 ,*, 李浩, 陈义珍, 柳展基, 刘任重, 王立国山东棉花研究中心 / 农业部黄淮海棉花遗传改良与栽培生理重点实验室, 山东济南 250100
,*, 李浩, 陈义珍, 柳展基, 刘任重, 王立国山东棉花研究中心 / 农业部黄淮海棉花遗传改良与栽培生理重点实验室, 山东济南 250100Identification of co-expressed modules of cotton genes responding to Verticillium dahliae infection by WGCNA
FU Ming-Chuan,*, LI Hao, CHEN Yi-Zhen, LIU Zhan-Ji, LIU Ren-Zhong, WANG Li-GuoCotton Research Center of Shandong Academy of Agricultural Sciences / Key Laboratory of Cotton Breeding and Cultivation in Huang-Huai-Hai Plain, Ministry of Agriculture, Jinan 250100, Shandong, China通讯作者:
收稿日期:2019-08-23接受日期:2020-01-15网络出版日期:2020-02-18
| 基金资助: |
Received:2019-08-23Accepted:2020-01-15Online:2020-02-18
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (1659KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
傅明川, 李浩, 陈义珍, 柳展基, 刘任重, 王立国. 利用WGCNA鉴定棉花抗黄萎病相关基因共表达网络[J]. 作物学报, 2020, 46(5): 668-679. doi:10.3724/SP.J.1006.2020.94124
FU Ming-Chuan, LI Hao, CHEN Yi-Zhen, LIU Zhan-Ji, LIU Ren-Zhong, WANG Li-Guo.
棉花(Gossypium spp.)是世界范围内重要的纤维及油料作物之一, 其在我国经济发展中也占有十分重要的地位。黄萎病(Verticillium wilt)作为土壤传播的维管束系统性病害, 是棉花生产中最严重的病害之一。过去通常认为黄萎病菌的致病机制是“导管堵塞”; 目前研究表明, 黄萎病菌分泌的毒素可能是其主要致病因素, 其中细胞壁降解酶、脂多糖蛋白复合物以及核蛋白可能是重要的致病成分。Wang等[1]研究证实, 黄萎病菌VdNEP蛋白是一种重要的黄萎病致病因子。通过研究高致病性菌株Vd080发现, VdCYC8基因与黄萎病菌的致病力和微菌核形成有关[2]。VdRGS1-8构成了黄萎病菌的RGS基因家族, 其中VdRGS1在孢子生成、菌丝生长、微菌核形成过程中发挥重要作用[3]。由于缺乏有效的黄萎病防治药剂, 选育和种植抗病品种是目前最经济有效、绿色环保的防治措施。但抗病基因资源缺乏、抗病机制不明, 导致棉花抗黄萎病育种工作进展缓慢[4]。因此, 挖掘棉花抗病基因、探究其抗病机制, 对于棉花抗病育种工作具有重要意义。Kawchuk等[5]利用图位克隆在番茄(Lycopersicon esculentum) Ve位点中分离出2个紧密相连的抗黄萎病主效基因Ve1和Ve2, 研究证实Ve基因可介导感病马铃薯品种(Solanum tuberosum)对黄萎病菌的抗性。进一步研究表明, 由于棉花黄萎病菌缺少ave1基因, 导致在棉花中过表达Ve1并不能明显增强其抗病性[6]。基因组学、转录组学及蛋白质组学的迅速发展, 为研究棉花抗黄萎病的分子机制提供了有利基础。Zhang等[7]通过对黄萎病菌侵染下的海岛棉(Gossypium barbadense)“Pima 90-53”幼苗根系进行转录组分析, 首次报道了“SA→NPR1→TGA→PR-1→抗病”代谢通路。目前, 在棉花中已鉴定出一些抗病相关基因。如GbERF2是海岛棉中一个ERF转录因子基因, 转GbERF2的烟草(Nicotiana tabacum)植株内抗病相关基因表达量升高, 并可提高转基因植株的抗病性[8]; GbSTK为一类丝氨酸/苏氨酸蛋白激酶基因, 参与调控多个抗逆相关信号通路, 从而在抵御病原菌侵染胁迫及氧化应激响应中发挥作用[9]; GhDSC1参与调控活性氧含量及JA信号通路相关基因的表达, 转GhDSC1基因的拟南芥(Arabidopsis thaliana)植株对黄萎病菌的抗性提高[10]。此外, GbTLP1[11]、GhPAO[12]、GhJAZ2[13]等基因也均在抗病响应中发挥重要作用。
目前, 棉花基因组测序工作已陆续完成, 这为从海量生物数据中挖掘有用信息提供了坚实基础。相比传统分子生物学方法, 借助生物信息学手段, 可更加快捷地定位目标基因, 挖掘与性状相关的关键基因。加权基因共表达网络分析(Weighted Gene Co-expression Network Analysis, WGCNA)以芯片或RNA-seq表达数据为基础, 通过幂指数加权构建无尺度拓扑重叠矩阵, 用以描述基因之间的相互关系, 并依此将表达模式相近的基因划分到一个基因表达模块中[14]。WGCNA多用来研究协同表达的基因模块与目标性状之间的生物学相关性, 并探索基因共表达网络中的核心基因。作为一种典型的系统生物学方法, WGCNA已广泛应用于植物学研究中。如Tan等[15]通过分析镉处理不同时间点的17个水稻 (Oryza sativa)转录组数据, 鉴定得到22个基因模块, 结合差异表达分析, 共挖掘到164个镉胁迫响应相关基因; Zou等[16]通过对2个棉花品系不同发育时期的纤维转录组数据进行WGCNA, 共鉴定得到5个纤维发育相关特异性模块, 并挖掘出模块内的核心基因; 通过对14份玉米(Zea mays)不同发育阶段的转录组数据进行WGCNA, 研究者鉴定得到14个组织特异性模块, 并对其中2个模块的基因互作网络进行了进一步研究, 从中挖掘到了ZCN8、ZCN7、COL1等开花相关核心基因[17]。
本研究以黄萎病菌侵染不同时间点的棉花转录组数据为材料, 对其进行差异表达分析; 通过构建加权基因共表达网络, 划分基因模块, 并筛选出抗病相关特异性模块; 经GO及KEGG富集分析, 探究模块功能; 根据基因在相应网络中的连通性, 鉴定出模块内的核心基因。本研究可为进一步理解棉花抗黄萎病的分子机制提供理论基础, 并为棉花抗病育种工作提供新的基因资源。
1 材料与方法
1.1 数据获取及差异表达分析
棉花在黄萎病菌侵染不同时间点的转录组数据下载自NCBI数据库(PRJNA234454)[18]。该数据以高抗黄萎病海岛棉品种“海7124”为材料, 对生长2周的幼苗根系使用Vd8孢子悬液侵染。分别选取侵染2、6、12、24、48和72 h的根尖, 提取总RNA, 并以未侵染材料(0 h)为对照。首先, 利用fastq-dump软件(https://ncbi.github.io/sra-tools/fastq-dump.html)将下载得到的SRA文件转换为fastq格式文件, 然后利用FastQC (http://www.bioinformatics.babraham. ac.uk/projects/fastqc/)对测序结果进行质量评估, 并通过Trimmomatic软件[19]进行质控, 将得到的clean data用于后续分析。以海岛棉基因组为参考基因组[20], 使用HISAT2[21]进行序列比对后, 利用featureCounts[22]计数, 得到每个基因在各个样本中的raw counts。将数据导入R中, 利用DESeq2[23]进行差异表达分析。本研究选取|log2FC| > 1 (log2FC代表处理与对照表达量比值的对数值)及padj<0.001 (padj代表校正后的p值)的基因作为差异表达基因。1.2 加权基因共表达网络构建
利用R程序中的WGCNA软件包[14]进行加权基因共表达网络构建。以标准化后的基因表达矩阵做为输入, 共21个转录组样本(7个时间点, 各3次重复)。通过计算每个基因在各个样本间表达水平的变异程度, 选取变异最大的前50%基因进行WGCNA。经过阈值筛选, 最终选择β=18对原始有尺度关系矩阵进行幂处理, 得到无尺度化邻接矩阵。为更好评估基因间表达模式的相关性, 进一步将邻接矩阵转化为拓扑重叠矩阵(Topological Overlap Matrix, TOM), 并利用拓扑相异矩阵(dissTOM=1-TOM), 采用动态剪切算法进行基因聚类及模块划分。模块内最少基因数为30 (minModuleSize=30), 相似模块合并阈值为0.25 (cutHeight=0.25), 网络类型为“signed” (type="signed"或networkType="signed", 依不同函数而定)。1.3 特异性模块筛选
对每个模块中的所有基因进行主成分分析(Principle Component Analysis, PCA), 将主成分1(PC1)的值称为该模块的模块特征向量(Module Eigengene, ME)。为筛选抗病相关特异性模块, 分别计算每个模块的模块特征向量与不同性状之间(此处为不同侵染时间)的相关系数r及相应p值。r>0代表正相关, r<0代表负相关。本研究选择|r|>0.70及p<0.001的模块作为特异性模块进一步分析。1.4 富集分析及代谢通路分析
利用R程序中的clusterProfiler软件包[24]进行GO (Gene Ontology)和KEGG (Kyoto Encyclopedia of Genes and Genomes)富集分析。阈值为p<0.01及q<0.05。1.5 转录因子分析
将各模块中的蛋白序列提交到PlantTFDB数据库(http://planttfdb.cbi.pku.edu.cn/prediction.php#)[25]分析预测, 从而得到每个模块中的转录因子。1.6 RNA提取及qRT-PCR
选取生长2周、长势一致的“海7124”幼苗, 采取浸根法用浓度1×107个mL-1的黄萎病菌Vd8孢子悬液侵染10 min。选取侵染后2、6、12、24、48、72 h和相应时间点未侵染幼苗根系, 利用TRIzol法提取总RNA, 取1 μg RNA反转录为cDNA。qPCR使用Thermo Fisher Scientific的QuantStudio 5实时荧光定量PCR系统, 选用Aidlab公司的2×Sybr Green qPCR Mix试剂盒, 荧光染料为SYBR Green, 内参基因为β-actin。反应程序95℃预变性3 min; 95℃ 15 s, 60℃ 15 s, 40个循环; 熔点曲线程序为95℃ 15 s, 60℃ 1 min, 95℃ 15 s。反应体系包含2×SYBR qPCR Mix 10 μL、DNA Template (稀释10倍) 0.8 μL、正向引物(10 μmol L-1) 0.4 μL、反向引物(10 μmol L-1) 0.4 μL、ddH2O 8.4 μL。使用2-ΔΔCt法分析基因相对表达量[26], 设置3次生物学重复。所用基因引物见表1。
Table 1
表1
表1qRT-PCR所用引物
Table 1
| 基因 Gene | 正向引物 Forward primer (5°-3°) | 反向引物 Reverse primer (5°-3°) |
|---|---|---|
| Gbar_D11G006810 | GGAGCTGTAGGATCATGCTCAGTG | GCCTTGCCATCCAAAATCCAGC |
| Gbar_A12G027410 | CGGCTTTCATAGGCAAGGTAGGG | AGCGTAACAAATGCCAATGCCG |
| Gbar_A04G004150 | CGTGCACTTTTCGGTCGTGATG | AGCTAGCCCTCTTGCTATCCCC |
| Gbar_D10G024280 | GTAGTTAACGCTGAACAACGTT | CCAACCTCCATACTCTTCTTCA |
| Gbar_A08G011260 | CAGTAATGTGAAGGCGGCCAGA | CAGTGCTCAACGACCTCGTCAT |
| Gbar_A13G004330 | GCTCTCAACAATGATGGGGTCCT | AGTTCCAGCTTTGCAAGACCGA |
| Gbar_A12G027680 | AACTCCGGTAAGTGGGTACCGT | AACGGCGGAGATCGAGTTGATG |
| Gbar_D03G017340 | AAACGAGAGAGAACGGCGAAGG | CTTAACGGCGAGAGTAACGGCA |
| Gbar_A07G022210 | GGAAATACCCACCGTGCAACCT | GAGCCAAGGGTGAGTGAGACAC |
| β-actin | GATTCCGTTGTCCAGAAGTCCT | TACGGTCTGCAATACCAGGGA |
新窗口打开|下载CSV
2 结果与分析
2.1 差异表达分析
通过分析棉花在黄萎病菌侵染不同时间点(0 h、2 h、6 h、12 h、24 h、48 h、72 h)的转录组数据, 最终共得到22,850个差异表达基因(|log2FC| > 1, padj < 0.001)。其中, 显著上调基因9398个, 显著下调基因13,171个, 另有281个基因在不同时间点表现出不同的上下调趋势。差异表达基因最多的时间点出现在侵染后24 h (14,679个), 最少的为2 h (10,729个), 所有时间点共有的差异表达基因为4685个(图1)。图1
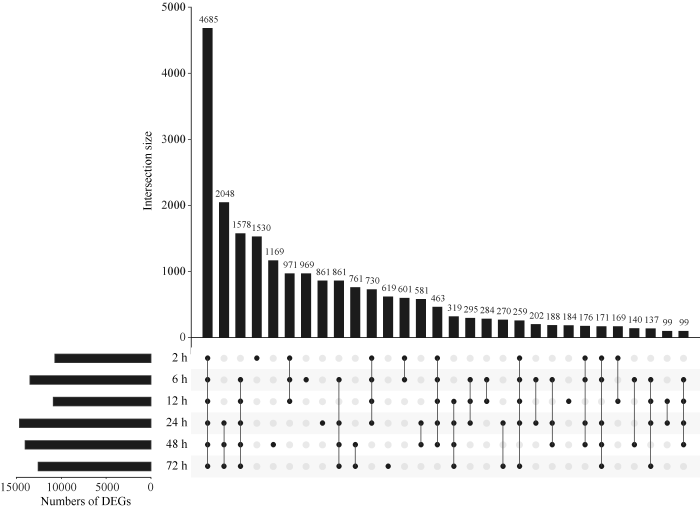 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1不同侵染时间点的差异表达基因
只有一个黑点的列代表某个数据集特有的差异基因; 有2个或以上用实线连接的黑点的列代表相应数据集之间共有的差异基因。
Fig. 1DEGs in different time points under infection
The column with one black point represents the DEGs only in the corresponding set; the column with two or more black points linked by a solid line represents the intersection among the sets.
2.2 加权基因共表达网络构建
对表达矩阵中变异较低的基因进行过滤, 最终选取35,647个基因进行WGCNA。当β=18时, 无尺度网络拟合指数R2>0.8, 平均连通性趋近于0, 表明用此值进行幂处理可以得到符合要求的无尺度网络, 因此选择β=18构建无尺度网络(图2)。采用动态剪切算法对基因进行聚类及模块划分, 通过计算每个模块的模块特征向量, 合并相似模块, 最终共得到18个基因共表达模块(图3)。模块内基因数量为61 (palevioletred2) ~ 8431 (black)个。各模块内差异表达基因所占比例为11% (thistle2) ~ 81% (paleturquoise)。值得注意的是, 除black及mediumpurple2模块外, 同一模块内的差异表达基因通常表现出相似的上下调模式, 如paleturquoise、turquoise模块内的差异基因均为显著下调, 而palevioletred2、thistle2模块内均为显著上调。图2
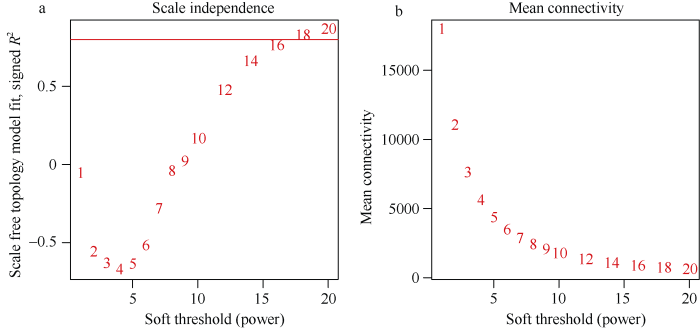 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2软阈值的选择
a: 不同软阈值下的无尺度网络拟合指数(R2), 红线代表R2=0.8。b: 不同软阈值下的平均连通性。
Fig. 2Determination of soft-thresholding power (β)
a: scale-free topology fit index as a function of the soft-thresholding power, the red line represents that R2 is equal to 0.8. b: mean connectivity as a function of the soft-thresholding power.
图3
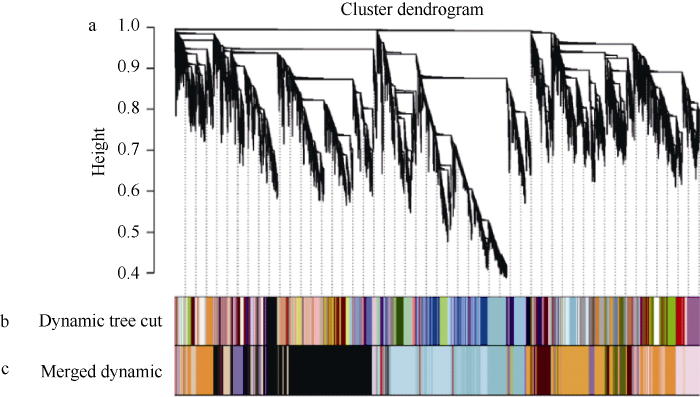 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3基因聚类树及模块划分
a: 基于拓扑相异矩阵构建的基因聚类树。b: 使用动态剪切算法得到的基因模块, 不同颜色代表不同模块。c: 合并相似模块后的模块划分结果。
Fig. 3Clustering dendrograms of genes and module detecting
a: gene clustering on TOM-based dissimilarity. b: module division by dynamic tree cut, different colors represent different modules. c: module division after merging similar modules.
转录因子是生物过程中一类重要的调控蛋白, 本研究也对每个模块内的转录因子进行了进一步分析。结果表明, 模块内转录因子数量分布为3 (mediumpurple2) ~ 590 (black)个, 在相应模块内所占比例为2% (mediumpurple2) ~ 19% (plum3), 不同模块内转录因子的分布类型存在差异, 但主要集中在ERF (9.27%)、MYB (8.09%)、bHLH (7.94%)、WRKY (7.03%)、C2H2 (5.74%)、NAC (5.36%)、bZIP (5.28%)等转录因子家族, 报道表明这些转录因子均参与调控植物的抗逆过程[27]。每个模块内差异表达的转录因子数占该模块内所有转录因子的比例为20% (thistle2) ~ 89% (navajowhite1), 数量显著偏高(χ2=92.24, p=2e-12), 进一步表明转录因子在抗逆调控过程中可能发挥重要作用。
2.3 抗病相关特异性模块鉴定
在18个基因模块中, 有5个与黄萎病菌侵染存在高度特异性(|r|>0.70, p<0.001)。其中, black (r=0.72, p=4e-04)、mediumpurple3 (r=0.78, p=5e-05)、darkolivegreen (r=0.76, p=9e-05)、plum3 (r=0.97, p=6e-12)模块分别与侵染2 h、6 h、48 h、72 h时间点正相关; mediumpurple2 (r= -0.82, p=8e-06)与侵染2 h时间点负相关(图4)。图4
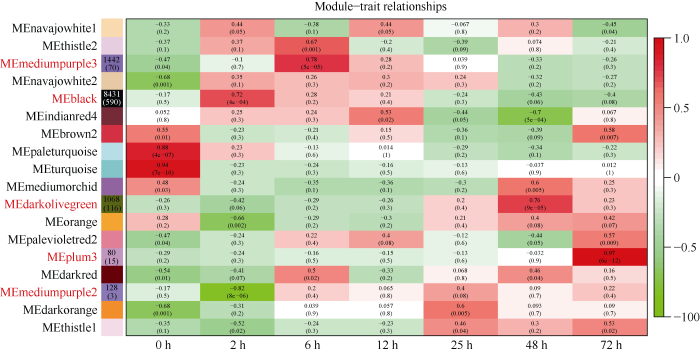 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4模块与性状相关性热图
每行代表一个模块, 每列代表一种性状。矩形框里的数字代表模块与性状之间的相关系数及相应p值。抗病相关特异性模块用红色标示, 其基因及转录因子数量标于左侧模块矩形框内。
Fig. 4Heat map of the correlation between modules and traits
Each row corresponds to a module, and each column corresponds to a trait. The correlation coefficient and the corresponding p-value are shown in each cell. The specific modules which significantly associated with V. dahliae infection are colored in red, and the corresponding numbers of genes and transcription factors are shown in the left cells.
