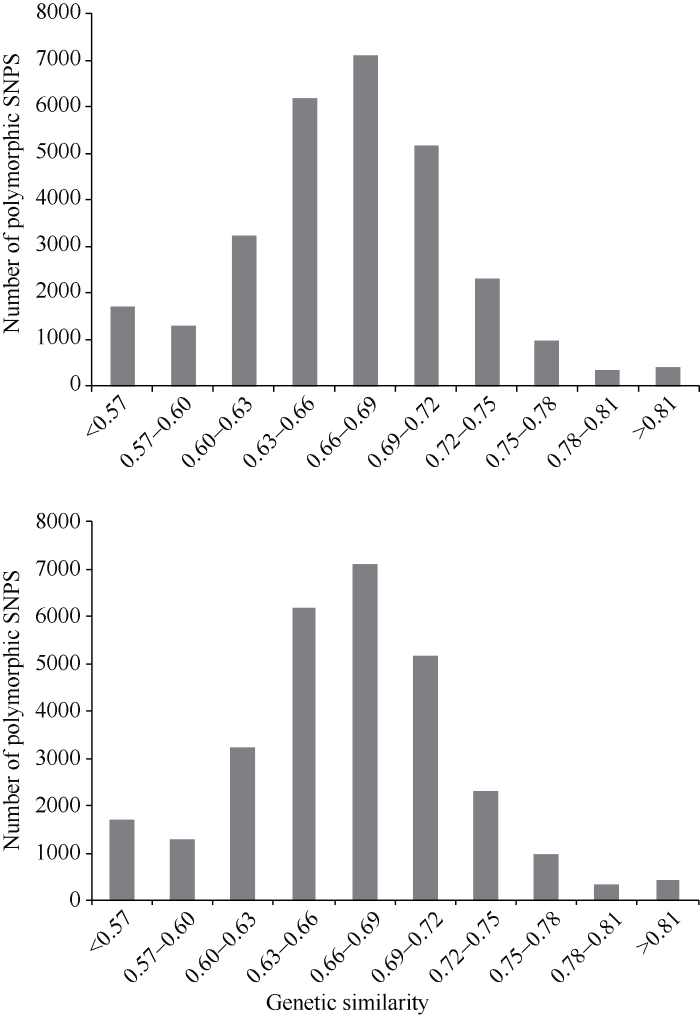 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3240个品种(系)间遗传相似系数的次数分布
Fig. 3Distribution of genetic similarity between 240 varieties (lines)
为了探讨我国不同麦区小麦品种(系)遗传多样性, 将240个品种(系)按育成区域分组, 在组内进行品种(系)间的遗传相似性分析。结果表明(表1), 西南麦区品种(系)间的平均相似系数最高, 为0.718, 其次为长江中下游麦区, 为0.708; 北部麦区、黄淮北片和黄淮南片品种(系)间的平均相似系数相对最低, 分别0.660、0.680和0.686; 国外品种(系)间的平均相似系数最低, 为0.552。
2.4 聚类分析
为探明240个品种(系)之间的亲缘关系, 根据品种间遗传相似系数, 采用非加权配对算术平均法(UPGMA)构建遗传关系树状图。聚类分析结果表明(图3), 240个品种(系)被划分为7个类群。第I类有56个品种(系), 主要包含长江中下游麦区品种(系) 38个和西南麦区品种(系) 14个。鄂麦11号是鄂麦18的亲本之一, 两者遗传相似系数较高为0.998; 扬麦11号与扬麦12号有共同的亲本扬麦158和85-853, 两者遗传相似系数较高为0.989; 宁麦16号与镇麦5号有共同的亲本宁麦9号, 两者遗传相似系数较高为0.986。
第II类和第III类分别有13个和7个品种(系), 第II类主要包含黄淮南片麦区品种(系) 9个和黄淮北片麦区品种(系)2个, 第III类主要包含黄淮南片麦区品种(系) 5个。
第IV类有34个品种(系), 主要包含黄淮南片麦区品种(系) 31个。其中偃展4110与新麦208有共同的亲本豫麦18, 两者遗传相似系数较高, 为0.992。
第V类有41个品种(系), 包含黄淮北片麦区品种(系) 29个、黄淮南片麦区品种(系) 7个和北部麦区品种(系) 5个。其中宁冬10号与宁冬11号均有国外品种血缘和共同的国内亲本北农2号, 两者的遗传相似系数较高, 为0.989。
第VI类含有17个品种(系), 包含国外品种(系)11个和宁春47号、宁春4号、宁春43号、宁麦13号、小偃6号鄂07901六个国内品种(系)。其中国内品种中宁春系列品种含有CIMMYT品种Sonoro-64的血缘, 宁麦13号含有日本品种西风的血缘, 小偃6号具有意大利小麦品种郑引4号(St2422/464)的血缘, 鄂07901含有CIMMYT品种的血缘。国外小麦品种Mayoor是SYN1的亲本之一, 两者遗传相似系数较高, 为0.861。
第VII类有72个品种(系), 该类品种分布比较广泛, 包含黄淮南片麦区品种(系) 35个、黄淮北片麦区品种(系) 13个、北部麦区品种(系) 11个、长江中下游麦区品种(系) 6个和西南麦区品种(系) 3个。豫麦70和豫麦70-36优系的相似系数为0.971, 豫麦49-197和豫麦49-168两个豫麦49改良优系的相似系数为0.998。
2.5 主成分分析
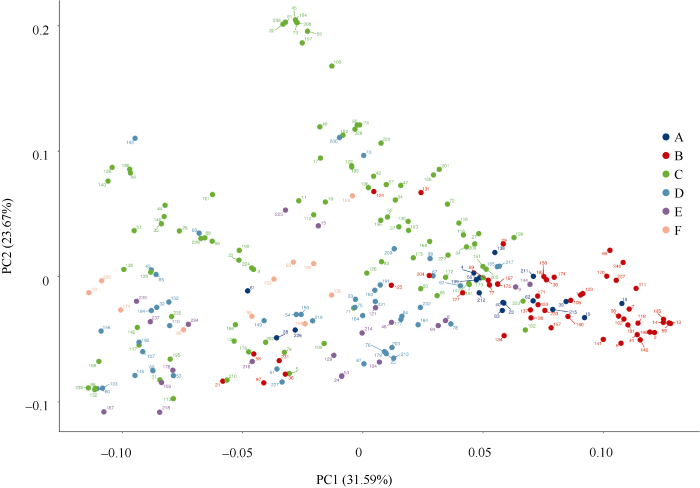
为反映不同群体间的遗传关系, 基于SNP标记对供试品种分组进行主坐标分析。结果表明(图5), 前2个坐标能够解释的遗传变异分别为31.59%和23.67%; 长江中下游麦区和西南麦区的大部分品种(系)聚在一起, 侧面验证了这2个地区的小麦品种(系)的遗传相似性较高(表1); 黄淮南片、黄淮北片和北部麦区的品种(系)在坐标图中分布广泛, 侧面验证了这些地区的小麦品种(系)的遗传相似性相对较低(表1); 供试材料中国外品种(系)的样品量虽少, 但分布也较为广泛, 故遗传相似性相对较低(表1)。品种(系)间的遗传相似性在主成分分析图中也得到了验证, 例如鄂麦11号(167)和鄂麦18号(175)、扬麦11号(185)和扬麦12号(14)、偃展4110 (3)和新麦208 (224)以及宁冬11号(24)和宁冬10号(50)等相似性较高的品种(图4), 在主成分分析图中也聚集在一起(图5)。
