 ,1,3,*, 杜雄明,1,2,*
,1,3,*, 杜雄明,1,2,*Analysis of differentially expressed genes and fiber development in Gossypium arboreum fuzzless mutant
WANG Xiao-Yang1, WANG Li-Yuan1, PAN Zhao-E1, HE Shou-Pu1, WANG Xiao1, GONG Wen-Fang,1,3,*, DU Xiong-Ming,1,2,*通讯作者:
收稿日期:2019-09-2接受日期:2019-12-26网络出版日期:2020-01-15
| 基金资助: |
Received:2019-09-2Accepted:2019-12-26Online:2020-01-15
| Fund supported: |
作者简介 About authors
E-mail:wangxiaoyang198806@126.com。

摘要
关键词:
Abstract
Keywords:
PDF (5451KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王晓阳, 王丽媛, 潘兆娥, 何守朴, 王骁, 龚文芳, 杜雄明. 亚洲棉短绒突变体纤维发育及其差异基因表达分析[J]. 作物学报, 2020, 46(5): 645-660. doi:10.3724/SP.J.1006.2020.94133
WANG Xiao-Yang, WANG Li-Yuan, PAN Zhao-E, HE Shou-Pu, WANG Xiao, GONG Wen-Fang, DU Xiong-Ming.
棉花是一种重要的经济作物, 种子是棉籽油的主要原料, 纤维更是一种重要的天然纺织资源。棉花纤维是高等植物中最长的单细胞突起, 由棉花胚珠表皮细胞产生。棉纤维的长度一般为30~40 mm [1]。棉花纤维根据形态可以分为长纤维和短绒2种不同的类型。具有重要经济价值的长纤维在开花前0DPA (day post anthesis)发育, 经过快速伸长, 纤维素生物合成和成熟, 最终形成成熟的长纤维, 通过轧花从种皮上收获并用于纺织产品[2,3]。短绒纤维起始于+3 ~ +5DPA, 长度不到5 mm, 轧花后不能从种皮上分离下来, 紧紧附着于种皮上, 对种子传播起重要作用[4]。胚珠上短纤维细胞和长纤维细胞的共存现象长期以来一直受到研究者的关注, 为研究植物细胞的分化提供了独特的模式体系。
棉纤维的发育受一系列基因调控, 这些基因与植物激素信号转导、细胞能量代谢、脂肪酸代谢、次生代谢等途径有关。目前关于调控纤维发育的基因和调控网络的信息还不太清楚。在棉花基因组测序之前, 许多研究者主要通过基因芯片技术筛选与棉花纤维发育有关的基因, 在分子水平上了解纤维发育的机制。如Shi等[5]通过cDNAs芯片分析技术发现了778个cDNAs优先在发育的纤维中表达。同年, Yang等[6]通过分析陆地棉及其光子突变体转录组, 鉴定出了受激素诱导的转录因子MIXTA、MYB5、GL2在纤维起始过程中高表达。随着棉花基因组测序的完成, 逐步完善了大量的棉花转基因技术, 使得通过转录组分析与纤维发育相关的基因与代谢通路成为可能[7,8,9]。鉴定出一些与纤维发育及调控细胞壁沉积与强度的候选基因[10]。通过分析0DPA和+10DPA的二倍体亚洲棉近等基因系有短绒无长纤维材料(Fl)和既有短绒又有长纤维材料(FL)的转录组发现, 一些参与植物激素信号转导、细胞能量代谢, 脂肪酸代谢、刺激代谢的转录因子如AP2- EREBP、AUX/IAA、bHLH、C2H2、C3H、HB、MYB、NAC、Orphans、PLATZ和WRKY在长纤维突变体+ 10DPA纤维中明显下调, 植物受体样激酶(plant receptor-like kinases, RLKs)、富含亮氨酸(Leucine Rich Repeats)的LRR家族蛋白和参与信号转导的编码丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)在突变体中也下调, 导致纤维生长减慢[11]。
虽然上述转录组分析的研究为棉纤维的起始过程提供了一些研究基础, 但纤维发育的关键基因的研究仍处于初级阶段, 长纤维和短绒纤维发育及生理过程仍然不清楚, 并且目前这些转录组研究主要集中在纤维伸长阶段, 关于短绒纤维起始的动态过程研究较少。