2.4 特异性模块富集分析
GO通路可分为生物过程(Biological Process, BP)、分子功能(Molecular Function, MF)及细胞组分(Cellular Component, CC) 3类。分析结果表明, black和mediumpurple3模块分别富集到41个和36个生物过程, 其中black模块主要富集到氮化合物转运(GO:0071705)、蛋白定位(GO:0045184)、蛋白转运(GO:0015031)等调控通路, 以及一些信号相关通路, 如信号转导(GO:0007165)、信号转导调控(GO:1902531)等(图5-a); mediumpurple3模块主要富集到一些代谢相关通路, 如氨基酸代谢(GO:0006520)、硫化物代谢(GO:0006790)、甘氨酸代谢(GO:0006544)等生物学过程(附表1)。此外, black、mediumpurple3、darkolivegreen、plum3及mediumpurple2模块分别富集到21、7、7、1和2个分子功能, 如蛋白质转运蛋白活性(GO:0008565)、钙离子结合(GO:0005509)(附表2); black和mediumpurple2模块分别富集到44和4个细胞组分, 最显著的分别为膜衣(GO:0030117)和蛋白酶体核心复合物(GO:0005839)(附表3)。图5
 新窗口打开|下载原图ZIP|生成PPT
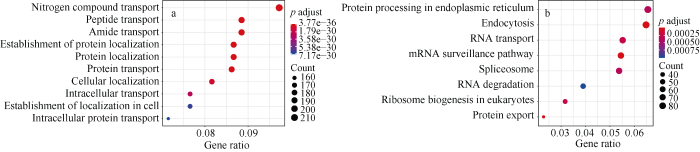
新窗口打开|下载原图ZIP|生成PPT图5Black模块的GO和KEGG富集分析
纵坐标代表GO条目或KEGG代谢通路, 横坐标代表富集到的基因在模块内所占比率。点的大小代表富集到的基因数量, 点的颜色代表多重校验后的p值大小。
Fig. 5GO functional and KEGG pathway enrichment analysis in the black module
The vertical axis represents the enriched GO term or KEGG pathway, and the horizontal axis represents the ratio of enriched genes in the module. The point size represents the gene number enriched in the pathway, and the color represents the p-value corrected for multiple testing.
Supplementary table 1
附表1
附表1特异性模块GO富集分析(生物过程)
Supplementary table 1
| 模块 Module | ID | 描述 Description | p值 p-value | q值 q-value |
|---|---|---|---|---|
| black | GO:0071705 | nitrogen compound transport | 6.27E-39 | 3.16E-36 |
| GO:0045184 | establishment of protein localization | 5.02E-38 | 1.12E-35 | |
| GO:0015031 | protein transport | 6.65E-38 | 1.12E-35 | |
| GO:0008104 | protein localization | 5.51E-37 | 5.61E-35 | |
| GO:0015833 | peptide transport | 5.57E-37 | 5.61E-35 | |
| GO:0042886 | amide transport | 9.91E-37 | 8.31E-35 | |
| GO:0051641 | cellular localization | 7.31E-33 | 5.25E-31 | |
| GO:0046907 | intracellular transport | 3.23E-32 | 2.03E-30 | |
| GO:0051649 | establishment of localization in cell | 1.02E-31 | 5.69E-30 | |
| GO:0006886 | intracellular protein transport | 1.19E-31 | 6.00E-30 | |
| GO:0071702 | organic substance transport | 2.57E-31 | 1.17E-29 | |
| GO:0034613 | cellular protein localization | 9.85E-31 | 3.81E-29 | |
| GO:0070727 | cellular macromolecule localization | 9.85E-31 | 3.81E-29 | |
| GO:0033036 | macromolecule localization | 2.30E-30 | 8.27E-29 | |
| GO:0016192 | vesicle-mediated transport | 4.18E-25 | 1.40E-23 | |
| GO:0048193 | Golgi vesicle transport | 1.92E-12 | 6.03E-11 | |
| GO:0007264 | small GTPase mediated signal transduction | 2.38E-09 | 7.04E-08 | |
| GO:0006888 | ER to Golgi vesicle-mediated transport | 1.75E-08 | 4.89E-07 | |
| GO:0007154 | cell communication | 2.02E-08 | 5.36E-07 | |
| GO:0007165 | signal transduction | 5.62E-07 | 1.35E-05 | |
| GO:0023052 | signaling | 5.62E-07 | 1.35E-05 | |
| GO:0035556 | intracellular signal transduction | 8.38E-07 | 1.92E-05 | |
| GO:0006605 | protein targeting | 1.56E-06 | 3.42E-05 | |
| GO:0051716 | cellular response to stimulus | 4.75E-06 | 9.96E-05 | |
| GO:0006890 | retrograde vesicle-mediated transport, Golgi to ER | 3.42E-05 | 6.88E-04 | |
| GO:1902531 | regulation of intracellular signal transduction | 4.55E-05 | 8.80E-04 | |
| GO:0006913 | nucleocytoplasmic transport | 5.42E-05 | 9.73E-04 | |
| GO:0051169 | nuclear transport | 5.42E-05 | 9.73E-04 | |
| GO:0009966 | regulation of signal transduction | 1.00E-04 | 1.63E-03 | |
| GO:0010646 | regulation of cell communication | 1.00E-04 | 1.63E-03 | |
| GO:0023051 | regulation of signaling | 1.00E-04 | 1.63E-03 | |
| GO:0009755 | hormone-mediated signaling pathway | 1.19E-04 | 1.71E-03 | |
| GO:0032870 | cellular response to hormone stimulus | 1.19E-04 | 1.71E-03 | |
| GO:0071310 | cellular response to organic substance | 1.19E-04 | 1.71E-03 | |
| GO:0071495 | cellular response to endogenous stimulus | 1.19E-04 | 1.71E-03 | |
| GO:0072594 | establishment of protein localization to organelle | 3.40E-04 | 4.76E-03 | |
| GO:0006904 | vesicle docking involved in exocytosis | 3.96E-04 | 5.25E-03 | |
| GO:0140029 | exocytic process | 3.96E-04 | 5.25E-03 | |
| GO:0072657 | protein localization to membrane | 4.49E-04 | 5.65E-03 | |
| GO:0090150 | establishment of protein localization to membrane | 4.49E-04 | 5.65E-03 | |
| mediumpurple3 | GO:0051640 | organelle localization | 4.71E-04 | 5.78E-03 |
| GO:0006520 | cellular amino acid metabolic process | 9.09E-10 | 2.99E-07 | |
| GO:1901605 | alpha-amino acid metabolic process | 1.50E-07 | 2.47E-05 | |
| GO:0006790 | sulfur compound metabolic process | 4.22E-07 | 4.64E-05 | |
| GO:0006544 | glycine metabolic process | 9.95E-06 | 8.19E-04 | |
| GO:0009069 | serine family amino acid metabolic process | 1.98E-05 | 9.92E-04 | |
| GO:0034660 | ncRNA metabolic process | 2.05E-05 | 9.92E-04 | |
| GO:0033866 | nucleoside bisphosphate biosynthetic process | 2.71E-05 | 9.92E-04 | |
| GO:0034030 | ribonucleoside bisphosphate biosynthetic process | 2.71E-05 | 9.92E-04 | |
| GO:0034033 | purine nucleoside bisphosphate biosynthetic process | 2.71E-05 | 9.92E-04 | |
| GO:0006418 | tRNA aminoacylation for protein translation | 4.26E-05 | 1.17E-03 | |
| GO:0043038 | amino acid activation | 4.26E-05 | 1.17E-03 | |
| GO:0043039 | tRNA aminoacylation | 4.26E-05 | 1.17E-03 | |
| GO:0006084 | acetyl-CoA metabolic process | 4.84E-05 | 1.23E-03 | |
| GO:0006732 | coenzyme metabolic process | 9.25E-05 | 1.98E-03 | |
| GO:0006555 | methionine metabolic process | 9.25E-05 | 1.98E-03 | |
| GO:0065008 | regulation of biological quality | 9.63E-05 | 1.98E-03 | |
| GO:0008652 | cellular amino acid biosynthetic process | 1.20E-04 | 1.99E-03 | |
| GO:0006637 | acyl-CoA metabolic process | 1.27E-04 | 1.99E-03 | |
| GO:0035383 | thioester metabolic process | 1.27E-04 | 1.99E-03 | |
| GO:0001505 | regulation of neurotransmitter levels | 1.32E-04 | 1.99E-03 | |
| GO:0042133 | neurotransmitter metabolic process | 1.32E-04 | 1.99E-03 | |
| GO:0006399 | tRNA metabolic process | 1.33E-04 | 1.99E-03 | |
| GO:1901606 | alpha-amino acid catabolic process | 1.48E-04 | 2.12E-03 | |
| GO:0051188 | cofactor biosynthetic process | 1.87E-04 | 2.57E-03 | |
| GO:0009108 | coenzyme biosynthetic process | 2.21E-04 | 2.71E-03 | |
| GO:0009066 | aspartate family amino acid metabolic process | 2.22E-04 | 2.71E-03 | |
| GO:0072350 | tricarboxylic acid metabolic process | 2.22E-04 | 2.71E-03 | |
| GO:0000096 | sulfur amino acid metabolic process | 3.21E-04 | 3.57E-03 | |
| GO:0033865 | nucleoside bisphosphate metabolic process | 3.40E-04 | 3.57E-03 | |
| GO:0033875 | ribonucleoside bisphosphate metabolic process | 3.40E-04 | 3.57E-03 | |
| GO:0034032 | purine nucleoside bisphosphate metabolic process | 3.40E-04 | 3.57E-03 | |
| GO:0044272 | sulfur compound biosynthetic process | 3.47E-04 | 3.57E-03 | |
| GO:0009063 | cellular amino acid catabolic process | 4.06E-04 | 4.05E-03 | |
| GO:0051186 | cofactor metabolic process | 4.87E-04 | 4.72E-03 | |
| GO:0017144 | drug metabolic process | 5.26E-04 | 4.95E-03 | |
| GO:0044283 | small molecule biosynthetic process | 6.99E-04 | 6.40E-03 |
新窗口打开|下载CSV
Supplementary table 2
附表 2
附表 2特异性模块GO富集分析(分子功能)
Supplementary table 2
| 模块 Module | ID | 描述 Description | p值 p-value | q值 q-value |
|---|---|---|---|---|
| black | GO:0008565 | protein transporter activity | 5.96E-10 | 1.93E-07 |
| GO:0001883 | purine nucleoside binding | 7.64E-08 | 3.59E-06 | |
| GO:0005525 | GTP binding | 7.64E-08 | 3.59E-06 | |
| GO:0032550 | purine ribonucleoside binding | 7.64E-08 | 3.59E-06 | |
| GO:0032561 | guanyl ribonucleotide binding | 7.64E-08 | 3.59E-06 | |
| GO:0001882 | nucleoside binding | 7.76E-08 | 3.59E-06 | |
| GO:0032549 | ribonucleoside binding | 7.76E-08 | 3.59E-06 | |
| GO:0005096 | GTPase activator activity | 2.09E-07 | 8.46E-06 | |
| GO:0019001 | guanyl nucleotide binding | 2.68E-07 | 9.66E-06 | |
| GO:0008047 | enzyme activator activity | 1.84E-06 | 5.97E-05 | |
| GO:0060589 | nucleoside-triphosphatase regulator activity | 2.32E-06 | 6.83E-05 | |
| GO:0030695 | GTPase regulator activity | 3.63E-06 | 9.81E-05 | |
| GO:0042887 | amide transmembrane transporter activity | 4.96E-05 | 1.24E-03 | |
| GO:0016838 | carbon-oxygen lyase activity, acting on phosphates | 5.61E-05 | 1.30E-03 | |
| GO:0003924 | GTPase activity | 1.94E-04 | 4.20E-03 | |
| GO:0010333 | terpene synthase activity | 2.07E-04 | 4.20E-03 | |
| GO:0000287 | magnesium ion binding | 2.22E-04 | 4.23E-03 | |
| GO:0098772 | molecular function regulator | 2.67E-04 | 4.80E-03 | |
| GO:0004743 | pyruvate kinase activity | 4.22E-04 | 6.52E-03 | |
| GO:0030955 | potassium ion binding | 4.22E-04 | 6.52E-03 | |
| GO:0031420 | alkali metal ion binding | 4.22E-04 | 6.52E-03 | |
| mediumpurple3 | GO:0004812 | aminoacyl-tRNA ligase activity | 9.02E-06 | 1.06E-03 |
| GO:0016875 | ligase activity, forming carbon-oxygen bonds | 9.02E-06 | 1.06E-03 | |
| GO:0140101 | catalytic activity, acting on a tRNA | 9.53E-05 | 5.36E-03 | |
| GO:0016874 | ligase activity | 1.00E-04 | 5.36E-03 | |
| GO:0046912 | transferase activity, transferring acyl groups, acyl groups converted into alkyl on transfer | 1.14E-04 | 5.36E-03 | |
| GO:0140098 | catalytic activity, acting on RNA | 1.41E-04 | 5.54E-03 | |
| GO:0050662 | coenzyme binding | 2.28E-04 | 7.69E-03 | |
| darkolivegreen | GO:0004650 | polygalacturonase activity | 1.16E-16 | 1.19E-14 |
| GO:0016798 | hydrolase activity, acting on glycosyl bonds | 2.98E-07 | 1.54E-05 | |
| GO:0004553 | hydrolase activity, hydrolyzing O-glycosyl compounds | 8.84E-07 | 3.04E-05 | |
| GO:0004190 | aspartic-type endopeptidase activity | 7.33E-05 | 1.51E-03 | |
| GO:0070001 | aspartic-type peptidase activity | 7.33E-05 | 1.51E-03 | |
| GO:0020037 | heme binding | 1.75E-04 | 2.58E-03 | |
| GO:0046906 | tetrapyrrole binding | 1.75E-04 | 2.58E-03 | |
| plum3 | GO:0005509 | calcium ion binding | 1.22E-07 | 1.93E-06 |
| mediumpurple2 | GO:0004298 | threonine-type endopeptidase activity | 0.00 | 0.01 |
| GO:0070003 | threonine-type peptidase activity | 0.00 | 0.01 |
新窗口打开|下载CSV
Supplementary table 3
附表3
附表3特异性模块GO富集分析(细胞组分)
Supplementary table 3
| 模块 Module | ID | 描述 Description | p值 p-value | q值 q-value |
|---|---|---|---|---|
| black | GO:0030117 | membrane coat | 1.97E-31 | 9.24E-30 |
| GO:0048475 | coated membrane | 1.97E-31 | 9.24E-30 | |
| GO:0098796 | membrane protein complex | 3.12E-22 | 9.74E-21 | |
| GO:0012505 | endomembrane system | 1.77E-21 | 4.15E-20 | |
| GO:0005798 | Golgi-associated vesicle | 4.90E-19 | 4.17E-18 | |
| GO:0012506 | vesicle membrane | 4.90E-19 | 4.17E-18 | |
| GO:0030120 | vesicle coat | 4.90E-19 | 4.17E-18 | |
| GO:0030659 | cytoplasmic vesicle membrane | 4.90E-19 | 4.17E-18 | |
| GO:0030660 | Golgi-associated vesicle membrane | 4.90E-19 | 4.17E-18 | |
| GO:0030662 | coated vesicle membrane | 4.90E-19 | 4.17E-18 | |
| GO:0044433 | cytoplasmic vesicle part | 4.90E-19 | 4.17E-18 | |
| GO:0005794 | Golgi apparatus | 8.11E-19 | 6.33E-18 | |
| GO:0044431 | Golgi apparatus part | 5.88E-18 | 4.24E-17 | |
| GO:0030135 | coated vesicle | 2.02E-17 | 1.12E-16 | |
| GO:0031410 | cytoplasmic vesicle | 2.02E-17 | 1.12E-16 | |
| GO:0031982 | vesicle | 2.02E-17 | 1.12E-16 | |
| GO:0097708 | intracellular vesicle | 2.02E-17 | 1.12E-16 | |
| GO:0098588 | bounding membrane of organelle | 4.47E-17 | 2.33E-16 | |
| GO:0031090 | organelle membrane | 4.82E-16 | 2.38E-15 | |
| GO:0098805 | whole membrane | 5.76E-13 | 2.70E-12 | |
| GO:0030126 | COPI vesicle coat | 2.09E-12 | 8.52E-12 | |
| GO:0030137 | COPI-coated vesicle | 2.09E-12 | 8.52E-12 | |
| GO:0030663 | COPI-coated vesicle membrane | 2.09E-12 | 8.52E-12 | |
| GO:0030118 | clathrin coat | 2.90E-09 | 1.13E-08 | |
| GO:0030133 | transport vesicle | 7.84E-09 | 2.82E-08 | |
| GO:0030658 | transport vesicle membrane | 7.84E-09 | 2.82E-08 | |
| GO:0030119 | AP-type membrane coat adaptor complex | 7.16E-08 | 2.40E-07 | |
| GO:0030131 | clathrin adaptor complex | 7.16E-08 | 2.40E-07 | |
| GO:0012507 | ER to Golgi transport vesicle membrane | 6.69E-07 | 2.02E-06 | |
| GO:0030127 | COPII vesicle coat | 6.69E-07 | 2.02E-06 | |
| GO:0030134 | COPII-coated ER to Golgi transport vesicle | 6.69E-07 | 2.02E-06 | |
| GO:0000139 | Golgi membrane | 1.76E-04 | 5.15E-04 | |
| GO:0031984 | organelle subcompartment | 3.23E-04 | 9.17E-04 | |
| GO:0098791 | Golgi subcompartment | 5.41E-04 | 1.49E-03 | |
| GO:0005783 | endoplasmic reticulum | 6.96E-04 | 1.86E-03 | |
| GO:0033177 | proton-transporting two-sector ATPase complex, proton-transporting domain | 1.77E-03 | 4.61E-03 | |
| GO:0030136 | clathrin-coated vesicle | 2.02E-03 | 4.86E-03 | |
| GO:0005905 | clathrin-coated pit | 2.28E-03 | 4.86E-03 | |
| GO:0012510 | trans-Golgi network transport vesicle membrane | 2.28E-03 | 4.86E-03 | |
| mediumpurple2 | GO:0030125 | clathrin vesicle coat | 2.28E-03 | 4.86E-03 |
| GO:0030130 | clathrin coat of trans-Golgi network vesicle | 2.28E-03 | 4.86E-03 | |
| GO:0030132 | clathrin coat of coated pit | 2.28E-03 | 4.86E-03 | |
| GO:0030140 | trans-Golgi network transport vesicle | 2.28E-03 | 4.86E-03 | |
| GO:0030665 | clathrin-coated vesicle membrane | 2.28E-03 | 4.86E-03 | |
| GO:0005839 | proteasome core complex | 0.00 | 0.00 | |
| GO:0000502 | proteasome complex | 0.00 | 0.00 | |
| GO:1905369 | endopeptidase complex | 0.00 | 0.00 | |
| GO:1905368 | peptidase complex | 0.00 | 0.00 |
新窗口打开|下载CSV
KEGG富集分析表明, black、mediumpurple3和darkolivegreen模块分别富集到8、10和1个KEGG代谢通路, 其中black模块主要富集到mRNA监测通路、内吞作用和蛋白质输出等通路(图5-b); mediumpurple3主要富集到氨基酸生物合成、柠檬酸循环和碳代谢等通路; darkolivegreen富集到黄酮类化合物生物合成代谢通路(附表4)。
Supplementary table 4
附表4
附表4特异性模块KEGG富集分析
Supplementary table 4
| 模块 Module | 描述 Description | p值 p-value | q值 q-value |
|---|---|---|---|
| black | mRNA surveillance pathway | 4.52E-09 | 4.75E-07 |
| Endocytosis | 1.71E-08 | 8.99E-07 | |
| Protein export | 3.16E-07 | 1.11E-05 | |
| RNA transport | 8.85E-06 | 1.96E-04 | |
| Ribosome biogenesis in eukaryotes | 9.32E-06 | 1.96E-04 | |
| Spliceosome | 2.04E-05 | 3.18E-04 | |
| Protein processing in endoplasmic reticulum | 2.12E-05 | 3.18E-04 | |
| RNA degradation | 6.77E-05 | 8.91E-04 | |
| mediumpurple3 | Biosynthesis of amino acids | 8.38E-09 | 7.41E-07 |
| Citrate cycle (TCA cycle) | 6.09E-06 | 2.32E-04 | |
| Carbon metabolism | 7.88E-06 | 2.32E-04 | |
| Proteasome | 1.13E-05 | 2.49E-04 | |
| Phenylalanine, tyrosine and tryptophan biosynthesis | 4.01E-05 | 7.09E-04 | |
| Phenylalanine metabolism | 6.54E-05 | 8.48E-04 | |
| Tyrosine metabolism | 6.72E-05 | 8.48E-04 | |
| Isoquinoline alkaloid biosynthesis | 1.74E-04 | 1.93E-03 | |
| Selenocompound metabolism | 5.32E-04 | 5.22E-03 | |
| Tropane, piperidine and pyridine alkaloid biosynthesis | 7.03E-04 | 6.21E-03 | |
| darkolivegreen | Flavonoid biosynthesis | 8.66E-10 | 5.20E-08 |
新窗口打开|下载CSV
2.5 核心基因鉴定及基因互作网络构建
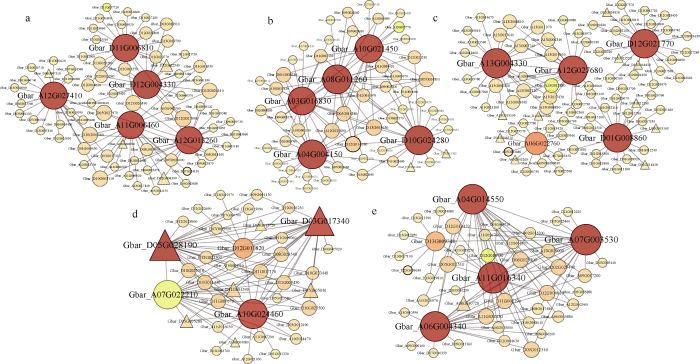
核心基因通常指模块内具有高连通性的基因, 本研究选取每个模块中kME值(Eigengene Connectivity)最高的前5个基因做为核心基因, 并利用核心基因及其互作基因绘制基因互作网络图(图6)。通过与拟南芥进行同源比对, 对核心基因及其互作基因进行了功能注释(表2, 附表5)。此外, 本研究选取了9个核心基因进行了qRT-PCR分析, 结果表明其表达模式与转录组数据基本一致(图7)。图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6抗病相关特异性模块内的基因共表达网络
点的大小和深浅代表该基因在网络中连通性的高低。转录因子用三角形表示, 其他基因用圆形表示。a: Black模块内的基因共表达网络。b: Mediumpurple3模块内的基因共表达网络。c: Darkolivegreen模块内的基因共表达网络。d: Plum3模块内的基因共表达网络。e: Mediumpurple2模块内的基因共表达网络。
Fig. 6Gene co-expression networks of the specific modules which significantly associated with V. dahliae infection
The genes with higher connectivity in the corresponding network are shown with larger circle sizes and darker colors. The transcription factors are indicated by triangles, with other genes being indicated by circles. a: Gene co-expression network of the black module. b: Gene co-expression network of the mediumpurple3 module. c: Gene co-expression network of the darkolivegreen module. d: Gene co-expression network of the plum3 module. e: Gene co-expression network of the mediumpurple2 module.