图4
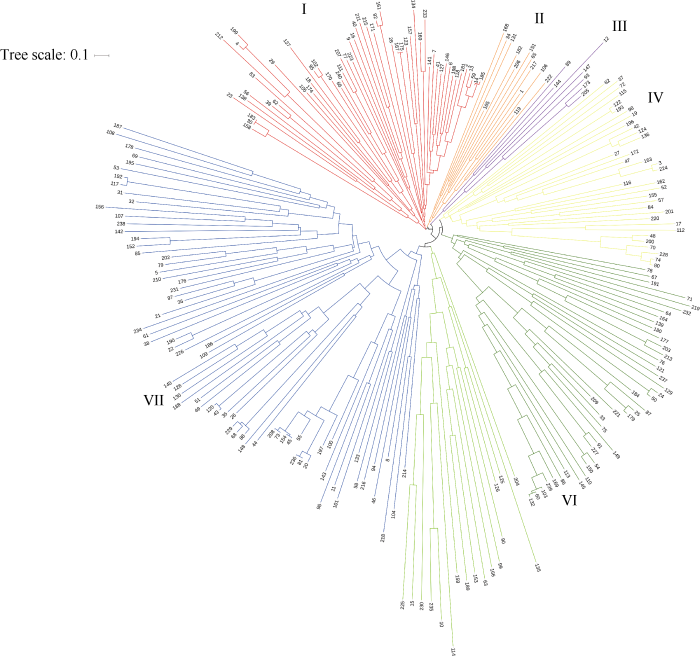 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4基于SNP标记的240个小麦品种(系)的聚类图
Fig. 4Clustering graph for 240 wheat varieties (lines) based on SNP markers analysis
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5基于SNP标记的240个小麦品种(系)主成分分析
A为西南麦区品种, B为长江中下游麦区品种, C为黄淮南片品种, D为黄淮北片品种, E为北部麦区品种, F为国外品种。
Fig. 5Principle component analysis (PCA) of 240 wheat varieties (lines) based on SNP markers
A: wheat varieties (lines) from Southwest China; B: wheat varieties (lines) from the middle and lower reaches of Yangtze River; C: wheat varieties (lines) from South Huang-Huai; D: wheat varieties (lines) from North Huang-Huai, China; E: wheat varieties (lines) from Northern China; E: wheat varieties (lines) from Foreign.
3 讨论
遗传多样性分析对植物种质资源的评价与利用、新品种培育以及新基因的定位克隆具有重要意义, 是植物遗传学、育种、保护和进化的重要组成部分[5]。以往的研究也发现半个多世纪的育种选择使中国小麦品种的遗传基础日趋狭窄[18]。由于品种审定制度和商业化育种对多出快出品种的要求, 小麦育种家过多关注产量、抗病性、品质等性状, 越来越多的杂交组合选配围绕部分大面积推广应用的高产品种[2]。以山东省2018年审定的小麦新品种为例, 在29个小麦新品种中, 单交育成品种有25个, 24个品种至少有一个亲本为大面积推广应用的高产品种, 其中矮抗58和良星99作为亲本之一育成的品种多达8个, 9个品种的亲本均为已育成品种。在小麦种质资源创新未取得突破性进展的情况下, 以现有品种(系)为亲本配制杂交组合的育种手段, 短期看来难以改观。本研究利用小麦高通量SNP芯片技术对包括矮抗58和良星99在内的我国主要小麦产区育成的代表性小麦品种(系)进行遗传多样性和亲缘关系研究, 可为现有条件下小麦新品种培育的亲本选配提供参考, 尽可能避免亲缘关系过近品种(系)间杂交, 有助于丰富育成品种的遗传多样性, 提高品种抵抗不良环境的能力。研究表明, 我国不同区域的小麦品种都不同程度存在亲缘关系较近和遗传多样性不足的情况[10,12,19-23]。