突变体特有的性状是研究基因表达的有利资源, 分析植物突变体加速了特定基因功能的发现。本研究所用的材料为二倍体亚洲棉短绒突变体GA0149有长纤维没有短绒(其光籽性状受单基因显性控制, 基因型为FZ)及其野生型GA0146既有长纤维又有短绒(基因型为fz), 经过连续十代的自花授粉, 突变体植株光籽性状稳定, 突变体和野生型材料在形态和发育上区别微乎其微, 除了光籽突变体种皮上没有短绒[8]。利用转录组测序技术分析突变体和野生型0DPA、+3DPA、+5DPA和+8DPA 4个时期的差异基因表达情况, 旨在探究棉花短绒起始的动态过程及其关键基因, 为棉花短绒起始的分子机制提供坚实基础。
1 材料与方法
1.1 试验材料
亚洲棉野生型毛子材料为中美棉DPL971 (fz), 短绒突变体材料为中美棉DPL972 (FZ), 均来自棉花种质资源中期库近10年的自交材料。2017年在中国农业科学院棉花研究所实验基地, 用标准的田间管理措施, 按3个重复种植每个材料。1.2 扫描电镜
将2个材料(fz和FZ) 0DPA、+3DPA的胚珠固定于含有2.5%戊二醛的0.1 mol L-1磷酸缓冲液中, 用真空泵抽真空15 min, 使胚珠完全浸没于固定液中, 4℃过夜后用0.1 mol L-1 pH 7.2磷酸缓冲液洗3次, 每次10 min; 接着, 用ddH2O清洗2次, 每次30 min; 用30% (v/v)、50% (v/v)、70% (v/v)、80% (v/v)、95% (v/v)乙醇对样品梯度脱水, 每个梯度脱水20 min; 用无水乙醇对样品进行3次脱水; 再用等体积的乙醇: 乙酸异戊酯置换15 min; 最后用100% 的乙酸异戊酯置换15 min; 将样品放于样品笼中用CO2干燥, 然后喷金, 用JSM-6390/LV扫描电镜观察。1.3 石蜡切片
将新鲜的0DPA、+3DPA的胚珠固定于含有4%的FAA (甲醛-冰乙酸-无水乙醇)固定液中, 抽真空, 保存于4℃冰箱中过夜, 用系列梯度浓度的乙醇(50%、70%、85%、95%和100%)对样品进行脱水处理, 每梯度30 min; 将脱水处理过的胚珠包埋于处理过的石蜡(Sigma-Aldrich)中, 60℃透蜡2 h; 然后将胚珠放于预热的包埋框中, 迅速倒入融化的石蜡, 过夜使石蜡凝固。用石蜡切片机, 切成8 μm 厚的连续切片, 移至42℃的水浴中, 使样品展平, 将切片放在载玻片上, 37℃干燥过夜。用番红固绿染色, 用带有数码相机的显微镜照相。1.4 文库构建与转录组测序
用北京天根公司的多糖多酚植物总RNA试剂盒提取FZ和fz 0DPA、+3DPA和+5DPA 的胚珠及+8DPA的纤维的RNA。将检测质量合格的RNA送至北京百迈客生物公司进行cDNA文库的构建, 借助 Illumina HiSeq 2000测序仪进行双末端测序, 获得原始数据。1.5 转录组原始数据的评估以及分析
将得到的原始数据转化为FASTQ形式进行保存, 通过fastp软件过滤掉低质量数据和接头序列, 得到高质量的reads, 即clean data.利用TopHat2/Bowtie2软件将 clean reads 比对到亚洲棉三代基因组上[8,12-13]。同时对比对到参考基因组的序列进行注释, 在比对过程中, 每个错配最多允许2个碱基。采用FPKM值(Fragments Per Kilobase of transcript per Million fragments mapped)来衡量基因表达[13]。根据FPKM值, 以错误发现率FDR<0.01, |log2 fold change| ≥ 1, P-value<0.01作为筛选突变体和野生型差异表达基因的标准。将获得的差异基因进行GO功能富集分析(WEGO:1.6 荧光定量Q-PCR验证
通过分析RNA-seq数据, 验证转录组数据的准确性, 随机挑选22个差异表达的基因, 利用qRT-PCR进行验证。利用在线软件NCBI-Primer (Table 1
表1
表1用于荧光定量PCR验证的基因和引物
Table 1
| 基因编号 Gene | 正向引物 Forward primer (5'-3') | 反向引物 Reverse primer (5'-3') | log2RT-QPCR | log2FPKM | 功能注释 Functional annotations |
|---|---|---|---|---|---|