Table 2
表2
表2抗病相关特异性模块中核心基因的功能注释
Table 2
| 模块 Module | 棉花基因ID Gene ID in G. barbadense | 拟南芥基因ID Gene ID in Arabidopsis | 基因功能 Gene function |
|---|---|---|---|
| black | Gbar_A11G006460 | AT3G26680 | Involved in a SNM-dependent recombinational repair process of oxidatively induced DNA damage |
| Gbar_D12G004330 | AT2G15910 | CSL zinc finger domain-containing protein | |
| Gbar_D11G006810 | AT3G26680 | Involved in a SNM-dependent recombinational repair process of oxidatively induced DNA damage | |
| Gbar_A12G013260 | AT5G26751 | Encodes a SHAGGY-related kinase involved in meristem organization | |
| Gbar_A12G027410 | AT5G51830 | Encodes one of the several Arabidopsis fructokinases | |
| mediumpurple3 | Gbar_A04G004150 | AT1G53430 | Leucine-rich repeat transmembrane protein kinase |
| Gbar_D10G024280 | AT3G55470 | Calcium-dependent lipid-binding (CaLB domain) family protein | |
| Gbar_A08G011260 | AT1G64300 | Protein kinase family protein | |
| Gbar_A03G016830 | AT5G50850 | Transketolase family protein | |
| Gbar_A10G021450 | AT3G52200 | Encodes a dihydrolipoamide S-acetyltransferase, a subunit of the mitochondrial pyruvate dehydrogenase complex | |
| darkolivegreen | Gbar_A06G022760 | AT1G78020 | FCS like zinc finger 6 is induced during energy starvation through SnRK1 signaling |
| Gbar_A13G004330 | AT4G34220 | Encodes a receptor like kinase involved in ABA-mediated seedling development and drought tolerance | |
| Gbar_A12G027680 | AT5G51550 | EXORDIUM like 3 | |
| Gbar_D01G008860 | AT5G56040 | Leucine-rich receptor-like protein kinase family protein | |
| Gbar_D12G021770 | AT2G45550 | Member of CYP76C | |
| plum3 | Gbar_A10G024460 | AT5G05960 | Bifunctional inhibitor/lipid-transfer protein/seed storage 2S albumin superfamily protein |
| Gbar_D03G017340 | AT5G47230 | Encodes a member of the ERF (ethylene response factor) subfamily B-3 of ERF/AP2 transcription factor family (ATERF-5) | |
| Gbar_A07G022210 | AT2G38830 | Ubiquitin-conjugating enzyme/RWD-like protein | |
| Gbar_D12G010820 | AT3G60220 | Encodes a putative RING-H2 zinc finger protein ATL4 (ATL4) | |
| Gbar_D05G028190 | AT3G56400 | Function as activator of SA-dependent defense genes and a repressor of JA-regulated genes | |
| mediumpurple2 | Gbar_A06G004340 | AT1G44790 | ChaC-like family protein |
| Gbar_D13G009340 | AT1G13580 | Encodes a ceramide synthase that together with LOH1 is essential for production of ceramides containing Very Long Chain Fatty acid VLCFA-Ceramides | |
| Gbar_A07G003530 | AT2G25080 | Encodes glutathione peroxidase | |
| Gbar_A04G014550 | AT1G70280 | NHL domain-containing protein | |
| Gbar_A11G016340 | AT2G30860 | Encodes glutathione transferase belonging to the phi class of GSTs |
新窗口打开|下载CSV
Supplementary table 5
附表5
附表5核心基因互作基因功能注释
Supplementary table 5
| 模块 Module | 棉花ID Gene ID in G. barbadense | 拟南芥ID Gene ID in Arabidopsis | 基因功能 Gene Function |
|---|---|---|---|
| black | Gbar_D03G018910 | AT5G36930 | Disease resistance protein (TIR-NBS-LRR class) family |
| black | Gbar_D11G029060 | AT3G05545 | RING/U-box superfamily protein |
| black | Gbar_A11G009020 | AT5G23850 | O-glucosyltransferase rumi-like protein (DUF821) |
| black | Gbar_D04G003120 | AT5G24080 | Protein kinase superfamily protein |
| black | Gbar_D01G005060 | AT3G47570 | Leucine-rich repeat protein kinase family protein |
| black | Gbar_D11G034710 | AT2G38920 | SPX (SYG1/Pho81/XPR1) domain-containing protein / zinc finger (C3HC4-type RING finger) protein-like protein |
| black | Gbar_D12G025730 | AT5G53620 | RNA polymerase II degradation factor |
| black | Gbar_A13G017840 | AT5G14230 | Ankyrin |
| black | Gbar_D01G019380 | AT4G24290 | MAC/Perforin domain-containing protein |
| black | Gbar_D13G004090 | AT2G15220 | Plant basic secretory protein (BSP) family protein |
| black | Gbar_D13G022900 | AT4G30060 | Core-2/I-branching beta-1,6-N-acetylglucosaminyltransferase family protein |
| black | Gbar_Scaffold3330G000020 | AT5G54130 | Calcium-binding endonuclease/exonuclease/phosphatase family |
| black | Gbar_A12G023730 | AT1G12460 | Leucine-rich repeat protein kinase family protein |
| black | Gbar_D09G023870 | AT1G06620 | Encodes a protein whose sequence is similar to a 2-oxoglutarate-dependent dioxygenase |
| black | Gbar_D03G017310 | AT5G53110 | RING/U-box superfamily protein |
| black | Gbar_D12G004330 | AT2G15910 | CSL zinc finger domain-containing protein |
| black | Gbar_D06G023370 | AT1G75290 | Encodes a protein whose sequence is similar to an isoflavone reductase |
| black | Gbar_D13G019590 | AT1G02460 | Pectin lyase-like superfamily protein |
| black | Gbar_D04G014850 | AT1G59650 | Encodes CW14 |
| black | Gbar_A12G008940 | AT4G37790 | Encodes homeobox protein HAT22, member of the HD-Zip II family |
| black | Gbar_D01G009090 | AT2G04240 | Encodes a small protein with an N-terminal trans-membrane domain and a RING-H2 zinc finger motif located at the C-terminus |
| black | Gbar_D01G000450 | AT2G16280 | Encodes KCS9, a member of the 3-ketoacyl-CoA synthase family involved in the biosynthesis of VLCFA (very long chain fatty acids) |
| black | Gbar_D02G010580 | AT5G66070 | E3 ubiquitin ligase that functions in negative regulation of ABA signaling. |
| black | Gbar_A13G012150 | AT3G05420 | Acyl-CoA binding protein with high affinity for oleoyl-CoA |
| black | Gbar_D12G014610 | AT1G50200 | Alanyl-tRNA synthetase |
| black | Gbar_D12G017990 | AT1G69830 | Encodes a plastid-localized α-amylase |
| black | Gbar_D13G008370 | AT1G26560 | Beta glucosidase 40 |
| black | Gbar_D05G027970 | AT1G35670 | Encodes a Ca(2)-dependent, calmodulin-independent protein kinase that is rapidly induced by drought and high-salt stress but not by low-temperature stress or heat stress |
| black | Gbar_D01G006740 | AT5G54590 | Encodes CRLK1 (440-amino acid in length) calcium/calmodulin-regulated receptor-like kinase crucial for cold tolerance |
| black | Gbar_D12G025650 | AT5G24530 | Encodes a putative 2OG-Fe(II) oxygenase that is defense-associated but required for susceptibility to downy mildew |
| black | Gbar_D13G021410 | AT4G33050 | Encodes a calmodulin-binding protein involved in stomatal movement |
| black | Gbar_D10G001060 | AT4G05520 | Encodes AtEHD2, one of the Arabidopsis Eps15 homology domain proteins involved in endocytosis (AtEHD1, At3g20290) |
| black | Gbar_D12G022540 | AT4G17500 | Encodes a member of the ERF (ethylene response factor) subfamily B-3 of ERF/AP2 transcription factor family (ATERF-1) |
| black | Gbar_D06G015340 | AT5G46330 | Encodes a leucine-rich repeat serine/threonine protein kinase that is expressed ubiquitousl |
| black | Gbar_D12G027320 | AT5G51830 | Encodes one of the several Arabidopsis fructokinases |
| black | Gbar_D10G003110 | AT2G29120 | Member of Putative ligand-gated ion channel subunit family |
| black | Gbar_D07G004760 | AT3G47420 | Encodes a Pi starvation-responsive protein AtPS3 |
| black | Gbar_D07G024150 | AT5G14760 | Encodes for L-aspartate oxidase involved in the early steps of NAD biosynthesis |
| black | Gbar_D12G020770 | AT5G23670 | Encodes the LCB2 subunit of serine palmitoyltransferase, an enzyme involved in sphingosine biosynthesis |
| black | Gbar_D13G005810 | AT5G23450 | Encodes a sphingosine kinase that specifically phosphorylates D-erythro-dihydrosphingosine (DHS), but not N-acetyl-DHS or D-threo-DHS |
| black | Gbar_D13G019790 | AT4G31080 | Encodes one of two LUNAPARK proteins in Arabidopsis |
| black | Gbar_D12G026490 | AT5G52870 | Encodes a member of the MAKR (MEMBRANE-ASSOCIATED KINASE REGULATOR) gene family |
| black | Gbar_D11G032600 | AT5G27450 | Encodes a protein with mevalonate kinase activity involved in the mevalonate pathway |
| black | Gbar_D06G007510 | AT5G57620 | MYB36 is a transcriptional regulator that acts to promote differentiation of the endodermis during root development |
| black | Gbar_A13G005180 | AT1G69850 | Encodes an inducible component of low-affinity nitrate uptake. mRNA found primarily in root hairs and the epidermis of roots |
| black | Gbar_D03G004100 | AT2G22480 | Phosphofructokinase 5 |
| black | Gbar_D04G003630 | AT3G52430 | Encodes a lipase-like gene that is important for salicylic acid signaling and function in resistance (R) gene-mediated and basal plant disease resistance |
| black | Gbar_D07G018560 | AT1G05000 | Encodes an atypical dual-specificity phosphatase |
| black | Gbar_D13G016920 | AT3G02800 | Encodes an atypical dual-specificity phosphatase |
| black | Gbar_D13G018380 | AT5G18480 | Encodes an IPC (inositol phosphorylceramide) glucuronosyltransferase |
| black | Gbar_D05G029400 | AT5G38280 | Putative receptor serine/threonine kinase PR5K (PR5K) mRNA, PR5-like receptor kinase |
| black | Gbar_D06G004430 | AT1G77720 | Encodes a predicted protein kinase based on sequence similarity |
| black | Gbar_A05G042760 | AT3G24560 | Novel gene involved in embryogenesis |
| black | Gbar_A09G027280 | AT5G01410 | Encodes a protein predicted to function in tandem with PDX2 to form glutamine amidotransferase complex with involved in vitamin B6 biosynthesis |
| black | Gbar_D01G003220 | AT1G11330 | S-locus lectin protein kinase family protein |
| black | Gbar_D13G023280 | AT2G42520 | P-loop containing nucleoside triphosphate hydrolases superfamily protein |
| black | Gbar_D12G013250 | AT5G26751 | Encodes a SHAGGY-related kinase involved in meristem organization |
| black | Gbar_D11G007720 | AT3G01090 | Encodes a SNF1-related protein kinase that physically interacts with SCF subunit SKP1/ASK1 and 20S proteosome subunit PAD1 |
| black | Gbar_D03G011160 | AT3G19930 | Encodes a sucrose hydrogen symporter that is induced by wounding |
| black | Gbar_D13G001830 | AT5G13820 | Encodes a protein that specifically binds plant telomeric DNA repeats |
| black | Gbar_D05G019900 | AT5G42710 | Hypothetical protein |
| black | Gbar_D02G004390 | AT3G23230 | Encodes a member of the ERF (ethylene response factor) subfamily B-3 of ERF/AP2 transcription factor family |
| black | Gbar_D11G017280 | AT3G02320 | N2,N2-dimethylguanosine tRNA methyltransferase |
| black | Gbar_D12G022400 | AT4G17610 | tRNA/rRNA methyltransferase (SpoU) family protein |
| black | Gbar_D13G025510 | AT2G30110 | Encodes a ubiquitin-activating enzyme (E1), involved in the first step in conjugating multiple ubiquitins to proteins targeted for degradation |
| darkolivegreen | Gbar_A07G025160 | AT5G58520 | Protein kinase superfamily protein |
| darkolivegreen | Gbar_D03G003590 | AT4G39790 | bZIP transcription factor, putative (DUF630 and DUF632) |
| darkolivegreen | Gbar_D03G001100 | AT4G36180 | Leucine-rich receptor-like protein kinase family protein |
| darkolivegreen | Gbar_D01G009920 | AT4G26830 | O-Glycosyl hydrolases family 17 protein |
| darkolivegreen | Gbar_D01G016760 | AT3G26430 | Encodes a functioning member of the GDS(L) lipase family with preference for long chain substrates that does not hydrolyze choline esters |
| darkolivegreen | Gbar_D03G001880 | AT5G66560 | Phototropic-responsive NPH3 family protein |
| darkolivegreen | Gbar_A13G025790 | AT1G55690 | Sec14p-like phosphatidylinositol transfer family protein |
| darkolivegreen | Gbar_D01G007300 | AT5G54855 | Pollen Ole e 1 allergen and extensin family protein |
| darkolivegreen | Gbar_A05G043880 | AT3G01790 | Ribosomal protein L13 family protein |
| darkolivegreen | Gbar_D13G016400 | AT1G61600 | DUF1262 family protein (DUF1262) |
| darkolivegreen | Gbar_D02G013480 | AT1G14190 | Glucose-methanol-choline (GMC) oxidoreductase family protein |
| darkolivegreen | Gbar_D13G010360 | AT2G37400 | Tetratricopeptide repeat (TPR)-like superfamily protein |
| darkolivegreen | Gbar_D01G007390 | AT1G50660 | Actin cytoskeleton-regulatory complex pan-like protein |
| darkolivegreen | Gbar_D03G014040 | AT5G60050 | BTB/POZ domain-containing protein |
| darkolivegreen | Gbar_D02G017350 | AT1G60590 | Pectin lyase-like superfamily protein |
| darkolivegreen | Gbar_D02G014970 | AT1G31050 | Basic helix-loop-helix (bHLH) DNA-binding superfamily protein |
| darkolivegreen | Gbar_D01G022140 | AT3G53190 | Pectin lyase-like superfamily protein |
| darkolivegreen | Gbar_D03G008970 | AT4G00910 | Aluminum activated malate transporter family protein |
| darkolivegreen | Gbar_D12G024850 | AT4G24060 | Dof-type zinc finger DNA-binding family protein |
| darkolivegreen | Gbar_A13G005740 | AT1G11120 | CTTNBP 2 amino-terminal-like protein |
| darkolivegreen | Gbar_D02G016040 | AT2G23520 | Pyridoxal phosphate (PLP)-dependent transferases superfamily protein |
| darkolivegreen | Gbar_D01G005690 | AT4G21700 | DUF2921 family protein, putative (DUF2921) |
| darkolivegreen | Gbar_D13G019080 | AT1G54820 | Protein kinase superfamily protein |
| darkolivegreen | Gbar_D03G002350 | AT5G67130 | PLC-like phosphodiesterases superfamily protein |
| darkolivegreen | Gbar_D03G012130 | AT5G24320 | Transducin/WD40 repeat-like superfamily protein |
| darkolivegreen | Gbar_A13G006430 | AT3G20015 | Eukaryotic aspartyl protease family protein |
| darkolivegreen | Gbar_D01G006010 | AT5G05520 | Outer membrane OMP85 family protein |
| darkolivegreen | Gbar_D11G035890 | AT2G38090 | Duplicated homeodomain-like superfamily protein |
| darkolivegreen | Gbar_D12G026880 | AT5G23760 | Copper transport protein family |
| darkolivegreen | Gbar_D02G017340 | AT1G23460 | Pectin lyase-like superfamily protein |
| darkolivegreen | Gbar_D03G008720 | AT3G61920 | UvrABC system protein C |
| darkolivegreen | Gbar_D02G019700 | AT4G28100 | Transmembrane protein |
| darkolivegreen | Gbar_D02G017750 | AT1G60010 | D-ribose-binding periplasmic protein |
| darkolivegreen | Gbar_D01G003090 | AT1G79030 | Chaperone DnaJ-domain superfamily protein |
| darkolivegreen | Gbar_D03G014810 | AT5G22930 | Enabled-like protein (DUF1635) |
| darkolivegreen | Gbar_D13G021850 | AT3G14470 | NB-ARC domain-containing disease resistance protein |
| darkolivegreen | Gbar_D13G007120 | AT1G25530 | Transmembrane amino acid transporter family protein |
| darkolivegreen | Gbar_D02G021450 | AT1G58120 | Hypothetical protein |
| darkolivegreen | Gbar_D01G005820 | AT5G17680 | Disease resistance protein (TIR-NBS-LRR class) |
| darkolivegreen | Gbar_A13G019670 | AT4G31330 | Transmembrane protein, putative (Protein of unknown function, DUF599) |
| darkolivegreen | Gbar_D02G014500 | AT3G02820 | Zinc knuckle (CCHC-type) family protein |
| darkolivegreen | Gbar_A13G023560 | AT1G07970 | Cytochrome B561, amino-terminal protein |
| darkolivegreen | Gbar_D02G008760 | AT1G22030 | BPS1-like protein |
| darkolivegreen | Gbar_D01G021510 | AT3G10120 | Hypothetical protein |
| darkolivegreen | Gbar_D02G019240 | AT4G28310 | Microtubule-associated protein |
| darkolivegreen | Gbar_A13G023180 | AT3G51950 | Zinc finger (CCCH-type) family protein / RNA recognition motif (RRM)-containing protein |
| darkolivegreen | Gbar_A13G017430 | AT5G17600 | RING/U-box superfamily protein |
| darkolivegreen | Gbar_D01G020700 | AT4G24380 | Dihydrofolate reductase |
| darkolivegreen | Gbar_Scaffold2990G000010 | AT3G22142 | Encodes a Protease inhibitor/seed storage/LTP family protein |
| darkolivegreen | Gbar_D02G009130 | AT1G09750 | Eukaryotic aspartyl protease family protein |
| darkolivegreen | Gbar_D03G000630 | AT2G27090 | bZIP transcription factor (DUF630 and DUF632) |
| darkolivegreen | Gbar_A13G008810 | AT2G03350 | DUF538 family protein (Protein of unknown function, DUF538) |
| darkolivegreen | Gbar_D02G001120 | AT1G05710 | Basic helix-loop-helix (bHLH) DNA-binding superfamily protein |
| darkolivegreen | Gbar_D02G025330 | AT1G09812 | Multidrug resistance protein |
| darkolivegreen | Gbar_D13G015880 | AT1G53050 | Protein kinase superfamily protein |
| darkolivegreen | Gbar_A13G011970 | AT3G05190 | D-aminoacid aminotransferase-like PLP-dependent enzymes superfamily protein |
| darkolivegreen | Gbar_D02G020480 | AT5G08540 | Ribosomal RNA small subunit methyltransferase J |
| darkolivegreen | Gbar_D13G000740 | AT5G05180 | Myosin heavy chain, striated protein |