蒲艳艳等[19]利用68对SSR引物对44个山东小麦品种进行遗传多样性分析品种间的平均遗传相似系数为0.694, 变幅0.593~0.918; 潘玉朋等[23]利用31对SSR引物对黄淮麦区大面积推广的25个小麦品种进行遗传多样性分析, 表明品种间的遗传相似系数为0.61, 变幅0.34~0.85; 刘丽华等[24]利用21对SSR对2009—2014年参加国家冬小麦区域试验的430个品系进行遗传多样性分析, 结果表明北部冬麦区旱地组参试品系的遗传多样性为0.73, 黄淮海冬麦区南片春水组参试品系的遗传多样性为0.63; 本研究利用高密度SNP芯片, 对国内的229份小麦品种(系)进行遗传相似性研究, 结果表明各大区品种(系)间遗传相似系数在0.660~0.718之间(表1), 与前人利用SSR标记做类似研究的结果相似[19,23-24]。本研究中, 西南地区因样品数量不足且均为四川小麦品种(系)外, 长江中下游麦区小麦品种(系)的相似性系数最高(0.712), 可能与长江中下游麦区小麦育种单位相对较少, 也可能与该区域育种单位引进利用其他地区种质资源相对有限、育成小麦品种(系)的类型较少有关。黄淮南片麦区、黄淮北片麦区和北部麦区的小麦品种(系)的相似性系数相对较低(0.660~0.686), 可能与这些区域小麦育种单位相对较多实力雄厚有关, 也可能与该区域育种单位积极引入外源优异种质资源有关, 如陕西育种过程中引入外源种质资源长穗偃麦草和地方品种[25], 相对丰富的遗传多样性也造就了陕西品种特别是西农系列品种相对较广的适应性。
我国幅员辽阔、生态环境多样, 不同麦区的品种经过长期演变和选择形成了相对独立的遗传特点, 与此同时本研究也发现(图4), 不同类组内大多含有来自不同区域或省份的育成品种(系), 且不同区域小麦品种(系)在主成分分析图中分布较广(图5), 说明我国各育种单位间种质资源交流较为频繁。国外品种(系)聚集在第VI类之中, 宁春系列的3个品种也属于第VI类(图4), 可能与宁春系列品种含有国外小麦品种的遗传物质有关[26]; 周麦系列品种(系)主要分布在第VII类, 新麦系列主要分布在第VI类, 郑麦系列品种广泛分布在第VII、IV、II和I类中, 可能与郑麦系列品种的遗传基础相对较广有关。江苏品种中扬麦和宁麦系列品种主要聚集在第I类中, 说明扬麦和宁麦系列的遗传关系相对较近。山东品种中济麦、烟农和山农系列品种交集一起聚集在第V类中, 说明3种系列的小麦品种遗传关系相对较近。湖北品种中鄂恩系列品种聚集在第VII类, 与陕西品种遗传关系较近; 鄂麦和襄麦系列品种属于第I类, 与江苏品种的遗传关系相对较近。陕西的西农和小偃系列在聚类图中分布较广, 说明两个系列的品种遗传基础较为丰富。四川的绵麦和川麦系列品种在聚类图中分布较广, 说明两个系列的品种遗传基础较为丰富。河北品种在聚类图中分布较广, 衡观系列品种(系)聚集分布在第VII类, 说明该系列品种的遗传关系相对较近。
本研究结果表明, 大部分品种(系)的聚类结果(图4)与其系谱来源及地域分布较为吻合, 也与笔者观测的田间性状相符。但研究中也发现, 部分品种遗传相似系数很高, 但系谱分析发现这些品种的亲本并无直接的亲缘关系。究其原因, 可能存在以下情况, 一是种子收集、繁殖、保存时出现偏差, 造成品种名称与实际材料不符的情况; 二是在同一生态区内, 育种家的长期定向选择, 造成选择的方向往往集中在某些位点上, 而造成另一些位点的丢失; 三是存在系谱记载不准确或不完善的可能[10]。下一步, 在确认种子无误的情况下, 可对这些品种(系)进行更为精细的田间观测, 也可利用其他分子标记或更高密度的SNP芯片对其进行分析, 已确定这些品种(系)的遗传相似性。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1155/2015/431487URLPMID:25874132 [本文引用: 2]