| Ga02G0173 | GTGCTCGGAAGAAAACGCTC | CGCACCTGCCAAAGTTGTTA | 1.31 | 1.10 | 谷胱甘肽转移酶 Glutathione S-transferase |
| Ga03G0324 | ACAGCAAAAGGCAATCGAGAAC | CGTGGGAAGCTATCGGCTTT | 2.85 | 1.65 | 谷胱甘肽转移酶 Glutathione S-transferase |
| Ga04G0014 | CTTTCGGGTCAGGTCGAAGG | GTTGTGTTACTCGGGGGACC | 3.52 | 2.89 | 细胞色素氧化酶 Cytochrome P450 |
| Ga05G3679 | GAGCCATCGCTAAGTTCTGGGC | TCCAACAATTTCAGGCACGCAA | -1.19 | -1.40 | 谷胱甘肽转移酶 Glutathione S-transferase |
| Ga05G3965 | CCTGCTATTGCCATCAACACTT | TTGTGCTGGACTTGTTCTTGCT | -2.00 | -1.24 | 分子伴侣 Molecular chaperone |
| Ga07G0568 | CAACGGCGAAAACAACACGGAA | TGCACTGGTCTCCTTCTGTGGA | 1.31 | 1.18 | 甲基转移酶Methyltransferase |
| Ga07G1530 | GGTGGTAACCCTAGCTTGTGGC | GCGCGTTATGTGTAACTTGCGA | 1.26 | 1.11 | 转录因子WD40 |
| Ga07G1742 | GCCGTGCTAACTTATTCCCTTGC | AGCACCCCAAGTAGTAACCAAGC | -1.54 | -1.20 | 钙联蛋白 Calnexin 1 |
| Ga07G2088 | TTGCATCAGAGCAGCCTGCATA | TCTGCTGCCCAAGATTTGCCAT | -1.54 | -1.32 | 转录因子 MYB108 |
| Ga07G2611 | GCAGAGGCAGCTTTAAGTTTGGG | GCATCCCACCAGGCCTAAGTTC | -1.61 | -1.55 | 网状蛋白 Reticulon-like protein |
| Ga08G0121 | TGTCTTCGGAACCCGGAGATGA | GGCATCCCACTAGCAGCATTGA | 4.47 | 6.69 | 蛋白受体激酶 Receptor-like protein kinase |
| Ga09G0409 | TACCGGTGCACTGATCGACCTA | TTCTGAGTCGACTGTTCGTCGC | 2.03 | 1.55 | 功能未知 Unknown |
| Ga09G0435 | TGCAGGGTTGTTAAGGTGTGGG | TGATTTCGTTATCGGTCCGCCC | 1.14 | 1.05 | 假定蛋白 Predicted protein |
| Ga09G0661 | CGCCATGCTTGCCATCTTCATC | CACTGAAGGCAGCTTGATCCGA | 1.28 | 1.31 | 还原酶 Dihydroflavonol-4-reductase |
| Ga10G0810 | ATGAGCCGCCCTATTGACTGCC | TGGTAGATGCCTTCGTTGCTGA | 2.24 | 2.51 | RNA加工 RNA processing |
| Ga10G1419 | CCAACAAGGGCCCAAGTGCTAT | TCTTGTTCCCTTGGCCGACTTG | -1.22 | -1.20 | NADH脱氢酶 NADH dehydrogenase |
| Ga11G2832 | TAAGATCGGTTCGGCTTCGG | ATGCTCCACCCTAACTTGCC | 2.01 | 2.45 | 假定蛋白 Hypothetical protein |
| Ga11G2833 | CACTTCTCGTGTGATGACCTGT | CATCTTCTAACGCTGGAATCTG | 3.70 | 5.20 | 谷胱甘肽转移酶 Glutathione S-transferase |