| darkolivegreen | Gbar_A13G008440 | AT1G10380 | Putative membrane lipoprotein |
| darkolivegreen | Gbar_D02G023270 | AT4G34480 | O-Glycosyl hydrolases family 17 protein |
| darkolivegreen | Gbar_D03G015720 | AT4G23500 | Pectin lyase-like superfamily protein |
| darkolivegreen | Gbar_D02G001660 | AT3G48400 | Cysteine/Histidine-rich C1 domain family protein |
| darkolivegreen | Gbar_D02G023930 | AT2G21290 | 30S ribosomal protein S31 |
| darkolivegreen | Gbar_D12G024650 | AT4G23895 | Pleckstrin homology (PH) domain-containing protein |
| darkolivegreen | Gbar_D12G023010 | AT4G16510 | YbaK/aminoacyl-tRNA synthetase-associated domain-containing protein |
| darkolivegreen | Gbar_D02G004850 | AT5G43960 | Nuclear transport factor 2 (NTF2) family protein with RNA binding (RRM-RBD-RNP motifs) domain-containing protein |
| darkolivegreen | Gbar_D03G009270 | AT1G33120 | Ribosomal protein L6 family |
| darkolivegreen | Gbar_D01G015160 | AT3G19850 | Phototropic-responsive NPH3 family protein |
| darkolivegreen | Gbar_D03G008670 | AT4G23930 | Late embryogenesis abundant (LEA) hydroxyproline-rich glycoprotein family |
| darkolivegreen | Gbar_D02G023430 | AT4G39210 | Encodes the large subunit of ADP-Glucose Pyrophosphorylase which catalyzes the first, rate limiting step in starch biosynthesis |
| darkolivegreen | Gbar_A13G019130 | AT1G22490 | Basic helix-loop-helix (bHLH) DNA-binding superfamily protein |
| darkolivegreen | Gbar_D11G036510 | AT1G70000 | Encodes a MYB-like Domain transcription factor that plays a positive role in anthocyanin accumulation in response to light and cytokinin via repression of MYBL2 |
| darkolivegreen | Gbar_D13G008600 | AT1G58520 | GDSL-like lipase/acylhydrolase superfamily protein |
| darkolivegreen | Gbar_D12G028470 | AT3G48210 | kinetochore protein |
| darkolivegreen | Gbar_D03G009120 | AT4G10850 | Nodulin MtN3 family protein |
| darkolivegreen | Gbar_A13G003860 | AT3G19100 | Encodes a protein kinase that positively regulates gibberellic acid (GA) signaling by inactivating the E3 ubiquitin ligase GARU |
| darkolivegreen | Gbar_D02G023340 | AT2G21660 | Encodes a small glycine-rich RNA binding protein that is part of a negative-feedback loop through which AtGRP7 regulates the circadian oscillations of its own transcript |
| darkolivegreen | Gbar_D01G008490 | AT4G14540 | nuclear factor Y, subunit B3 |
| darkolivegreen | Gbar_D01G011180 | AT1G47250 | Encodes 20S proteasome subunit PAF2 (PAF2). |
| darkolivegreen | Gbar_D03G005030 | AT1G65060 | Encodes an isoform of 4-coumarate:CoA ligase (4CL), which is involved in the last step of the general phenylpropanoid pathway |
| darkolivegreen | Gbar_D02G024250 | AT2G16700 | Encodes actin depolymerizing factor 5 (ADF5). |
| darkolivegreen | Gbar_D02G024080 | AT4G34860 | Plant neutral invertase family protein |
| darkolivegreen | Gbar_D11G036750 | AT1G68560 | Encodes a bifunctional alpha-l-arabinofuranosidase/beta-d-xylosidase that belongs to family 3 of glycoside hydrolases |
| darkolivegreen | Gbar_D01G012240 | AT5G49630 | Is a high affinity amino acid transporter capable of transporting aspartate and tryptophan |
| darkolivegreen | Gbar_A13G017880 | AT5G12380 | Annexin 8 |
| darkolivegreen | Gbar_D01G021800 | AT3G23620 | BRIX domain containing protein, similar to RNA biogenesis factors in yeast. Binds rRNA and likely also functions in RNA biogenesis in Arabidopsis |
| darkolivegreen | Gbar_D03G012140 | AT5G24330 | Encodes a SET-domain protein, a H3K27 monomethyltransferases required for chromatin structure and gene silencing |
| darkolivegreen | Gbar_D01G012780 | AT2G26430 | Encodes an ania-6a type arginine-rich cyclin which confers tolerance to LiCl and NaCl when expressed in yeast. |
| darkolivegreen | Gbar_D12G027500 | AT4G25320 | AT hook motif DNA-binding family protein |
| darkolivegreen | Gbar_D13G014580 | AT3G13080 | Encodes an ATP-dependent MRP-like ABC transporter able to transport glutathione-conjugates as well as chlorophyll catabolites |
| darkolivegreen | Gbar_D03G001120 | AT4G38960 | BBX19 is a B-box containing transcriptional regulator involved in photomporphogenesis and flowering. |
| darkolivegreen | Gbar_D02G019950 | AT3G21890 | B-box type zinc finger family protein |
| darkolivegreen | Gbar_D02G025320 | AT5G65700 | Encodes a CLAVATA1-related receptor kinase-like protein required for both shoot and flower meristem function |
| darkolivegreen | Gbar_D02G025580 | AT4G20270 | Encodes a CLAVATA1-related receptor kinase-like protein required for both shoot and flower meristem function. It has a broad expression pattern and is involved in vascular strand development in the leaf, control of leaf shape, size and symmetry, male gametophyte development and ovule specification and function. The mRNA is cell-to-cell mobile. |
| darkolivegreen | Gbar_D02G024640 | AT3G12500 | Encodes a basic chitinase involved in ethylene/jasmonic acid mediated signalling pathway during systemic acquired resistance based on expression analyses. |
| darkolivegreen | Gbar_D03G003360 | AT5G65640 | bHLH093/NFL encodes a bHLH transcription factor involved in GA mediated control of flowering time |
| darkolivegreen | Gbar_D02G020910 | AT5G64570 | Encodes a beta-d-xylosidase that belongs to family 3 of glycoside hydrolases. |
| darkolivegreen | Gbar_D13G020070 | AT4G31910 | Encodes an acyltransferase that can modify brassinosteroids (BRs) by acylation and may modulate endogenous BR levels. |
| darkolivegreen | Gbar_D03G005940 | AT3G16300 | Uncharacterized protein family (UPF0497) |
| darkolivegreen | Gbar_D12G022380 | AT5G04770 | Encodes a member of the cationic amino acid transporter (CAT) subfamily of amino acid polyamine choline transporters |
| darkolivegreen | Gbar_A13G017960 | AT1G55850 | Encodes a protein similar to cellulose synthase The mRNA is cell-to-cell mobile. |
| darkolivegreen | Gbar_D04G001300 | AT5G05270 | Chalcone-flavanone isomerase family protein |
| darkolivegreen | Gbar_Scaffold769G000030 | AT1G75820 | Putative receptor kinase with an extracellular leucine-rich domain |
| darkolivegreen | Gbar_D02G004490 | AT5G43330 | Predicted to encode a cytosolic malate dehydrogenase |
| darkolivegreen | Gbar_D01G000430 | AT2G16370 | Encodes a bifunctional dihydrofolate reductase - thymidylate synthase gene |
| darkolivegreen | Gbar_A13G004830 | AT5G48490 | Encodes a protein with similarity to a lipid transfer protein that may contribute to systemic acquired resistance (SAR). |
| darkolivegreen | Gbar_D05G039960 | AT3G13310 | Chaperone DnaJ-domain superfamily protein |
| darkolivegreen | Gbar_A13G003970 | AT2G15690 | Encodes an atypical PPR-DYW protein |
| darkolivegreen | Gbar_D12G028430 | AT1G18330 | EARLY-PHYTOCHROME-RESPONSIVE1 |
| darkolivegreen | Gbar_D01G004350 | AT5G20480 | Encodes a predicted leucine-rich repeat receptor kinase (LRR-RLK). Functions as the receptor for bacterial PAMP (pathogen associated molecular patterns). |
| darkolivegreen | Gbar_A13G017640 | AT5G17710 | Chloroplast GrpE protein involved in chloroplastic response to heat stress and the correct oligomerization of the photosynthesis-related LHCII complex. |
| darkolivegreen | Gbar_D03G006920 | AT1G21390 | Embryo defective 2170 |
| darkolivegreen | Gbar_D12G027570 | AT5G51550 | EXORDIUM like 3 |
| darkolivegreen | Gbar_A13G000700 | AT2G40610 | Member of Alpha-Expansin Gene Family |
| darkolivegreen | Gbar_D02G012070 | AT2G43280 | Encodes one of four FRS (FAR1-RELATED SEQUENCE) factor-like genes in Arabidopsis |
| darkolivegreen | Gbar_D13G016510 | AT1G53520 | Encodes a plastid stroma localized fatty acid binding protein involved in fatty acid metabolism. |
| darkolivegreen | Gbar_A06G022760 | AT1G78020 | FCS like zinc finger 6 is induced during energy starvation through SnRK1 signaling |
| darkolivegreen | Gbar_A13G006340 | AT5G23310 | Fe superoxide dismutase |
| darkolivegreen | Gbar_D13G024310 | AT2G27510 | ferredoxin 3 |
| darkolivegreen | Gbar_D02G000800 | AT2G43800 | Localizes to plasmodesmata (PD) through its transmembrane domain and is required for normal intercellular trafficking |
| darkolivegreen | Gbar_D03G001360 | AT1G47260 | Encodes mitochondrial gamma carbonic anhydrase. Component of the NADH dehydrogenase complex. |
| darkolivegreen | Gbar_A13G019010 | AT1G54690 | Encodes HTA3, a histone H2A protein. H2AX is a meiosis-specific isoform of histone H2A |
| darkolivegreen | Gbar_D01G022110 | AT2G36830 | Encodes a tonoplast intrinsic protein, which functions as water channel |
| darkolivegreen | Gbar_D02G018320 | AT1G68360 | Encodes a nuclear localized member of the C2H2 family of TFIIIA transcription factors |
| darkolivegreen | Gbar_D13G009120 | AT1G05200 | Encodes a putative glutamate receptor GLR3 with dual localization in plastid and plasma membrane. |
| darkolivegreen | Gbar_D01G015560 | AT1G70710 | endo-1,4-beta-glucanase. Involved in cell elongation. |
| darkolivegreen | Gbar_D13G002820 | AT4G39010 | Cellulase involved in cell wall modification during valve dehiscence. |
| darkolivegreen | Gbar_D02G018880 | AT1G28130 | Encodes an IAA-amido synthase that conjugates Asp and other amino acids to auxin in vitro |
| darkolivegreen | Gbar_D01G005250 | AT3G15095 | Encodes HCF243 (high chlorophyll fluorescence), a chloroplast-localized protein involved in the D1 protein stability of the photosystem II complex1. |
| darkolivegreen | Gbar_A13G002560 | AT5G03260 | LAC11 is a putative laccase, a member of laccase family of genes (17 members in Arabidopsis). |
| darkolivegreen | Gbar_D12G028450 | AT5G50150 | LOTR1 protein has an unknown function. It contains both DUF4409 and DUF239 domains |
| darkolivegreen | Gbar_D01G021830 | AT2G36530 | Involved in light-dependent cold tolerance and encodes an enolase |
| darkolivegreen | Gbar_D02G011760 | AT3G11710 | lysyl-tRNA synthetase 1 |
| darkolivegreen | Gbar_D03G009750 | AT3G47520 | Encodes a protein with NAD-dependent malate dehydrogenase activity, located in chloroplasts |
| darkolivegreen | Gbar_D02G004910 | AT4G08850 | MIK1 encodes a receptor kinase that forms a complex with MDIS1/MIK2 and binds LURE1, the female pollen guidance chemi-attractant |
| darkolivegreen | Gbar_D03G008120 | AT1G64080 | Encodes a member of the MAKR (MEMBRANE-ASSOCIATED KINASE REGULATOR) gene family |
| darkolivegreen | Gbar_D13G023490 | AT5G12970 | Calcium-dependent lipid-binding (CaLB domain) plant phosphoribosyltransferase family protein |
| darkolivegreen | Gbar_D03G013750 | AT3G46130 | Encodes a putative transcription factor (MYB48) that functions to regulate flavonol biosynthesis primarily in cotyledons. |
| darkolivegreen | Gbar_D11G035630 | AT5G14750 | Encodes a MyB-related protein containing R2 and R3 repeats, involved in root and hypocotyl epidermal cell fate determination |
| darkolivegreen | Gbar_D13G007380 | AT2G02820 | Encodes a putative transcription factor (MYB88), involved in stomata development, double loss of MYB88 and FLP (MYB124) activity results in a failure of guard mother cells (GMCs) to adopt the guard cell fate, thus they continue to divide resulting in abnormal stomata consisting of clusters of numerous guard cell-like cells |
| darkolivegreen | Gbar_D02G015320 | AT1G70750 | Myosin-binding protein |
| darkolivegreen | Gbar_D03G017750 | AT4G17980 | Encodes ANAC071, a transcription factor involved in cell proliferation in incised inflorescence stems. |
| darkolivegreen | Gbar_A11G035350 | AT5G16000 | NSP-interacting kinase (NIK1), receptor-like kinase, involved in defense response against geminivirus |
| darkolivegreen | Gbar_A13G010550 | AT3G25560 | NSP-interacting kinase 2 |
| darkolivegreen | Gbar_D11G035920 | AT3G09070 | Encodes a polarly localised membrane-associated protein that regulates phloem differentiation entry. |
| darkolivegreen | Gbar_D12G028320 | AT1G73220 | Encodes Organic Cation Transporter 1 (OCT1), likely to be involved in polyamine transport. |
| darkolivegreen | Gbar_D03G016160 | AT4G11650 | osmotin-like protein |
| darkolivegreen | Gbar_D02G000480 | AT5G01840 | Encodes a member of the plant specific ovate protein family |
| darkolivegreen | Gbar_D12G027290 | AT5G51890 | encodes peroxidase involved in the lignification of tracheary elements (TE) in roots |
| darkolivegreen | Gbar_D01G021840 | AT3G52960 | Thioredoxin superfamily protein |
| darkolivegreen | Gbar_A13G018460 | AT1G08650 | Encodes a phosphoenolpyruvate carboxylase kinase that is expressed at highest levels in leaves |
| darkolivegreen | Gbar_D01G022280 | AT2G37170 | A member of the plasma membrane intrinsic protein subfamily PIP2 |
| darkolivegreen | Gbar_D13G008460 | AT1G18650 | Encodes a member of the X8-GPI family of proteins |
| darkolivegreen | Gbar_D02G006410 | AT1G48100 | Pectin lyase-like superfamily protein |
| darkolivegreen | Gbar_D02G018990 | AT1G03860 | prohibitin 2 |
| darkolivegreen | Gbar_A13G004330 | AT4G34220 | Encodes a receptor like kinase involved in ABA-mediated seedling development and drought tolerance |
| darkolivegreen | Gbar_D13G003440 | AT3G18990 | Essential for the complete repression of FLC in vernalized plants |
| darkolivegreen | Gbar_D03G016800 | AT4G22790 | Encodes a plasma membrane localized MATE type transporter that is involved in CO2 signaling during stomatal aperture regulation |
| darkolivegreen | Gbar_D13G013740 | AT1G79380 | Encodes a ubiquitin ligase that is an essential upstream modulator of JA signaling in response to various stimuli. |
| darkolivegreen | Gbar_D13G009470 | AT3G26420 | Zinc finger-containing glycine-rich RNA-binding protein |
| darkolivegreen | Gbar_D03G015290 | AT2G30280 | Encodes RDM4, a transcriptional regulator functioning in RNA-directed DNA methylation and plant development. |
| darkolivegreen | Gbar_D01G017350 | AT2G16600 | Encodes cytosolic cyclophilin ROC3. The mRNA is cell-to-cell mobile. |
| darkolivegreen | Gbar_D03G003950 | AT5G10350 | Encodes a nuclear-localized RNA recognition motif-containing protein that forms homodimers |
| darkolivegreen | Gbar_A13G019760 | AT4G31580 | Encodes a Serine/arginine-rich (SR) protein RSZp22 |
| darkolivegreen | Gbar_D03G015520 | AT5G38410 | Encodes a member of the Rubisco small subunit (RBCS) multigene family |
| darkolivegreen | Gbar_D01G013700 | AT1G17020 | Encodes a novel member of the Fe(II)/ascorbate oxidase gene family; senescence-related gene. |
| darkolivegreen | Gbar_D01G008320 | AT3G13110 | Encodes a mitochondrial serine O-acetyltransferase involved in sulfur assimilation and cysteine biosynthesis. Expressed in the vascular system. |
| darkolivegreen | Gbar_D02G010370 | AT1G75520 | A member of SHI gene family. Arabidopsis thaliana has ten members that encode proteins with a RING finger-like zinc finger motif |
| darkolivegreen | Gbar_D01G014830 | AT2G01940 | Encodes a transcription factor that, together with IDD14 and IDD16, regulates auxin biosynthesis and transport and thus aerial organ morphogenesis and gravitropic responses |
| darkolivegreen | Gbar_A13G000980 | AT3G52490 | Encodes a member of an eight-gene family (SMAX1 and SMAX1-like) that has weak similarity to AtHSP101, a ClpB chaperonin required for thermo tolerance. |
| darkolivegreen | Gbar_D01G008860 | AT5G56040 | Leucine-rich receptor-like protein kinase family protein |
| darkolivegreen | Gbar_D01G019160 | AT1G58100 | Encodes TCP8, belongs to the TCP transcription factor family known to bind site II elements in promoter regions. |
| darkolivegreen | Gbar_D12G023520 | AT2G45680 | TCP family transcription factor |
| darkolivegreen | Gbar_D02G008770 | AT1G77920 | bZIP transcription factor family protein |
| darkolivegreen | Gbar_D05G039710 | AT5G55860 | WEB1/PMI2 related protein involved in mecahno transduction |
| darkolivegreen | Gbar_A13G002180 | AT3G55120 | Catalyzes the conversion of chalcones into flavanone |
| darkolivegreen | Gbar_D01G024260 | AT2G37025 | TRF-like 8 |
| darkolivegreen | Gbar_D13G008210 | AT1G07250 | UDP-glucosyl transferase 71C4 |