The importance of plant genetic diversity (PGD) is now being recognized as a specific area since exploding population with urbanization and decreasing cultivable lands are the critical factors contributing to food insecurity in developing world. Agricultural scientists realized that PGD can be captured and stored in the form of plant genetic resources (PGR) such as gene bank, DNA library, and so forth, in the biorepository which preserve genetic material for long period. However, conserved PGR must be utilized for crop improvement in order to meet future global challenges in relation to food and nutritional security. This paper comprehensively reviews four important areas; (i) the significance of plant genetic diversity (PGD) and PGR especially on agriculturally important crops (mostly field crops); (ii) risk associated with narrowing the genetic base of current commercial cultivars and climate change; (iii) analysis of existing PGD analytical methods in pregenomic and genomic era; and (iv) modern tools available for PGD analysis in postgenomic era. This discussion benefits the plant scientist community in order to use the new methods and technology for better and rapid assessment, for utilization of germplasm from gene banks to their applied breeding programs. With the advent of new biotechnological techniques, this process of genetic manipulation is now being accelerated and carried out with more precision (neglecting environmental effects) and fast-track manner than the classical breeding techniques. It is also to note that gene banks look into several issues in order to improve levels of germplasm distribution and its utilization, duplication of plant identity, and access to database, for prebreeding activities. Since plant breeding research and cultivar development are integral components of improving food production, therefore, availability of and access to diverse genetic sources will ensure that the global food production network becomes more sustainable. The pros and cons of the basic and advanced statistical tools available for measuring genetic diversity are briefly discussed and their source links (mostly) were provided to get easy access; thus, it improves the understanding of tools and its practical applicability to the researchers.
[本文引用: 3]
[本文引用: 3]
DOI:10.3724/SP.J.1006.2011.00202URL [本文引用: 1]
近10年我国小麦育种研究在3个方面取得新进展:育成一批高产优质多抗新品种,周8425B、鲁麦14和普通小麦-簇毛麦6VS/6AL易位系在全国小麦育种中发挥了重要作用,育种技术研究也取得重要进展。但育种工作也存在4个主要问题。从育种角度评述分子标记辅助育种中连锁标记和功能标记的研究现状和存在的主要问题,并提出今后的重点领域。概括小麦品质研究中与育种密切相关的实用技术和方法,即面包、面条和饼干品质育种中的品质评价方法和选择指标,建议今后加强5个方面的工作。对未来小麦育种4个重要问题做了分析,提出国内进一步加强高产潜力研究的初步设想,建议加大持久抗性的研究力度,重视抗旱、抗热及适应性等与气候变化相关性状的研究,还分析了种业商业化等问题。
DOI:10.3724/SP.J.1006.2011.00202URL [本文引用: 1]
近10年我国小麦育种研究在3个方面取得新进展:育成一批高产优质多抗新品种,周8425B、鲁麦14和普通小麦-簇毛麦6VS/6AL易位系在全国小麦育种中发挥了重要作用,育种技术研究也取得重要进展。但育种工作也存在4个主要问题。从育种角度评述分子标记辅助育种中连锁标记和功能标记的研究现状和存在的主要问题,并提出今后的重点领域。概括小麦品质研究中与育种密切相关的实用技术和方法,即面包、面条和饼干品质育种中的品质评价方法和选择指标,建议今后加强5个方面的工作。对未来小麦育种4个重要问题做了分析,提出国内进一步加强高产潜力研究的初步设想,建议加大持久抗性的研究力度,重视抗旱、抗热及适应性等与气候变化相关性状的研究,还分析了种业商业化等问题。
URL [本文引用: 1]
骨干亲本形成的遗传基础研究已经成为作物优异种质分子评价的重要部分,对作物育种具有非常重要的实践意义。为此,就小麦骨干亲本的遗传多样性、重要基因组区段及有利基因发掘的研究进行了阐述,并介绍了以连锁不平衡研究为基础开始在植物系谱群体进行数量性状研究的几种策略。
URL [本文引用: 1]
骨干亲本形成的遗传基础研究已经成为作物优异种质分子评价的重要部分,对作物育种具有非常重要的实践意义。为此,就小麦骨干亲本的遗传多样性、重要基因组区段及有利基因发掘的研究进行了阐述,并介绍了以连锁不平衡研究为基础开始在植物系谱群体进行数量性状研究的几种策略。
DOI:10.3390/d6040665URL [本文引用: 2]
In: Carena M J, ed.