| darkolivegreen | Gbar_D02G002150 | AT1G05530 | Encodes a protein with glucosyltransferase activity with high sequence homology to UGT1 (AT1G05560) |
| darkolivegreen | Gbar_D03G008130 | AT3G16520 | UDP-glucosyl transferase 88A1 |
| darkolivegreen | Gbar_D01G020070 | AT4G08300 | Nodulin MtN21-like transporter family protein |
| darkolivegreen | Gbar_D02G017680 | AT1G70260 | Encodes an endoplasmic reticulum (ER)-localized nodulin MtN21-like transporter family protein that negatively regulates resistance against biotrophic pathogens but not the necrotrophic pathogen, B. cinerea, possibly by regulating ROS production, cell death and PR1 expression. |
| darkolivegreen | Gbar_A13G002730 | AT2G01770 | Encodes an iron transporter required for iron sequestration into vacuoles |
| darkolivegreen | Gbar_D03G003330 | AT2G22680 | Zinc finger (C3HC4-type RING finger) family protein |
| darkolivegreen | Gbar_D13G002720 | AT2G01830 | Histidine kinase: cytokinin-binding receptor that transduces cytokinin signals across the plasma membrane |
| darkolivegreen | Gbar_D13G017670 | AT3G03660 | Encodes a WUSCHEL-related homeobox gene family member with 65 amino acids in its homeodomain |
| darkolivegreen | Gbar_D13G014440 | AT1G55910 | Member of Putative zinc transporter ZIP2 - like family |
| mediumpurple2 | Gbar_D13G017100 | AT2G36780 | UDP-Glycosyltransferase superfamily protein |
| mediumpurple2 | Gbar_D12G013470 | AT4G02010 | Protein kinase superfamily protein |
| mediumpurple2 | Gbar_D05G025460 | AT1G33810 | Zinc finger/BTB domain protein |
| mediumpurple2 | Gbar_A05G035880 | AT5G14390 | Alpha/beta-Hydrolases superfamily protein |
| mediumpurple2 | Gbar_A10G019000 | AT2G40800 | Import inner membrane translocase subunit |
| mediumpurple2 | Gbar_A11G026760 | AT1G60690 | NAD(P)-linked oxidoreductase superfamily protein |
| mediumpurple2 | Gbar_D06G023300 | AT5G60710 | Zinc finger (C3HC4-type RING finger) family protein |
| mediumpurple2 | Gbar_A08G010720 | AT4G25610 | C2H2-like zinc finger protein |
| mediumpurple2 | Gbar_D02G025440 | AT2G34670 | Benzoyl-CoA reductase subunit C, putative (DUF630 and DUF632) |
| mediumpurple2 | Gbar_D05G005440 | AT2G24230 | Leucine-rich repeat protein kinase family protein |
| mediumpurple2 | Gbar_D09G012540 | AT3G57080 | Non-catalytic subunit unique to Nuclear DNA-dependent RNA polymerase V; homologous to budding yeast RPB5. |
| mediumpurple2 | Gbar_D13G019810 | AT4G31300 | Encodes 20S proteasome subunit PBA1 (PBA1). PBA1 acts as a plant caspase-3-like enzyme. |
| mediumpurple2 | Gbar_D13G011390 | AT5G28840 | Encodes a protein with GDP-D-mannose 3',5'-epimerase activity. The enzyme is involved in ascorbate biosynthesis |
| mediumpurple2 | Gbar_A11G029250 | AT1G56450 | 20S proteasome beta subunit PBG1 (PBG1) mRNA, complete cds |
| mediumpurple2 | Gbar_D10G008010 | AT1G80070 | Encodes a factor that influences pre-mRNA splicing and is required for embryonic development |
| mediumpurple2 | Gbar_A12G004970 | AT4G20070 | The gene encoding Arabidopsis thaliana Allantoate Amidohydrolase (AtAAH) which catalyzes the allantoate deiminase reaction |
| mediumpurple2 | Gbar_D05G011510 | AT3G21180 | Encodes an autoinhibited Ca(2)-ATPase that contains an N-terminal calmodulin binding autoinhibitory domain. |
| mediumpurple2 | Gbar_D08G006200 | AT3G63520 | Encodes a protein with 9-cis-epoxycarotenoid dioxygenase activity |
| mediumpurple2 | Gbar_D01G010230 | AT5G42310 | Encodes a member of the Arabidopsis PPR family |
| mediumpurple2 | Gbar_D01G008710 | AT5G47750 | D6PK family kinase involved in pulse-induced phototropism and continuous light-induced hypocotyl phototropism, minor contribution to time-dependent phototropism. |
| mediumpurple2 | Gbar_D07G010350 | AT1G07810 | Encodes an ER-type Ca2+pumping ATPase. The mRNA is cell-to-cell mobile. |
| mediumpurple2 | Gbar_D08G017480 | AT1G31930 | Encodes XLG3 (extra-large G protein 3) that shows significant similarity to the G protein alpha subunit in its C terminal region |
| mediumpurple2 | Gbar_A07G003530 | AT2G25080 | Encodes glutathione peroxidase. The mRNA is cell-to-cell mobile. |
| mediumpurple2 | Gbar_A11G016340 | AT2G30860 | Encodes glutathione transferase belonging to the phi class of GSTs |
| mediumpurple2 | Gbar_A09G017200 | AT5G02790 | Glutathione S-transferase family protein |
| mediumpurple2 | Gbar_D11G012220 | AT3G06720 | Encodes importin alpha involved in nuclear import |
| mediumpurple2 | Gbar_A13G010590 | AT1G13580 | Encodes a ceramide synthase that together with LOH1 is essential for production of ceramides containing Very Long Chain Fatty acid VLCFA-Ceramides. |
| mediumpurple2 | Gbar_D02G015200 | AT1G23260 | MMZ1/UEV1A encodes a protein that may play a role in DNA damage responses and error-free post-replicative DNA repair by participating in lysine-63-based polyubiquitination reactions |
| mediumpurple2 | Gbar_D09G013360 | AT3G57880 | Required for maintenance of inflorescence and shoot SAMs and normal development of the derived vascular cambium, functions in the SAM to promote continuous organogenesis, affects SAM development through STM, where it affects intracellular localization of STM in SAM cells in the peripheral region and prevents STM localization toward the cell wall of SAM cells in the peripheral region. |
| mediumpurple2 | Gbar_A06G015880 | AT4G04880 | Adenosine/AMP deaminase family protein |
| mediumpurple2 | Gbar_A06G015500 | AT1G53840 | Encodes a pectin methylesterase |
| mediumpurple2 | Gbar_A09G009100 | AT5G13640 | Arabidopsis phospholipid:diacylglycerol acyltransferase (PDAT) |
| mediumpurple2 | Gbar_A12G002960 | AT2G14260 | Encodes proline iminopeptidase |
| mediumpurple2 | Gbar_A11G032430 | AT1G08540 | Enodes a subunit of chloroplast RNA polymerase, confers the ability to recognize promoter sequences on the core enzyme. SIG1 is induced by red and blue light. |
| mediumpurple2 | Gbar_D12G004810 | AT3G12800 | Short-chain dehydrogenase-reductase B |
| mediumpurple2 | Gbar_D13G009630 | AT5G16270 | Encodes a SCC1/REC8 ortholog that may be involved in mitosis and may represent a mitotic cohesin |
| mediumpurple2 | Gbar_D12G018730 | AT1G76160 | SKU5 similar 5 |
| mediumpurple2 | Gbar_D13G012850 | AT1G73720 | Encodes SMU1, a protein involved in RNA splicing. |
| mediumpurple2 | Gbar_D11G002190 | AT1G63800 | ubiquitin-conjugating enzyme 5 |
| mediumpurple2 | Gbar_A11G026970 | AT3G04870 | Involved in the biosynthesis of carotenes and xanthophylls, reduces zeta-carotene to lycopene. |
| mediumpurple3 | Gbar_D13G023330 | AT1G58170 | Disease resistance-responsive (dirigent-like protein) family protein |
| mediumpurple3 | Gbar_A11G019370 | AT2G23450 | Protein kinase superfamily protein |
| mediumpurple3 | Gbar_A04G004150 | AT1G53440 | Leucine-rich repeat transmembrane protein kinase |
| mediumpurple3 | Gbar_A08G011260 | AT1G64300 | Protein kinase family protein |
| mediumpurple3 | Gbar_A13G015520 | AT5G55340 | MBOAT (membrane bound O-acyl transferase) family protein |
| mediumpurple3 | Gbar_D10G024280 | AT3G55470 | Calcium-dependent lipid-binding (CaLB domain) family protein |
| mediumpurple3 | Gbar_D03G018360 | AT4G19010 | Encodes for a 4-coumarate-CoA ligase involved in the biosynthesis of the benzenoid ring of ubiquinone from phenylalanine. |
| mediumpurple3 | Gbar_D11G004420 | AT4G23340 | 2-oxoglutarate (2OG) and Fe(II)-dependent oxygenase superfamily protein |
| mediumpurple3 | Gbar_A06G011840 | AT1G22750 | Transmembrane protein |
| mediumpurple3 | Gbar_D13G003580 | AT2G15780 | Cupredoxin superfamily protein |
| mediumpurple3 | Gbar_D13G003030 | AT2G21520 | Sec14p-like phosphatidylinositol transfer family protein |
| mediumpurple3 | Gbar_D05G014810 | AT4G19880 | Glutathione S-transferase family protein |
| mediumpurple3 | Gbar_D12G011070 | AT2G45300 | Encodes 3-phosphoshikimate 1-carboxyvinyltransferase / 5-enolpyruvylshikimate-3-phosphate / EPSP synthase involved in chorismate biosynthesis |
| mediumpurple3 | Gbar_A12G016890 | AT1G05210 | Transmembrane protein 97, Putative |
| mediumpurple3 | Gbar_A07G021160 | AT3G07270 | GTP cyclohydrolase I |
| mediumpurple3 | Gbar_D13G022570 | AT1G52820 | 2-oxoglutarate (2OG) and Fe(II)-dependent oxygenase superfamily protein |
| mediumpurple3 | Gbar_A09G027420 | AT1G08230 | Codes for a H+driven, high affinity gamma-aminobutyric acid (GABA) transporter |
| mediumpurple3 | Gbar_A09G023570 | AT1G60680 | NAD(P)-linked oxidoreductase superfamily protein |
| mediumpurple3 | Gbar_D12G016630 | AT1G14590 | Nucleotide-diphospho-sugar transferase family protein |
| mediumpurple3 | Gbar_A11G014520 | AT5G48630 | Cyclin family protein |
| mediumpurple3 | Gbar_D09G004390 | AT4G23030 | MATE efflux family protein |
| mediumpurple3 | Gbar_D05G016380 | AT2G44350 | Encodes a mitochrondrion targeted citrate synthase, the first enzyme of the tricarboxylic acid cycle, catalyzing the condensation of acetyl-CoA and oxaloacetate, finally yielding citrate and CoA. |
| mediumpurple3 | Gbar_D12G014080 | AT3G62830 | Encodes an isoform of UDP-glucuronic acid decarboxylase, which is predicted to be membrane-bound by PSORT. |
| mediumpurple3 | Gbar_D01G008230 | AT5G25180 | Putative cytochrome P450 |
| mediumpurple3 | Gbar_D05G020260 | AT4G31940 | The gene encodes a cytochrome P450 enzyme, CYP82C |
| mediumpurple3 | Gbar_D13G011760 | AT3G21430 | DNA binding protein |
| mediumpurple3 | Gbar_D11G023600 | AT2G22250 | Encodes a prokaryotic-type plastidic aspartate aminotransferase with glutamate/aspartate-prephenate aminotransferase (PAT) activity |
| mediumpurple3 | Gbar_A12G004610 | AT2G34660 | Encodes a multidrug resistance-associated protein that is MgATP-energized glutathione S-conjugate pump |
| mediumpurple3 | Gbar_D11G011340 | AT2G37280 | Encodes an ATP-binding cassette (ABC) transporter |
| mediumpurple3 | Gbar_D10G026940 | AT3G57330 | Lesion mimic phenotype when mutation in the gene is combined with a mutation in ACA4 |
| mediumpurple3 | Gbar_D13G022050 | AT5G20230 | Encodes a Al-stress-induced gene. Along with TCF, it promotes lignin biosynthesis in response to cold stress |
| mediumpurple3 | Gbar_D11G036850 | AT5G06200 | Uncharacterized protein family (UPF0497) |
| mediumpurple3 | Gbar_D04G019360 | AT2G44050 | 6,7-dimethyl-8-ribityllumazine synthase / DMRL synthase / lumazine synthase / riboflavin synthase [Arabidopsis thaliana] |
| mediumpurple3 | Gbar_D11G018340 | AT1G65930 | Encodes a NADP+isocitrate dehydrogenase that is believed to function in the cytosol |
| mediumpurple3 | Gbar_D05G036060 | AT3G23550 | MATE efflux family protein |
| mediumpurple3 | Gbar_A07G006230 | AT5G08170 | Porphyromonas-type peptidyl-arginine deiminase family protein |
| mediumpurple3 | Gbar_D10G026400 | AT5G05580 | Encodes a temperature sensitive plastidic fatty acid desaturase. |
| mediumpurple3 | Gbar_A09G017280 | AT3G09270 | Encodes glutathione transferase belonging to the tau class of GSTs |
| mediumpurple3 | Gbar_A12G023890 | AT4G11820 | Encodes a protein with hydroxymethylglutaryl-CoA synthase activity which was characterized by phenotypical complementation of the S. cerevisiae mutant. |
| mediumpurple3 | Gbar_D11G013690 | AT4G02670 | Indeterminate(ID)-domain 12 |
| mediumpurple3 | Gbar_A10G001130 | AT4G05530 | Encodes a peroxisomal member of the short-chain dehydrogenase/reductase (SDR) family of enzymes |
| mediumpurple3 | Gbar_D07G009160 | AT3G22160 | JAV1 is a repressor of jasmonate-mediated defense responses. |
| mediumpurple3 | Gbar_D13G003050 | AT5G11880 | Meso-diaminopimelate decarboxylase which catalyzes the decarboxylation of mesodiaminopimelate |
| mediumpurple3 | Gbar_A10G014630 | AT2G43120 | Encodes a member of the functionally diverse cupin protein superfamily that is involved in susceptibility to the bacterial plant pathogen Ralstonia solanacearum |
| mediumpurple3 | Gbar_D05G025610 | AT4G10050 | Esterase/lipase/thioesterase family protein |
| mediumpurple3 | Gbar_D02G009200 | AT5G26780 | Encodes a protein with serine hydroxymethyltransferase activity which is thought to be localized in the mitochondrial matrix |
| mediumpurple3 | Gbar_D04G005270 | AT5G39950 | Encodes a cytosolic thioredoxin that reduces disulfide bridges of target proteins by the reversible formation of a disulfide bridge between two neighboring Cys residues present in the active site |
| mediumpurple3 | Gbar_D12G029450 | AT5G17990 | Encodes the tryptophan biosynthetic enzyme phosphoribosylanthranilate transferase (PAT1, called trpD in bacteria) |
| plum3 | Gbar_A12G021030 | AT5G07610 | F-box family protein |
| plum3 | Gbar_D12G026230 | AT5G53220 | Hypothetical protein |
| plum3 | Gbar_D12G022690 | AT4G16720 | Ribosomal protein L23/L15e family protein |
| plum3 | Gbar_A10G024460 | AT5G05960 | Bifunctional inhibitor/lipid-transfer protein/seed storage 2S albumin superfamily protein |
| plum3 | Gbar_D10G023440 | AT4G10720 | Ankyrin repeat family protein |
| plum3 | Gbar_D01G011220 | AT1G19530 | DNA polymerase epsilon catalytic subunit A |
| plum3 | Gbar_D11G009060 | AT2G45590 | Protein kinase superfamily protein |
| plum3 | Gbar_D07G007700 | AT1G53540 | HSP20-like chaperones superfamily protein |
| plum3 | Gbar_D07G005520 | AT5G50790 | Encodes a member of the SWEET sucrose efflux transporter family proteins. |
| plum3 | Gbar_D13G015090 | AT3G25585 | Amino alcohol phosphor transferase (AAPT2) |
| plum3 | Gbar_D11G009450 | AT2G45760 | Encodes a protein that is similar to BONZAI1-binding protein BAP1. |
| plum3 | Gbar_D05G023580 | AT1G24620 | Encodes an EF-hand calcium-binding protein family member |
| plum3 | Gbar_D09G001430 | AT5G39670 | Calmodulin like protein involved in negative regulation of pattern triggered immunity. |
| plum3 | Gbar_A11G016050 | AT2G32530 | Encodes a gene similar to cellulose synthase |
| plum3 | Gbar_A10G004080 | AT1G29160 | Encodes a DOF transcription factor involved in seed coat development. |
| plum3 | Gbar_D11G011430 | AT1G48300 | Cytosolic iron-sulfur protein |
| plum3 | Gbar_D07G005040 | AT5G51190 | Encodes a member of the ERF (ethylene response factor) subfamily B-3 of ERF/AP2 transcription factor family |
| plum3 | Gbar_D03G017340 | AT5G47230 | Encodes a member of the ERF (ethylene response factor) subfamily B-3 of ERF/AP2 transcription factor family (ATERF-5) |
| plum3 | Gbar_D11G017170 | AT3G51550 | Encodes a synergid-expressed, plasma-membrane localized receptor-like kinase that accumulates asymetrically in the synergid membrnane at the filiform apparatus and mediates male-female gametophyte interactions during pollen tube reception |
| plum3 | Gbar_D08G023540 | AT5G27320 | Encodes a gibberellin (GA) receptor ortholog of the rice GA receptor gene (OsGID1) |
| plum3 | Gbar_D13G019370 | AT5G20090 | MPC1 negatively regulates ABA enhanced slow anion channel function during stomatal closure. |
| plum3 | Gbar_D08G017720 | AT5G16770 | Member of the R2R3 factor gene family. |
| plum3 | Gbar_D03G012490 | AT3G46980 | Encodes an inorganic phosphate transporter (PHT4;3). |
| plum3 | Gbar_D12G022500 | AT4G16820 | Encodes a lipase that hydrolyzes phosphatidylcholine, glycolipids as well as triacylglycerols. |
| plum3 | Gbar_D05G005280 | AT2G25180 | Encodes an Arabidopsis response regulator (ARR) protein that acts in concert with other type-B ARRs in the cytokinin signaling pathway |
| plum3 | Gbar_D12G010820 | AT3G60220 | Encodes a putative RING-H2 zinc finger protein ATL4 (ATL4). |
| plum3 | Gbar_D11G004590 | AT2G18060 | Encodes a NAC-domain transcription factor that is expressed in developing vessels and protoxylem |
| plum3 | Gbar_D05G028190 | AT3G56400 | Function as activator of SA-dependent defense genes and a repressor of JA-regulated genes |
新窗口打开|下载CSV
图7
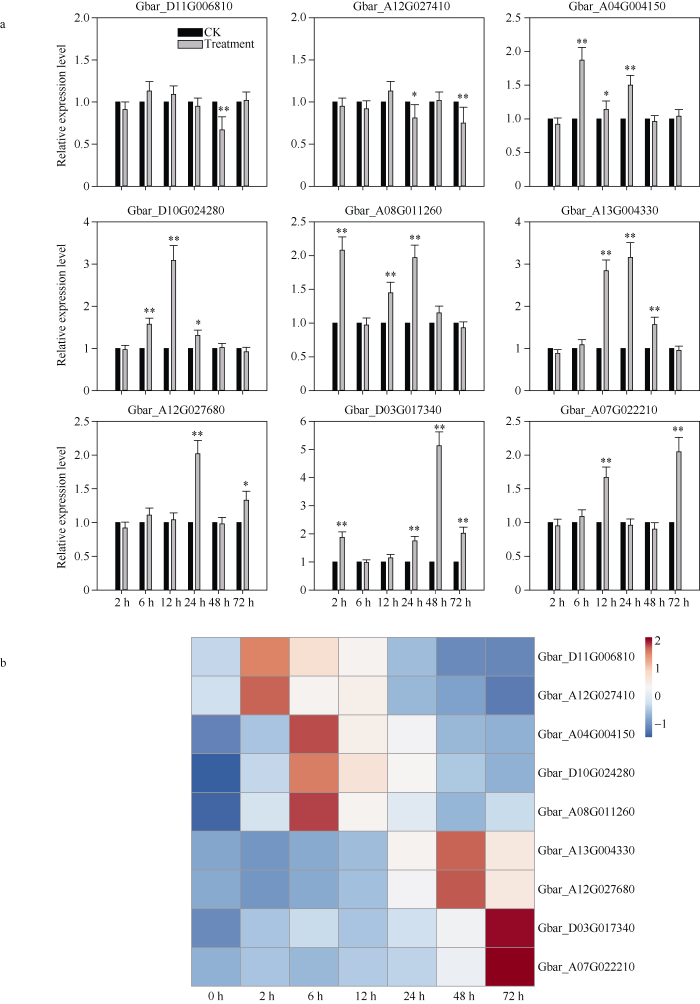 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7核心基因表达分析
a: qRT-PCR分析, 上方垂直线代
Fig. 7Expression analysis of the hub genes
a: expression analysis by qRT-PCR, the bars indicate standard deviation of three replications. b: heat map of the hub genes.