[本文引用: 1]
DOI:10.1186/1471-2148-13-169URLPMID:23937410 [本文引用: 1]
Patterns of genetic diversity between and within natural plant populations and their driving forces are of great interest in evolutionary biology. However, few studies have been performed on the genetic structure and population divergence in wild emmer wheat using a large number of EST-related single nucleotide polymorphism (SNP) markers.
DOI:10.1186/1471-2164-11-727URLPMID:21190581 [本文引用: 1]
Single nucleotide polymorphisms (SNPs) are ideally suited for the construction of high-resolution genetic maps, studying population evolutionary history and performing genome-wide association mapping experiments. Here, we used a genome-wide set of 1536 SNPs to study linkage disequilibrium (LD) and population structure in a panel of 478 spring and winter wheat cultivars (Triticum aestivum) from 17 populations across the United States and Mexico.
DOI:10.1007/s00122-013-2065-1URL [本文引用: 2]
Modern genomics approaches rely on the availability of high-throughput and high-density genotyping platforms. A major breakthrough in wheat genotyping was the development of an SNP array. In this study, we used a diverse panel of 172 elite European winter wheat lines to evaluate the utility of the SNP array for genomic analyses in wheat germplasm derived from breeding programs. We investigated population structure and genetic relatedness and found that the results obtained with SNP and SSR markers differ. This suggests that additional research is required to determine the optimum approach for the investigation of population structure and kinship. Our analysis of linkage disequilibrium (LD) showed that LD decays within approximately 5–10?cM. Moreover, we found that LD is variable along chromosomes. Our results suggest that the number of SNPs needs to be increased further to obtain a higher coverage of the chromosomes. Taken together, SNPs can be a valuable tool for genomics approaches and for a knowledge-based improvement of wheat.
DOI:10.3724/SP.J.1006.2015.00197URL [本文引用: 4]
为了解河南省近年小麦品种的遗传基础,利用Illumina 90k iSelect SNP标记技术对豫麦34及该省2000—2013年审定的小麦品种共96个进行全基因组扫描,分析了其遗传多样性和遗传基础。结果表明,在所有SNP位点中,多态性比率为47.39% (38 661/81 587),多态性标记在基因组间分布呈现B>A>D。96个品种亲缘关系较近,两两遗传相似系数的平均值为0.719,变幅为0.552~0.998,且94.3%的品种间遗传相似系数在0.652~0.812之间;按UPGMA法将96个品种划分为7个类群。综合SNP和系谱分析,近10年河南省审定的96个小麦品种遗传多样性不够丰富,多数品种亲缘关系较近,在育种中迫切需要引入新的种质资源,拓宽遗传背景。
DOI:10.3724/SP.J.1006.2015.00197URL [本文引用: 4]
为了解河南省近年小麦品种的遗传基础,利用Illumina 90k iSelect SNP标记技术对豫麦34及该省2000—2013年审定的小麦品种共96个进行全基因组扫描,分析了其遗传多样性和遗传基础。结果表明,在所有SNP位点中,多态性比率为47.39% (38 661/81 587),多态性标记在基因组间分布呈现B>A>D。96个品种亲缘关系较近,两两遗传相似系数的平均值为0.719,变幅为0.552~0.998,且94.3%的品种间遗传相似系数在0.652~0.812之间;按UPGMA法将96个品种划分为7个类群。综合SNP和系谱分析,近10年河南省审定的96个小麦品种遗传多样性不够丰富,多数品种亲缘关系较近,在育种中迫切需要引入新的种质资源,拓宽遗传背景。
DOI:10.1371/journal.pone.0167821URLPMID:28099442 [本文引用: 1]
Until now, little attention has been paid to the geographic distribution and evaluation of genetic diversity of durum wheat from the Central Fertile Crescent (modern-day Turkey and Syria). Turkey and Syria are considered as primary centers of wheat diversity, and thousands of locally adapted wheat landraces are still present in the farmers' small fields. We planned this study to evaluate the genetic diversity of durum wheat landraces from the Central Fertile Crescent by genotyping based on DArTseq and SNP analysis. A total