*: p < 0.05, **: p < 0.01.
3 讨论
本研究以黄萎病菌侵染下海岛棉幼苗根系RNA-seq数据为基础, 对其进行了差异表达分析, 共鉴定出22,850个差异表达基因。Chen等[18]利用该转录组数据共得到17,517个差异表达基因, 结果产生差异的主要原因是在分析过程中使用了不同的参考基因组及分析软件。Chen等[18]以雷蒙德氏棉(Gossypium ramondii)基因组为参考基因组, 比对率约为76%, 本研究以海岛棉基因组为参考基因组, 比对率约为94%, 因此比对效果更好。Zhang等[28] 通过分析黄萎病菌侵染24 h下陆地棉(Gossypium hirsutum)幼苗根的转录组数据, 共鉴定到4794个差异表达基因, 其中一些基因在本研究中具有相似的差异表达模式(如GhFLS2、GhLRRC、GhDBTNBT等), 但也有部分基因在2个研究中表现出相反的上下调变化趋势(如GhWRKY29、GhSLP、GhJAZ等), 暗示这些基因在海岛棉和陆地棉的抗病过程中可能发挥不同作用。通过WGCNA方法, 本研究共鉴定到5个(black、mediumpurple3、darkolivegreen、plum3、mediumpurple2)抗病相关特异性模块。富集分析结果表明, 特异性模块可得到具有相关生物学意义的功能富集和代谢通路富集结果。如GO富集分析中, black模块可富集到刺激响应(GO:0051716)、激素介导的信号通路(GO:0009755)、激素刺激应答(GO:0032870)等抗逆相关细胞过程; plum3模块富集到钙离子结合分子功能(GO:0005509), 而研究表明钙离子是植物防御应答过程中一类非常重要的第二信使[29]; KEGG富集分析中, darkolivegreen模块可富集到黄酮类化合物生物合成代谢通路, 此类化合物是植物中所特有的一类多功能复合物, 在植株抵御生物/非生物胁迫过程中发挥重要作用[30]。
通过计算模块内基因的连通性, 可推测该基因在网络中的位置及重要程度。结合已经报道过的棉花中抗黄萎病相关基因发现, 这些基因在相应模块内都具有较高的连通性。如black模块中的GbWRKY1基因(Gbar_A04G014110, kME=0.89), 可通过激活JAZ1基因的表达介导棉花抗病过程[31]; mediumpurple2模块中的GbRVd (Gbar_D11G031930, kME=0.89)为一类NBS-LRR (nucleotide-binding site-leucine rich repeat)基因, 在棉花抗黄萎病侵染过程中发挥重要作用[32]。此外, darkolivegreen模块中的Gbar_D01G017350、plum3模块中的Gbar_D05G028190在相应模块内都具有较高连通性, 其在陆地棉中的同源基因GhCYP-3及GhWRKY70均为抗病相关基因[33,34]。其他非抗病相关特异性模块中也包含一些高连通性的抗病基因, 如turquoise模块中的GbMYB108 (Gbar_A11G011280, kME= 0.90)[35]、GbNRX1 (Gbar_D02G017380, kME=0.95)[36]及mediumorchid模块中的GbSBT1 (Gbar_D06G 000060, kME=0.91)[37]基因等。
选取特异性模块中连通性最高的前5个基因为核心基因, 推测其可能在抗病过程中发挥重要作用。这些基因在棉花中的功能大多尚不明确, 而它们在拟南芥中的同源基因部分已报道为抗逆相关基因。如plum3模块中的Gbar_D03G017340为一个ERF转录因子家族基因, 其在拟南芥中的同源基因为AtERF5, 该基因可通过调控JA/ET信号通路响应病原菌胁迫[38]; darkolivegreen模块中Gbar_ A13G004330的同源基因AtRDK1参与调控ABA信号转导, 在植株非生物胁迫应答中发挥作用[39]; black模块中Gbar_A12G013260在拟南芥中的同源基因为ASKα, 该基因通过激活G6PD蛋白调节细胞内氧化还原反应平衡, 从而在植株响应逆境胁迫中发挥重要作用[40]。
与核心基因连接度较高的基因, 同样可能在逆境胁迫应答中发挥作用。如在darkolivegreen模块中, Gbar_A12G027680的高连接度基因Gbar_D02G 017680在拟南芥中的同源基因为RTP1, 该基因可通过调节活性氧含量、细胞死亡过程及PR1基因的表达参与响应病原菌侵染胁迫[41]; 在plum3模块中, Gbar_A07G022210的高连接度基因Gbar_D05G 005280在拟南芥中的同源基因编码一个细胞分裂素信号通路相关转录因子ARR12, 参与调控植株的干旱胁迫应答[42]; 在black模块中, Gbar_A11G006460的高连接度基因Gbar_D04G 003630在拟南芥中的同源基因编码PAD4蛋白, 可与另一个抗病相关蛋白EDS1组成复合体共同调控SA信号通路, 进而在病原菌胁迫应答中发挥作用[43]。此外, 结合差异表达分析结果表明, 以上基因基本都发生了显著上下调, 从而在表达水平上也说明它们可能在抗病过程中发挥作用。
本研究构建的网络中核心基因在棉花中的具体生物学功能目前大多尚不明确, 后续可进一步通过过量表达、VIGS、基因敲除等生物技术手段对其开展深入研究。
4 结论
通过构建权重基因共表达网络共鉴定到5个抗病相关特异性模块, 并揭示了它们的生物学意义。以连通性为指标, 揭示了特异性模块中可能在抗病过程中发挥重要作用的关键基因。本研究结果为进一步理解棉花抗病过程的分子机制提供了理论指导。附表 请见网络版: 1) 本刊网站http://zwxb. chinacrops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical-zuowxb. aspx。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1128/AEM.70.8.4989-4995.2004URLPMID:15294839 [本文引用: 1]

Verticillium wilt is a vascular disease of cotton. The causal fungus, Verticillium dahliae, secretes elicitors in culture. We have generated approximately 1,000 5'-terminal expressed sequence tags (ESTs) from a cultured mycelium of V. dahliae. A number of ESTs were found to encode proteins harboring putative signal peptides for secretion, and their cDNAs were isolated. Heterologous expression led to the identification of a protein with elicitor activities. This protein, named V. dahliae necrosis- and ethylene-inducing protein (VdNEP), is composed of 233 amino acids and has high sequence identities with fungal necrosis- and ethylene-inducing proteins. Infiltration of the bacterially expressed His-VdNEP into Nicotiana benthamiana leaves resulted in necrotic lesion formation. In Arabidopsis thaliana, the fusion protein also triggered production of reactive oxygen species and induced the expression of PR genes. When added into suspension cultured cells of cotton (Gossypium arboreum), the fusion protein elicited the biosynthesis of gossypol and related sesquiterpene phytoalexins at low concentrations, and it induced cell death at higher concentrations. On cotton cotyledons and leaves, His-VdNEP induced dehydration and wilting, similar to symptoms caused by a crude preparation of V. dahliae elicitors. Northern blotting showed a low level of VdNEP expression in the mycelium during culture. These data suggest that VdNEP is a wilt-inducing factor and that it participates in cotton-V. dahliae interactions.
DOI:10.1371/journal.pone.0144020URLPMID:26633180 [本文引用: 1]
Verticillium dahliae is the primary causal agent for Verticillium wilt disease on a diverse array of economically important crops, including cotton. In previous research, we obtained the low-pathogenicity mutant T286 from the T-DNA insertional mutant library of the highly virulent isolate Vd080 derived from cotton. In this study, the target disrupted gene VdCYC8 was identified by TAIL-PCR, encoding a homolog of CYC8 proteins involved in glucose repression. The deletion mutant ΔCYC8 exhibited several developmental deficiencies, including reduced microsclerotia formation, reduced sporulation, and slower growth. Moreover, compared with the wild type strain Vd080, the pathogenicity of strain ΔCYC8 was significantly decreased on cotton seedlings. However, the complementary mutants ΔCYC8-C led to restoration of the wild type phenotype or near wild type levels of virulence on cotton. Interestingly, pathogenicity of the strains was correlated with VdCYC8 gene expression levels in complemented mutants. Gene expression analyses in the wild type strain Vd080, the ΔCYC8-45 strain, and complemented strain ΔCYC8-C26 indicated that VdCYC8 regulates the transcription levels of several genes in V. dahliae that have roles in melanin and production.
DOI:10.1111/pbi.12900URLPMID:29431919 [本文引用: 1]
Verticillium wilt (VW), caused by soil-borne fungi of the genus Verticillium, is a serious disease affecting a wide range of plants and leading to a constant and major challenge to agriculture worldwide. Cotton (Gossypium hirsutum) is the world's most important natural textile fibre and oil crop. VW of cotton is a highly devastating vascular disease; however, few resistant germplasms have been reported in cotton. An increasing number of studies have shown that RNA interference (RNAi)-based host-induced gene silencing (HIGS) is an effective strategy for improving plant resistance to pathogens by silencing genes essential for the pathogenicity of these pathogens. Here, we have identified and characterized multifunctional regulators of G protein signalling (RGS) in the Verticillium dahliae virulence strain, Vd8. Of eight VdRGS genes, VdRGS1 showed the most significant increase in expression in V.?dahliae after treating with the roots of cotton seedlings. Based on the phenotype detection of VdRGS1 deletion and complementation mutants, we found that VdRGS1 played crucial roles in spore production, hyphal development, microsclerotia formation and pathogenicity. Tobacco rattle virus-mediated HIGS in cotton plants silenced VdRGS1 transcripts in invaded V.?dahliae strains and enhanced broad-spectrum resistance to cotton VW. Our data demonstrate that VdRGS1 is a conserved and essential gene for V.?dahliae virulence. HIGS of VdRGS1 provides effective control against V.?dahliae infection and could obtain the durable disease resistance in cotton and in other VW-susceptible host crops by developing the stable transformants.
DOI:10.11963/1002-7807.zhqzhq.20170825URL [本文引用: 1]
棉花黄萎病是影响我国棉花生产可持续发展的主要障碍之一。近年来,国内外在棉花黄萎病菌的遗传多样性及致病机制、棉花抗病机制、棉花黄萎病的预警技术及综合防控等方面均取得新的研究进展,尤其是进一步明确了棉花黄萎病菌的侵染过程和分子调控机制;系统研究了我国主产棉区棉花黄萎病菌的遗传多样性与致病力和地理来源的关系,首次建立了病原菌的信息档案库。并对我国棉花黄萎病未来的研究方向进行了展望。
DOI:10.11963/1002-7807.zhqzhq.20170825URL [本文引用: 1]
棉花黄萎病是影响我国棉花生产可持续发展的主要障碍之一。近年来,国内外在棉花黄萎病菌的遗传多样性及致病机制、棉花抗病机制、棉花黄萎病的预警技术及综合防控等方面均取得新的研究进展,尤其是进一步明确了棉花黄萎病菌的侵染过程和分子调控机制;系统研究了我国主产棉区棉花黄萎病菌的遗传多样性与致病力和地理来源的关系,首次建立了病原菌的信息档案库。并对我国棉花黄萎病未来的研究方向进行了展望。
DOI:10.1073/pnas.091114198URLPMID:11331751 [本文引用: 1]
In tomato, Ve is implicated in race-specific resistance to infection by Verticillium species causing crop disease. Characterization of the Ve locus involved positional cloning and isolation of two closely linked inverted genes. Expression of individual Ve genes in susceptible potato plants conferred resistance to an aggressive race 1 isolate of Verticillium albo-atrum. The deduced primary structure of Ve1 and Ve2 included a hydrophobic N-terminal signal peptide, leucine-rich repeats containing 28 or 35 potential glycosylation sites, a hydrophobic membrane-spanning domain, and a C-terminal domain with the mammalian E/DXXXLphi or YXXphi endocytosis signals (phi is an amino acid with a hydrophobic side chain). A leucine zipper-like sequence occurs in the hydrophobic N-terminal signal peptide of Ve1 and a Pro-Glu-Ser-Thr (PEST)-like sequence resides in the C-terminal domain of Ve2. These structures suggest that the Ve genes encode a class of cell-surface glycoproteins with receptor-mediated endocytosis-like signals and leucine zipper or PEST sequences.
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/1471-2164-14-637URLPMID:24053558 [本文引用: 1]
Verticillium wilt, caused by the fungal pathogen Verticillium dahliae, is the most severe disease in cotton (Gossypium spp.), causing great lint losses worldwide. Disease management could be achieved in the field if genetically improved, resistant plants were used. However, the interaction between V. dahliae and cotton is a complicated process, and its molecular mechanism remains obscure. To understand better the defense response to this pathogen as a means for obtaining more tolerant cultivars, we monitored the transcriptome profiles of roots from resistant plants of G. barbadense cv. Pima90-53 that were challenged with V. dahliae.
DOI:10.1016/j.gene.2006.12.019URLPMID:17321073 [本文引用: 1]
ERF transcription factors can bind GCC boxes or non-GCC cis elements to regulate biotic and abiotic stress responses. Here, we report that an ERF transcription factor gene (GbERF2) was cloned by suppression subtraction hybridization from sea-island cotton after Verticillium dahliae attack. The GbERF2 cDNA has a total length of 1143 bp with an open reading frame of 597 bp. The genomic sequence of GbERF2 contains an intron of 515 bp. The gene encodes a predicated polypeptide of 198 amino acids with a molecular weight of 22.5 kDa and a calculated pI of 9.82. The GbERF2 protein has a highly conserved ERF domain while the nucleotide and amino acid sequences have low homology with other ERF plant proteins. An RNA blot revealed that GbERF2 is constitutively expressed in different tissues, but is higher in the leaves. High levels of GbERF2 transcripts rapidly accumulated when the plants were exposed to exogenous ethylene treatment and V. dahliae infection, while there was only a slight accumulation in response to salt, cold, drought and water stresses. In contrast, GbERF2 transcripts declined in response to exogenous abscisic acid (ABA) treatment. GbERF2 transgenic tobacco plants constitutively accumulated higher levels of pathogenesis-related gene transcripts, such as PR-1b, PR2 and PR4. The resistance of transgenic tobacco to fungal infection by Alternaria longipes was enhanced. However, the resistance to bacterial infection by Pseudomonas syringae pv. tabaci was not improved. These results show that GbERF2 plays an important role in response to ethylene stress and fungal attack in cotton.
DOI:10.1007/s00299-013-1481-7URLPMID:23912851 [本文引用: 1]
Overexpression of a cotton defense-related gene GbSTK in Arabidopsis resulted in enhancing pathogen infection and oxidative stress by activating multiple defense-signaling pathways.