of 39,568 DArTseq and 20,661 SNP markers were used to characterize the genetic characteristic of 91 durum wheat land races. Clustering based on Neighbor joining analysis, principal coordinate as well as Bayesian model implemented in structure, clearly showed that the grouping pattern is not associated with the geographical distribution of the durum wheat due to the mixing of the Turkish and Syrian landraces. Significant correlation between DArTseq and SNP markers was observed in the Mantel test. However, we detected a non-significant relationship between geographical coordinates and DArTseq (r = -0.085) and SNP (r = -0.039) loci. These results showed that unconscious farmer selection and lack of the commercial varieties might have resulted in the exchange of genetic material and this was apparent in the genetic structure of durum wheat in Turkey and Syria. The genomic characterization presented here is an essential step towards a future exploitation of the available durum wheat genetic resources in genomic and breeding programs. The results of this study have also depicted a clear insight about the genetic diversity of wheat accessions from the Central Fertile Crescent.
[本文引用: 2]
[本文引用: 2]
DOI:10.3389/fpls.2017.02115URLPMID:29312381 [本文引用: 1]
Since the introduction of wheat into Australia by the First Fleet settlers, germplasm from different geographical origins has been used to adapt wheat to the Australian climate through selection and breeding. In this paper, we used 482 cultivars, representing the breeding history of bread wheat in Australia since 1840, to characterize their diversity and population structure and to define the geographical ancestral background of Australian wheat germplasm. This was achieved by comparing them to a global wheat collection using in-silico chromosome painting based on SNP genotyping. The global collection involved 2,335 wheat accessions which was divided into 23 different geographical subpopulations. However, the whole set was reduced to 1,544 accessions to increase the differentiation and decrease the admixture among different global subpopulations to increase the power of the painting analysis. Our analysis revealed that the structure of Australian wheat germplasm and its geographic ancestors have changed significantly through time, especially after the Green Revolution. Before 1920, breeders used cultivars from around the world, but mainly Europe and Africa, to select potential cultivars that could tolerate Australian growing conditions. Between 1921 and 1970, a dependence on African wheat germplasm became more prevalent. Since 1970, a heavy reliance on International Maize and Wheat Improvement Center (CIMMYT) germplasm has persisted. Combining the results from linkage disequilibrium, population structure and in-silico painting revealed that the dependence on CIMMYT materials has varied among different Australian States, has shrunken the germplasm effective population size and produced larger linkage disequilibrium blocks. This study documents the evolutionary history of wheat breeding in Australia and provides an understanding for how the wheat genome has been adapted to local growing conditions. This information provides a guide for industry to assist with maintaining genetic diversity for long-term selection gains and to plan future breeding programs.