DOI:10.1111/mpp.12797URLPMID:30957942 [本文引用: 1]
Improving genetic resistance is a preferred method to manage Verticillium wilt of cotton and other hosts. Identifying host resistance is difficult because of the dearth of resistance genes against this pathogen. Previously, a novel candidate gene involved in Verticillium wilt resistance was identified by a genome-wide association study using a panel of Gossypium hirsutum accessions. In this study, we cloned the candidate resistance gene from cotton that encodes a protein sharing homology with the TIR-NBS-LRR receptor-like defence protein DSC1 in Arabidopsis thaliana (hereafter named GhDSC1). GhDSC1 expressed at higher levels in response to Verticillium wilt and jasmonic acid (JA) treatment in resistant cotton cultivars as compared to susceptible cultivars and its product was localized to nucleus. The transfer of GhDSC1 to Arabidopsis conferred Verticillium resistance in an A. thaliana dsc1 mutant. This resistance response was associated with reactive oxygen species (ROS) accumulation and increased expression of JA-signalling-related genes. Furthermore, the expression of GhDSC1 in response to Verticillium wilt and JA signalling in A. thaliana displayed expression patterns similar to GhCAMTA3 in cotton under identical conditions, suggesting a coordinated DSC1 and CAMTA3 response in A. thaliana to Verticillium wilt. Analyses of GhDSC1 sequence polymorphism revealed a single nucleotide polymorphism (SNP) difference between resistant and susceptible cotton accessions, within the P-loop motif encoded by GhDSC1. This SNP difference causes ineffective activation of defence response in susceptible cultivars. These results demonstrated that GhDSC1 confers Verticillium resistance in the model plant system of A. thaliana, and therefore represents a suitable candidate for the genetic engineering of Verticillium wilt resistance in cotton.
DOI:10.1016/j.bbrc.2010.01.069URLPMID:20097164 [本文引用: 1]
For the first time, a sea-island cotton (Gossypium barbadense L.) thaumatin-like protein gene (GbTLP1) with a potential role in secondary cell wall development has been overexpressed in tobacco to elucidate its function. The presence of the transgene was verified by Southern blotting and higher expression levels of GbTLP1 in transgenic tobacco plants were revealed by reverse-transcription and quantitative real-time polymerase chain reaction analyses. Transgenic plants with constitutively higher expression of the GbTLP1 showed enhanced resistance against different stress agents, particularly, its performance against Verticillium dahliae was exceptional. Transgenic tobacco plants also exhibited considerable resistance against Fusarium oxysporum and some abiotic stresses including salinity and drought. In this experiment, transgenic plants without GbTLP1 expression were also used as controls, which behaved similar to non-transgenic control plants. Overexpression of GbTLP1 had no significant deleterious effect on plant growth except that flowering was delayed for 3-5 weeks. The apparent pleiotropic effect of this novel gene has given us insight to the plant defense mechanism.
DOI:10.1111/tpj.12941URLPMID:26221980 [本文引用: 1]
Verticillium dahliae is a destructive, soil-borne fungal pathogen that causes vascular wilt disease in many economically important crops worldwide. A polyamine oxidase (PAO) gene was identified and cloned by screening suppression subtractive hybridisation and cDNA libraries of cotton genotypes tolerant to Verticillium wilt and was induced early and strongly by inoculation with V.?dahliae and application of plant hormone. Recombinant cotton polyamine oxidase (GhPAO) was found to catalyse the conversion of spermine (Spm) to spermidine (Spd) in?vitro. Constitutive expression of GhPAO in Arabidopsis thaliana produced improved resistance to V.?dahliae and maintained putrescine, Spd and Spm at high levels. Hydrogen peroxide (H2 O2 ), salicylic acid and camalexin (a phytoalexin) levels were distinctly increased in GhPAO-overexpressing Arabidopsis plants during V.?dahliae infection when compared with wild-type plants, and Spm and camalexin efficiently inhibited growth of V.?dahliae in?vitro. Spermine promoted the accumulation of camalexin by inducing the expression of mitogen-activated protein kinases and cytochrome P450 proteins in Arabidopsis and cotton plants. The three polyamines all showed higher accumulation in tolerant cotton cultivars than in susceptible cotton cultivars after inoculation with V.?dahliae. GhPAO silencing in cotton significantly reduced the Spd level and increased the Spm level, leading to enhanced susceptibility to infection by V.?dahliae, and the levels of H2 O2 and camalexin were distinctly lower in GhPAO-silenced cotton plants after V.?dahliae infection. Together, these results suggest that GhPAO contributes to resistance of the plant against V.?dahliae through the mediation of Spm and camalexin signalling.
DOI:10.1111/mpp.12575URLPMID:28665036 [本文引用: 1]
Plants have evolved effective mechanisms to protect themselves against multiple stresses, and employ jasmonates (JAs) as vital defence signals to defend against pathogen infection. The accumulation of JA, induced by signals from biotic and abiotic stresses, results in the degradation of Jasmonate-ZIM-domain (JAZ) proteins, followed by the de-repression of JAZ-repressed transcription factors (such as MYC2) to activate defence responses and developmental processes. Here, we characterized a JAZ family protein, GhJAZ2, from cotton (Gossypium hirsutum) which was induced by methyl jasmonate (MeJA) and inoculation of Verticillium dahliae. The overexpression of GhJAZ2 in cotton impairs the sensitivity to JA, decreases the expression level of JA-response genes (GhPDF1.2 and GhVSP) and enhances the susceptibility to V.?dahliae and insect herbivory. Yeast two-hybrid and bimolecular fluorescence complementation assays showed that GhJAZ2 may be involved in the regulation of cotton disease resistance by interaction with further disease-response proteins, such as pathogenesis-related protein GhPR10, dirigent-like protein GhD2, nucleotide-binding site leucine-rich repeat (NBS-LRR) disease-resistant protein GhR1 and a basic helix-loop-helix transcription factor GhbHLH171. Unlike MYC2, overexpression of GhbHLH171 in cotton activates the JA synthesis and signalling pathway, and improves plant tolerance to the fungus V.?dahliae. Molecular and genetic evidence shows that GhJAZ2 can interact with GhbHLH171 and inhibit its transcriptional activity and, as a result, can restrain the JA-mediated defence response. This study provides new insights into the molecular mechanisms of GhJAZ2 in the regulation of the cotton defence response.
.
DOI:10.1186/1471-2105-9-559URLPMID:19114008 [本文引用: 2]
Correlation networks are increasingly being used in bioinformatics applications. For example, weighted gene co-expression network analysis is a systems biology method for describing the correlation patterns among genes across microarray samples. Weighted correlation network analysis (WGCNA) can be used for finding clusters (modules) of highly correlated genes, for summarizing such clusters using the module eigengene or an intramodular hub gene, for relating modules to one another and to external sample traits (using eigengene network methodology), and for calculating module membership measures. Correlation networks facilitate network based gene screening methods that can be used to identify candidate biomarkers or therapeutic targets. These methods have been successfully applied in various biological contexts, e.g. cancer, mouse genetics, yeast genetics, and analysis of brain imaging data. While parts of the correlation network methodology have been described in separate publications, there is a need to provide a user-friendly, comprehensive, and consistent software implementation and an accompanying tutorial.
.
DOI:10.1186/s12870-017-1143-yURLPMID:29115926 [本文引用: 1]
The migration of cadmium (Cd) from contaminated soil to rice is a cause for concern. However, the molecular mechanism underlying the response of rice roots to various Cd stresses remains to be clarified from the viewpoint of the co-expression network at a system-wide scale.
DOI:10.3390/genes10020119URLPMID:30736327 [本文引用: 1]
Upland cotton (Gossypium hirsutum) is grown for its elite fiber. Understanding differential gene expression patterns during fiber development will help to identify genes associated with fiber quality. In this study, we used two recombinant inbred lines (RILs) differing in fiber quality derived from an intra-hirsutum population to explore expression profiling differences and identify genes associated with high-quality fiber or specific fiber-development stages using RNA sequencing. Overall, 72/27, 1137/1584, 437/393, 1019/184, and 2555/1479 differentially expressed genes were up-/down-regulated in an elite fiber line (L1) relative to a poor-quality fiber line (L2) at 10, 15, 20, 25, and 30 days post-anthesis, respectively. Three-hundred sixty-three differentially expressed genes (DEGs) between two lines were colocalized in fiber strength (FS) quantitative trait loci (QTL). Short Time-series Expression Miner (STEM) analysis discriminated seven expression profiles; gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation were performed to identify difference in function between genes unique to L1 and L2. Co-expression network analysis detected five modules highly associated with specific fiber-development stages, especially for high-quality fiber tissues. The hub genes in each module were identified by weighted gene co-expression network analysis. Hub genes encoding actin 1, Rho GTPase-activating protein with PAK-box, TPX2 protein, bHLH transcription factor, and leucine-rich repeat receptor-like protein kinase were identified. Correlation networks revealed considerable interaction among the hub genes, transcription factors, and other genes.
DOI:10.3724/SP.J.1006.2019.83053URL [本文引用: 1]
权重基因共表达网络分析(weighted gene co-expression network analysis, WGCNA)是系统生物学的一种研究方法, 在挖掘生物学数据与特定性状之间的生物学关系方面具有十分重要的作用。本研究利用玉米(Zea mays L.)自交系B73的14份不同发育阶段的转录组数据, 筛选掉低表达丰度的基因, 最终得到了22,426个高表达的基因用于创建基因表达矩阵; 利用不同组织作为性状, 创建表型矩阵。然后利用R软件中的WGCNA包建立了共表达网络, 共得到20个模块。本研究将与组织相关性高于0.65的模块定义为组织特异性模块, 最终鉴定到14个组织特异性模块。利用在线网站Agrigo对组织特异性模块中的基因进行GO (gene ontology)富集分析, 发现14个模块中均可以得到富集种类。开花作为玉米生育周期中的一个重要生理过程, 不仅代表着植物从营养生长到生殖生长的转变, 也关系到产量、株高和抗逆性等农艺性状。本研究发现8个组织特异性模块中的基因可以富集到与开花调控的代谢通 。此外, 有17个已经报道过的开花时间调控基因存在于共表达模块中, 并且主要分布在Blue模块和Darkmagenta模块, 因此本研究重点关注了这2个模块内部的基因调控网络。本研究通过计算不同组织中的基因表达丰度, 并联合权重基因共表达网络分析的方法, 鉴定到了具有生物学意义的共表达基因模块, 挖掘到了数个开花相关的模块, 有助于揭示玉米开花调控的遗传机制。
DOI:10.3724/SP.J.1006.2019.83053URL [本文引用: 1]
权重基因共表达网络分析(weighted gene co-expression network analysis, WGCNA)是系统生物学的一种研究方法, 在挖掘生物学数据与特定性状之间的生物学关系方面具有十分重要的作用。本研究利用玉米(Zea mays L.)自交系B73的14份不同发育阶段的转录组数据, 筛选掉低表达丰度的基因, 最终得到了22,426个高表达的基因用于创建基因表达矩阵; 利用不同组织作为性状, 创建表型矩阵。然后利用R软件中的WGCNA包建立了共表达网络, 共得到20个模块。本研究将与组织相关性高于0.65的模块定义为组织特异性模块, 最终鉴定到14个组织特异性模块。利用在线网站Agrigo对组织特异性模块中的基因进行GO (gene ontology)富集分析, 发现14个模块中均可以得到富集种类。开花作为玉米生育周期中的一个重要生理过程, 不仅代表着植物从营养生长到生殖生长的转变, 也关系到产量、株高和抗逆性等农艺性状。本研究发现8个组织特异性模块中的基因可以富集到与开花调控的代谢通 。此外, 有17个已经报道过的开花时间调控基因存在于共表达模块中, 并且主要分布在Blue模块和Darkmagenta模块, 因此本研究重点关注了这2个模块内部的基因调控网络。本研究通过计算不同组织中的基因表达丰度, 并联合权重基因共表达网络分析的方法, 鉴定到了具有生物学意义的共表达基因模块, 挖掘到了数个开花相关的模块, 有助于揭示玉米开花调控的遗传机制。
DOI:10.1186/s12870-015-0508-3URLPMID:26084488 [本文引用: 3]
Gossypium raimondii is a Verticillium wilt-resistant cotton species whose genome encodes numerous disease resistance genes that play important roles in the defence against pathogens. However, the characteristics of resistance gene analogues (RGAs) and Verticillium dahliae response loci (VdRLs) have not been investigated on a global scale. In this study, the characteristics of RGA genes were systematically analysed using bioinformatics-driven methods. Moreover, the potential VdRLs involved in the defence response to Verticillium wilt were identified by RNA-seq and correlations with known resistance QTLs.
DOI:10.1093/bioinformatics/btu170URLPMID:24695404 [本文引用: 1]
Although many next-generation sequencing (NGS) read preprocessing tools already existed, we could not find any tool or combination of tools that met our requirements in terms of flexibility, correct handling of paired-end data and high performance. We have developed Trimmomatic as a more flexible and efficient preprocessing tool, which could correctly handle paired-end data.
DOI:10.1038/s41588-018-0282-xURLPMID:30510239 [本文引用: 1]
Allotetraploid cotton species (Gossypium hirsutum and Gossypium barbadense) have long been cultivated worldwide for natural renewable textile fibers. The draft genome sequences of both species are available but they are highly fragmented and incomplete1-4. Here we report reference-grade genome assemblies and annotations for G. hirsutum accession Texas Marker-1 (TM-1) and G. barbadense accession 3-79 by integrating single-molecule real-time sequencing, BioNano optical mapping and high-throughput chromosome conformation capture techniques. Compared with previous assembled draft genomes1,3, these genome sequences show considerable improvements in contiguity and completeness for regions with high content of repeats such as centromeres. Comparative genomics analyses identify extensive structural variations that probably occurred after polyploidization, highlighted by large paracentric/pericentric inversions in 14 chromosomes. We constructed an introgression line population to introduce favorable chromosome segments from G. barbadense to G. hirsutum, allowing us to identify 13 quantitative trait loci associated with superior fiber quality. These resources will accelerate evolutionary and functional genomic studies in cotton and inform future breeding programs for fiber improvement.
.
DOI:10.1038/nmeth.3317URLPMID:25751142 [本文引用: 1]
HISAT (hierarchical indexing for spliced alignment of transcripts) is a highly efficient system for aligning reads from RNA sequencing experiments. HISAT uses an indexing scheme based on the Burrows-Wheeler transform and the Ferragina-Manzini (FM) index, employing two types of indexes for alignment: a whole-genome FM index to anchor each alignment and numerous local FM indexes for very rapid extensions of these alignments. HISAT's hierarchical index for the human genome contains 48,000 local FM indexes, each representing a genomic region of ~64,000 bp. Tests on real and simulated data sets showed that HISAT is the fastest system currently available, with equal or better accuracy than any other method. Despite its large number of indexes, HISAT requires only 4.3 gigabytes of memory. HISAT supports genomes of any size, including those larger than 4 billion bases.
.
DOI:10.1093/bioinformatics/btt656URLPMID:24227677 [本文引用: 1]
Motivation: Next-generation sequencing technologies generate millions of short sequence reads, which are usually aligned to a reference genome. In many applications, the key information required for downstream analysis is the number of reads mapping to each genomic feature, for example to each exon or each gene. The process of counting reads is called read summarization. Read summarization is required for a great variety of genomic analyses but has so far received relatively little attention in the literature.
Results: We present featureCounts, a read summarization program suitable for counting reads generated from either RNA or genomic DNA sequencing experiments. featureCounts implements highly efficient chromosome hashing and feature blocking techniques. It is considerably faster than existing methods (by an order of magnitude for gene-level summarization) and requires far less computer memory. It works with either single or paired-end reads and provides a wide range of options appropriate for different sequencing applications.
.
DOI:10.1186/s13059-014-0550-8URLPMID:25516281 [本文引用: 1]
In comparative high-throughput sequencing assays, a fundamental task is the analysis of count data, such as read counts per gene in RNA-seq, for evidence of systematic changes across experimental conditions. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. We present DESeq2, a method for differential analysis of count data, using shrinkage estimation for dispersions and fold changes to improve stability and interpretability of estimates. This enables a more quantitative analysis focused on the strength rather than the mere presence of differential expression. The DESeq2 package is available at http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html webcite.
DOI:10.1089/omi.2011.0118URLPMID:22455463 [本文引用: 1]
Increasing quantitative data generated from transcriptomics and proteomics require integrative strategies for analysis. Here, we present an R package, clusterProfiler that automates the process of biological-term classification and the enrichment analysis of gene clusters. The analysis module and visualization module were combined into a reusable workflow. Currently, clusterProfiler supports three species, including humans, mice, and yeast. Methods provided in this package can be easily extended to other species and ontologies. The clusterProfiler package is released under Artistic-2.0 License within Bioconductor project. The source code and vignette are freely available at http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html.
DOI:10.1093/nar/gkw982URLPMID:27924042 [本文引用: 1]
With the goal of providing a comprehensive, high-quality resource for both plant transcription factors (TFs) and their regulatory interactions with target genes, we upgraded plant TF database PlantTFDB to version 4.0 (http://planttfdb.cbi.pku.edu.cn/). In the new version, we identified 320 370 TFs from 165 species, presenting a more comprehensive genomic TF repertoires of green plants. Besides updating the pre-existing abundant functional and evolutionary annotation for identified TFs, we generated three new types of annotation which provide more directly clues to investigate functional mechanisms underlying: (i) a set of high-quality, non-redundant TF binding motifs derived from experiments; (ii) multiple types of regulatory elements identified from high-throughput sequencing data; (iii) regulatory interactions curated from literature and inferred by combining TF binding motifs and regulatory elements. In addition, we upgraded previous TF prediction server, and set up four novel tools for regulation prediction and functional enrichment analyses. Finally, we set up a novel companion portal PlantRegMap (http://plantregmap.cbi.pku.edu.cn) for users to access the regulation resource and analysis tools conveniently.
DOI:10.1006/meth.2001.1262URLPMID:11846609 [本文引用: 1]
The two most commonly used methods to analyze data from real-time, quantitative PCR experiments are absolute quantification and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control. The 2(-Delta Delta C(T)) method is a convenient way to analyze the relative changes in gene expression from real-time quantitative PCR experiments. The purpose of this report is to present the derivation, assumptions, and applications of the 2(-Delta Delta C(T)) method. In addition, we present the derivation and applications of two variations of the 2(-Delta Delta C(T)) method that may be useful in the analysis of real-time, quantitative PCR data.
DOI:10.1007/s11240-017-1320-6URL [本文引用: 1]
DOI:10.1371/journal.pone.0181609URLPMID:28767675 [本文引用: 1]
Verticillium wilt is a devastating disease of cotton, which is caused by the soil-borne fungus Verticillium dahliae (V. dahliae). Although previous studies have identified some genes or biological processes involved in the interaction between cotton and V. dahliae, its underlying molecular mechanism remains unclear, especially in G. hirsutum. In the present study, we obtained an overview of transcriptome characteristics of resistant upland cotton (G. hirsutum) after V. dahliae infection at 24 h post-inoculation (hpi) via a high-throughput RNA-sequencing technique. A total of 4,794 differentially expressed genes (DEGs) were identified, including 820 up-regulated genes and 3,974 down-regulated genes. The enrichment analysis showed that several important processes were induced upon V. dahliae infection, such as plant hormone signal transduction, plant-pathogen interaction, phenylpropanoid-related and ubiquitin-mediated signals. Moreover, we investigated some key regulatory gene families involved in the defense response, such as receptor-like protein kinases (RLKs), WRKY transcription factors and cytochrome P450 (CYPs), via virus-induced gene silencing (VIGS). GhSKIP35, a partner of SKP1 protein, was involved in ubiquitin-mediated signal. Over-expression of GhSKIP35 in Arabidopsis improved its tolerance to Verticillium wilt in transgenic plants. Collectively, global transcriptome analysis and functional gene characterization provided significant insights into the molecular mechanisms of G. hirsutum-V. dahliae interaction and offered a number of candidate genes as potential sources for breeding wilt-tolerance in cotton.
DOI:10.1111/j.1469-8137.2006.01777.xURLPMID:16866934 [本文引用: 1]
In plant cells, the calcium ion is a ubiquitous intracellular second messenger involved in numerous signalling pathways. Variations in the cytosolic concentration of Ca2+ ([Ca2+]cyt) couple a large array of signals and responses. Here we concentrate on calcium signalling in plant defence responses, particularly on the generation of the calcium signal and downstream calcium-dependent events participating in the establishment of defence responses with special reference to calcium-binding proteins.
DOI:10.1016/j.molp.2015.03.012URLPMID:25840349 [本文引用: 1]
Phenylpropanoid-derived compounds represent a diverse family of secondary metabolites that originate from phenylalanine. These compounds have roles in plant growth and development, and in defense against biotic and abiotic stress. Many of these compounds are also beneficial to human health and welfare. V-myb myeloblastosis viral oncogene homolog (MYB) proteins belong to a large family of transcription factors and are key regulators of the synthesis of phenylpropanoid-derived compounds. This review summarizes the current understanding of MYB proteins and their roles in the regulation of phenylpropanoid metabolism in plants.
DOI:10.1104/pp.114.246694URLPMID:25301887 [本文引用: 1]
Plants have evolved an elaborate signaling network to ensure an appropriate level of immune response to meet the differing demands of developmental processes. Previous research has demonstrated that DELLA proteins physically interact with JASMONATE ZIM-DOMAIN1 (JAZ1) and dynamically regulate the interaction of the gibberellin (GA) and jasmonate (JA) signaling pathways. However, whether and how the JAZ1-DELLA regulatory node is regulated at the transcriptional level in plants under normal growth conditions or during pathogen infection is not known. Here, we demonstrate multiple functions of cotton (Gossypium barbadense) GbWRKY1 in the plant defense response and during development. Although GbWRKY1 expression is induced rapidly by methyl jasmonate and infection by Verticillium dahliae, our results show that GbWRKY1 is a negative regulator of the JA-mediated defense response and plant resistance to the pathogens Botrytis cinerea and V. dahliae. Under normal growth conditions, GbWRKY1-overexpressing lines displayed GA-associated phenotypes, including organ elongation and early flowering, coupled with the down-regulation of the putative targets of DELLA. We show that the GA-related phenotypes of GbWRKY1-overexpressing plants depend on the constitutive expression of Gossypium hirsutum GhJAZ1. We also show that GhJAZ1 can be transactivated by GbWRKY1 through TGAC core sequences, and the adjacent sequences of this binding site are essential for binding specificity and affinity to GbWRKY1, as revealed by dual-luciferase reporter assays and electrophoretic mobility shift assays. In summary, our data suggest that GbWRKY1 is a critical regulator mediating the plant defense-to-development transition during V. dahliae infection by activating JAZ1 expression.
DOI:10.1016/j.gene.2015.09.046URLPMID:26407869 [本文引用: 1]
Most of the disease resistance genes already characterized in plants encode nucleotide-binding site-leucine rich repeat (NBS-LRR) proteins that have key roles in resistance to Verticillium dahliae. Using a cDNA library and RACE protocols, we cloned a coiled-coil (CC)-NBS-LRR-type gene, GbRVd, from a resistant tetraploid cotton species, Gossypium barbadense (RVd=Resistance to V. dahliae). We also applied RT-qPCR and VIGS technologies to analyze how expression of GbRVd was induced upon attack by V. dahliae. Its 2862-bp ORF encodes a predicted protein containing 953 amino acid residues, with a predicted molecular weight of 110.17kDa and an isoelectric point of 5.87. GbRVd has three domains - CC, NBS, and LRR - and is most closely related to Gossypium raimondii RVd (88% amino acid identity). Profiling demonstrated that GbRVd is constitutively expressed in all tested tissues, and transcript levels are especially high in the leaves. In plants inoculated with V. dahliae, GbRVd was significantly up-regulated when compared with the control, with expression peaking at 48h post-inoculation. Silencing of GbRVd in cotton through VIGS dramatically down-regulated SA, NO, and H2O2 production, resulting in greater susceptibility to V. dahliae. Taken together, these results suggest that GbRVd has an important role in protecting G. barbadense against infection by V. dahliae.
DOI:10.1186/s12870-019-1848-1URLPMID:31226952 [本文引用: 1]
Cyclophilins (CYPs), belonging to the peptidyl prolyl cis/trans isomerase (PPIase) superfamily, play important roles during plant responses to biotic and abiotic stresses.
DOI:10.3389/fpls.2020.00069URLPMID:32158454 [本文引用: 1]
Verticillium wilt caused by Verticillium dahliae is a destructive cotton disease causing severe yield and quality losses worldwide. WRKY transcription factors play important roles in plant defense against pathogen infection. However, little has been reported on the functions of WRKYs in cotton's resistance to V. dahliae. Here, we identified 5, 5, and 10 WRKY70 genes in Gossypium arboreum, Gossypium raimondii, and Gossypium hirsutum, respectively, and investigated the expression profiles of all GhWRKY70 genes in various cotton tissues and in response to hormone treatment or V. dahliae infection. Reverse transcription-quantitative PCR analysis showed that GhWRKY70D13 was expressed higher in roots and stems than in other tissues, and up-regulated after V. dahliae inoculation. Knock-down of GhWRKY70D13 improved resistance to V. dahliae in both resistant and susceptible cotton cultivars. Comparative analysis of transcriptomes generated from wild-type and stable RNAi (RNA interference) plant with down-regulated GhWRKY70D13 showed that genes involved in ethylene (ET) and jasmonic acid (JA) biosynthesis and signaling were significantly upregulated in the GhWRKY70D13 RNAi plants. Consistently, the contents of 1-aminocyclopropane-1-carboxylic (ACC), JA, and JA-isoleucine levels were significantly higher in the GhWRKY70D13 RNAi plants than in wild-type. Following V. dahliae infection, the levels of ACC and JA decreased in the GhWRKY70D13 RNAi plants but still significantly higher (for ACC) than that in wild-type or at the same level (for JA) as in non-infected wild-type plants. Collectively, our results suggested that GhWRKY70D13 negatively regulates cotton's resistance to V. dahliae mainly through its effect on ET and JA biosynthesis and signaling pathways.
DOI:10.1093/jxb/erw016URLPMID:26873979 [本文引用: 1]
Accumulating evidence indicates that plant MYB transcription factors participate in defense against pathogen attack, but their regulatory targets and related signaling processes remain largely unknown. Here, we identified a defense-related MYB gene (GhMYB108) from upland cotton (Gossypium hirsutum) and characterized its functional mechanism. Expression of GhMYB108 in cotton plants was induced by Verticillium dahliae infection and responded to the application of defense signaling molecules, including salicylic acid, jasmonic acid, and ethylene. Knockdown of GhMYB108 expression led to increased susceptibility of cotton plants to V. dahliae, while ecotopic overexpression of GhMYB108 in Arabidopsis thaliana conferred enhanced tolerance to the pathogen. Further analysis demonstrated that GhMYB108 interacted with the calmodulin-like protein GhCML11, and the two proteins form a positive feedback loop to enhance the transcription of GhCML11 in a calcium-dependent manner. Verticillium dahliae infection stimulated Ca(2+) influx into the cytosol in cotton root cells, but this response was disrupted in both GhCML11-silenced plants and GhMYB108-silenced plants in which expression of several calcium signaling-related genes was down-regulated. Taken together, these results indicate that GhMYB108 acts as a positive regulator in defense against V. dahliae infection by interacting with GhCML11. Furthermore, the data also revealed the important roles and synergetic regulation of MYB transcription factor, Ca(2+), and calmodulin in plant immune responses.
DOI:10.1104/pp.15.01930URLPMID:26869704 [本文引用: 1]
Examining the proteins that plants secrete into the apoplast in response to pathogen attack provides crucial information for understanding the molecular mechanisms underlying plant innate immunity. In this study, we analyzed the changes in the root apoplast secretome of the Verticillium wilt-resistant island cotton cv Hai 7124 (Gossypium barbadense) upon infection with Verticillium dahliae Two-dimensional differential gel electrophoresis and matrix-assisted laser desorption/ionization tandem time-of-flight mass spectrometry analysis identified 68 significantly altered spots, corresponding to 49 different proteins. Gene ontology annotation indicated that most of these proteins function in reactive oxygen species (ROS) metabolism and defense response. Of the ROS-related proteins identified, we further characterized a thioredoxin, GbNRX1, which increased in abundance in response to V. dahliae challenge, finding that GbNRX1 functions in apoplastic ROS scavenging after the ROS burst that occurs upon recognition of V. dahliae Silencing of GbNRX1 resulted in defective dissipation of apoplastic ROS, which led to higher ROS accumulation in protoplasts. As a result, the GbNRX1-silenced plants showed reduced wilt resistance, indicating that the initial defense response in the root apoplast requires the antioxidant activity of GbNRX1. Together, our results demonstrate that apoplastic ROS generation and scavenging occur in tandem in response to pathogen attack; also, the rapid balancing of redox to maintain homeostasis after the ROS burst, which involves GbNRX1, is critical for the apoplastic immune response.
DOI:10.1371/journal.pone.0153988URLPMID:27088499 [本文引用: 1]
Verticillium wilt is a disastrous vascular disease in plants caused by Verticillium dahliae. Verticillium pathogens secrete various disease-causing effectors in cotton. This study identified a subtilase gene GbSBT1 from Gossypium babardense and investigated the roles against V. dahliae infection. GbSBT1 gene expression is responsive to V. dahliae defense signals, jasmonic acid, and ethylene treatments. Moreover, the GbSBT1 protein is mainly localized in the cell membrane and moves into the cytoplasm following jasmonic acid and ethylene treatments. Silencing GbSBT1 gene expression through virus-induced GbSBT1 gene silencing reduced the tolerance of Pima-90 (resistant genotype), but not facilitated the infection process of V. dahliae in Coker-312 (sensitive genotype). Moreover, the ectopically expressed GbSBT1 gene enhanced the resistance of Arabidopsis to Fusarium oxysporum and V. dahliae infection and activated the expression levels of defense-related genes. Furthermore, pull-down, yeast two-hybrid assay, and BiFC analysis revealed that GbSBT1 interacts with a prohibitin (PHB)-like protein expressed in V. dahliae pathogens during infection. In summary, GbSBT1 recognizes the effector PHB protein secreted from V. dahliae and is involved in Verticillium-induced resistance in cotton.
DOI:10.1371/journal.pone.0035995URLPMID:22563431 [本文引用: 1]
The ethylene response factor (ERF) family in Arabidopsis thaliana comprises 122 members in 12 groups, yet the biological functions of the majority remain unknown. Of the group IX ERFs, the IXc subgroup has been studied the most, and includes ERF1, ERF14 and ORA59, which play roles in plant innate immunity. Here we investigate the biological functions of two members of the less studied IXb subgroup: ERF5 and ERF6. In order to identify potential targets of these transcription factors, microarray analyses were performed on plants constitutively expressing either ERF5 or ERF6. Expression of defense genes, JA/Et-responsive genes and genes containing the GCC box promoter motif were significantly upregulated in both ERF5 and ERF6 transgenic plants, suggesting that ERF5 and ERF6 may act as positive regulators of JA-mediated defense and potentially overlap in their function. Since defense against necrotrophic pathogens is generally mediated through JA/Et-signalling, resistance against the fungal necrotroph Botrytis cinerea was examined. Constitutive expression of ERF5 or ERF6 resulted in significantly increased resistance. Although no significant difference in susceptibility to B. cinerea was observed in either erf5 or erf6 mutants, the erf5 erf6 double mutant showed a significant increase in susceptibility, which was likely due to compromised JA-mediated gene expression, since JA-induced gene expression was reduced in the double mutant. Taken together these data suggest that ERF5 and ERF6 play positive but redundant roles in defense against B. cinerea. Since mutual antagonism between JA/Et and salicylic acid (SA) signalling is well known, the UV-C inducibility of an SA-inducible gene, PR-1, was examined. Reduced inducibilty in both ERF5 and ERF6 constitutive overexepressors was consistent with suppression of SA-mediated signalling, as was an increased susceptibility to avirulent Pseudomonas syringae. These data suggest that ERF5 and ERF6 may also play a role in the antagonistic crosstalk between the JA/Et and SA signalling pathways.
DOI:10.1016/j.molp.2016.11.011URLPMID:27923613 [本文引用: 1]
Abscisic acid (ABA) is a major phytohormone involved in important stress-related and developmental plant processes. Membrane-delimited ABA signal transduction plays an important role in early ABA signaling, but the molecular mechanisms connecting core signaling components to the plasma membrane?remain unclear. Plants have evolved a large number of receptor-like kinases (RLKs) to modulate diverse biological processes by perceiving extracellular stimuli and activating downstream signaling responses. In this study, a putative leucine-rich repeat-RLK gene named RECEPTOR DEAD KINASE1 (AtRDK1) was identified and characterized in Arabidopsis thaliana. RDK1 promoter-GUS analysis revealed that RDK1 is expressed ubiquitously in the various tissues in Arabidopsis, and its expression is mainly induced by ABA. In the presence of ABA, RDK1-deficient rdk1-1 and rdk1-2 lines showed significant resistance in cotyledon greening and root growth, whereas RDK1-overexpressing lines showed enhanced sensitivity. Consistently, the expression of ABA-responsive genes was significantly downregulated in rdk1 mutant seedlings, which were also hypersensitive to drought stress with increased water loss. Interestingly, RDK1 was found to be an atypical kinase localized to the plasma membrane and did not require its kinase activity during ABA-mediated inhibition of seedling development. Accordingly, RDK1 interacted in the plasma membrane with type 2C protein phosphatase ABSCISIC ACID INSENSITIVE1 (ABI1); this interaction was further enhanced by exogenous application of ABA, suggesting that RDK1-mediated recruitment of ABI1 onto the plasma membrane is important for ABA signaling. Taken together, these results reveal an important role for RDK1 in plant responses to abiotic stress conditions in an ABA-dependent manner.
DOI:10.1105/tpc.112.101279URLPMID:22885737 [本文引用: 1]
Diverse stresses such as high salt conditions cause an increase in reactive oxygen species (ROS), necessitating a redox stress response. However, little is known about the signaling pathways that regulate the antioxidant system to counteract oxidative stress. Here, we show that a Glycogen Synthase Kinase3 from Arabidopsis thaliana (ASKα) regulates stress tolerance by activating Glc-6-phosphate dehydrogenase (G6PD), which is essential for maintaining the cellular redox balance. Loss of stress-activated ASKα leads to reduced G6PD activity, elevated levels of ROS, and enhanced sensitivity to salt stress. Conversely, plants overexpressing ASKα have increased G6PD activity and low levels of ROS in response to stress and are more tolerant to salt stress. ASKα stimulates the activity of a specific cytosolic G6PD isoform by phosphorylating the evolutionarily conserved Thr-467, which is implicated in cosubstrate binding. Our results reveal a novel mechanism of G6PD adaptive regulation that is critical for the cellular stress response.
DOI:10.1111/nph.13707URLPMID:26484750 [本文引用: 1]
Oomycete pathogens cause serious damage to a wide spectrum of plants. Although host pathogen recognition via pathogen effectors and cognate plant resistance proteins is well established, the genetic basis of host factors that mediate plant susceptibility to oomycete pathogens is relatively unexplored. Here, we report on RTP1, a nodulin-related MtN21 family gene in Arabidopsis that mediates susceptibility to Phytophthora parasitica. RTP1 was identified by screening a T-DNA insertion mutant population and encoded an endoplasmic reticulum (ER)-localized protein. Overexpression of RTP1 rendered Arabidopsis more susceptible, whereas RNA silencing of RTP1 led to enhanced resistance to P.?parasitica. Moreover, an RTP1 mutant, rtp1-1, displayed localized cell death, increased reactive oxygen species (ROS) production and accelerated PR1 expression, compared to the wild-type Col-0, in response to P.?parasitica infection. rtp1-1 showed a similar disease response to the bacterial pathogen Pseudomonas syringae pv. tomato (Pst) DC3000, including increased disease resistance, cell death and ROS production. Furthermore, rpt1-1 exhibited resistance to the fungal pathogen Golovinomyces cichoracearum, but not to the necrotrophic pathogen Botrytis cinerea. Taken together, these results suggest that RTP1 negatively regulates plant resistance to biotrophic pathogens, possibly by regulating ROS production, cell death progression and PR1 expression.
DOI:10.1073/pnas.1600399113URLPMID:26884175 [本文引用: 1]
In this study, we used a loss-of-function approach to elucidate the functions of three Arabidopsis type B response regulators (ARRs)--namely ARR1, ARR10, and ARR12--in regulating the Arabidopsis plant responses to drought. The arr1,10,12 triple mutant showed a significant increase in drought tolerance versus WT plants, as indicated by its higher relative water content and survival rate on drying soil. This enhanced drought tolerance of arr1,10,12 plants can be attributed to enhanced cell membrane integrity, increased anthocyanin biosynthesis, abscisic acid (ABA) hypersensitivity, and reduced stomatal aperture, but not to altered stomatal density. Further drought-tolerance tests of lower-order double and single mutants indicated that ARR1, ARR10, and ARR12 negatively and redundantly control plant responses to drought, with ARR1 appearing to bear the most critical function among the three proteins. In agreement with these findings, a comparative genome-wide analysis of the leaves of arr1,10,12 and WT plants under both normal and dehydration conditions suggested a cytokinin (CK) signaling-mediated network controlling plant adaptation to drought via many dehydration/drought- and/or ABA-responsive genes that can provide osmotic adjustment and protection to cellular and membrane structures. Expression of all three ARR genes was repressed by dehydration and ABA treatments, inferring that plants down-regulate these genes as an adaptive mechanism to survive drought. Collectively, our results demonstrate that repression of CK response, and thus CK signaling, is one of the strategies plants use to cope with water deficit, providing novel insight for the design of drought-tolerant plants by genetic engineering.
DOI:10.1016/j.molp.2018.05.007URLPMID:29842929 [本文引用: 1]
In plant immunity, pathogen-activated intracellular nucleotide binding/leucine rich repeat (NLR) receptors mobilize disease resistance pathways, but the downstream signaling mechanisms remain obscure. Enhanced disease susceptibility 1 (EDS1) controls transcriptional reprogramming in resistance triggered by Toll-Interleukin1-Receptor domain (TIR)-family NLRs (TNLs). Transcriptional induction of the salicylic acid (SA) hormone defense sector provides one crucial barrier against biotrophic pathogens. Here, we present genetic and molecular evidence that in Arabidopsis?an EDS1 complex with its partner PAD4?inhibits MYC2, a master regulator of SA-antagonizing jasmonic acid (JA) hormone pathways. In the TNL immune response, EDS1/PAD4 interference with MYC2 boosts the SA defense sector independently of EDS1-induced SA synthesis, thereby effectively blocking actions of a potent bacterial JA mimic, coronatine (COR). We show that antagonism of MYC2 occurs after COR has been sensed inside the nucleús but before or coincident with MYC2 binding to a target promoter, pANAC019. The stable interaction of PAD4 with MYC2 in planta is competed by EDS1-PAD4 complexes. However, suppression of MYC2-promoted genes requires EDS1 together with PAD4, pointing to an essential EDS1-PAD4 heterodimer activity in MYC2 inhibition. Taken together, these results uncover an immune receptor signaling circuit that intersects with hormone pathway crosstalk to reduce bacterial pathogen growth.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}