DOI:10.1017/S1479262115000325URL [本文引用: 1]
DOI:10.1007/s00122-017-3010-5URLPMID:29103142 [本文引用: 1]
High-throughput genotyping of Swiss bread wheat and spelt accessions revealed differences in their gene pools and identified bread wheat landraces that were not used in breeding. Genebanks play a pivotal role in preserving the genetic diversity present among old landraces and wild progenitors of modern crops and they represent sources of agriculturally important genes that were lost during domestication and in modern breeding. However, undesirable genes that negatively affect crop performance are often co-introduced when landraces and wild crop progenitors are crossed with elite cultivars, which often limit the use of genebank material in modern breeding programs. A detailed genetic characterization is an important prerequisite to solve this problem and to make genebank material more accessible to breeding. Here, we genotyped 502 bread wheat and 293 spelt accessions held in the Swiss National Genebank using a 15K wheat SNP array. The material included both spring and winter wheats and consisted of old landraces and modern cultivars. Genome- and sub-genome-wide analyses revealed that spelt and bread wheat form two distinct gene pools. In addition, we identified bread wheat landraces that were genetically distinct from modern cultivars. Such accessions were possibly missed in the early Swiss wheat breeding program and are promising targets for the identification of novel genes. The genetic information obtained in this study is appropriate to perform genome-wide association studies, which will facilitate the identification and transfer of agriculturally important genes from the genebank into modern cultivars through marker-assisted selection.
DOI:10.1073/pnas.81.24.8014URLPMID:6096873 [本文引用: 1]
Spacer-length (sl) variation in ribosomal RNA gene clusters (rDNA) was surveyed in 502 individual barley plants, including samples from 50 accessions of cultivated barley, 25 accessions of its wild ancestor, and five generations of composite cross II (CCII), an experimental population of barley. In total, 17 rDNA sl phenotypes, made up of 15 different rDNA sl variants, were observed. The 15 rDNA sl variants comprise a complete ladder in which each variant differs in length from adjacent variants by approximately equal to 115 nucleotide pairs. Studies of four rDNA sl variants in an F2 population showed that these variants are located at two unlinked loci, Rrn1 and Rrn2, each with two codominant alleles. Using wheat-barley addition lines, we determined that Rrn1 and Rrn2 are located on chromosomes 6 and 7, respectively. The nonrandom distribution of sl variants between loci suggests that genetic exchange occurs much less frequently between than within the two loci, which demonstrates that Rrn1 and Rrn2 are useful as new genetic markers. Frequencies of rDNA sl phenotypes and variants were monitored over 54 generations in CCII. A phenotype that was originally infrequent in CCII ultimately became predominant, whereas the originally most frequent phenotype decreased drastically in frequency, and all other phenotypes originally present disappeared from the population. We conclude that the sl variants and/or associated loci are under selection in CCII.
DOI:10.1111/pbi.12183URL [本文引用: 1]
High-density single nucleotide polymorphism (SNP) genotyping arrays are a powerful tool for studying genomic patterns of diversity, inferring ancestral relationships between individuals in populations and studying marker-trait associations in mapping experiments. We developed a genotyping array including about 90 000 gene-associated SNPs and used it to characterize genetic variation in allohexaploid and allotetraploid wheat populations. The array includes a significant fraction of common genome-wide distributed SNPs that are represented in populations of diverse geographical origin. We used density-based spatial clustering algorithms to enable high-throughput genotype calling in complex data sets obtained for polyploid wheat. We show that these model-free clustering algorithms provide accurate genotype calling in the presence of multiple clusters including clusters with low signal intensity resulting from significant sequence divergence at the target SNP site or gene deletions. Assays that detect low-intensity clusters can provide insight into the distribution of presence-absence variation (PAV) in wheat populations. A total of 46 977 SNPs from the wheat 90K array were genetically mapped using a combination of eight mapping populations. The developed array and cluster identification algorithms provide an opportunity to infer detailed haplotype structure in polyploid wheat and will serve as an invaluable resource for diversity studies and investigating the genetic basis of trait variation in wheat.
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 3]
[本文引用: 3]
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}