| Ga11G3625 | ATGCTTCTCAACTGCGCAGGAA | GCTGTTGTCGAACTTTATGGGGC | 2.45 | 2.86 | 谷胱甘肽转移酶 Glutathione S-transferase |
| Ga11G3670 | GACCGATGAAGAGGAACCCAGC | CCATGTGGTGTCCCGGTATGAC | -2.88 | -1.78 | NADH脱氢酶 NADH dehydrogenase |
| Ga12G0113 | TCGACGTCGCCCTGTTTTTCAT | CTGACGTGGACGCCATGTTGAT | 1.53 | 1.58 | 假定蛋白 Hypothetical protein |
| Ga10G0013 | TACCAGTTGAAGCCGAAGGCTG | CCATTGTAAGGCCTGCGCTTTG | 2.18 | 1.81 | 甲基转移酶N-methyltransferase |
新窗口打开|下载CSV
2 结果与分析
2.1 二倍体亚洲棉短绒纤维起始的动态学观察
前人研究结果表明, 长纤维细胞起始于开花当天(0DPA), 在纤维发育早期阶段如在+3DPA时, 长纤维已经突破表皮细胞, 长度约为2 mm, 而短纤维细胞还未突破表皮, 但是胚珠表皮细胞开始出现排列不规则现象。为了鉴定野生型毛子fz和突变体FZ短绒纤维起始的差异, 取这2个材料的0和+3DPA的胚珠, 用扫描电镜观察发现, 在0DPA时野生型和突变体都有明显的纤维突起, 但是野生型纤维明显长于突变体, 而且纤维数目多于突变体; 当胚珠发育到+3DPA时, fz和FZ长纤维长度基本一致, 但纤维数目fz多于FZ, 由于胚珠表面长纤维的干扰, 无法观察到胚珠表面的短纤维(图1-A)。为了进一步验证短绒在细胞水平上的突起, 在电子显微镜下观察fz和FZ 0DPA和+3DPA的胚珠的石蜡切片, 如图1-B所示, 在野生型 fz 胚珠表面能观察到长纤维和短纤维细胞, 其中红色箭头所指位置为长纤维, 而黑色箭头所指位置为短纤维细胞。在0DPA时野生型纤维细胞开始隆起, 而突变体只有少量突起; 随着胚珠的发育 fz 和 FZ 突起细胞数目都有所增加, 但是在+3DPA时, 短绒纤维开始在 fz 胚珠表面出现, 而 FZ 胚珠在相同的发育阶段没有短绒细胞突起, 表明短纤维细胞在FZ中起始失败。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1野生型fz和短绒突变体FZ材料中短纤维发育过程的形态学观察
A: 野生型、短绒突变体的种子以及扫描电镜下看到的短绒纤维起始; B: 石蜡切片所示短绒纤维起始。红色箭头所示长纤维, 黑色箭头所示短纤维细胞。
Fig. 1Morphology analysis of fuzz-fiber development in normal wilt-type fz (upper) and fuzz mutant FZ (lower)
A: the seeds of wild type, natural fuzz mutant and fuzz fiber initiation under scanning electron microscope; B: paraffin section showing fuzz fiber initiation. Red arrow indicates lint fiber, and black arrow indicates fuzz fiber cells.
2.2 转录组数据质量评估
为了进一步理解短绒发育的动态过程和鉴定与短绒发育相关的基因, 选取突变体FZ和野生型材料fz 4个发育时期的胚珠和纤维(0DPA、+3DPA、+5 DPA、+8DPA)的RNA, 每个材料3个生物学重复, 构建了24个RNA文库。24个测序文库获得的约3.2亿条原始数据经去接头和过滤后, 产生出约1.6亿条clean reads。每个文库平均得到65,841,318个高质量的reads (表2)。大约有88%的clean reads可成功比对到已发表的亚洲棉三代基因组上。比对到参考基因组上唯一位置和多个位置的比例分别约为76%~85%、3.53%~6.00%。比对到基因组正链的数目与比对到负链的数目几乎相当。另外每个样本的Q30比例均大于86%, GC含量在35%~65%衡量指标之间。
Table 2
表2
表2样品测序数据与参考基因组序列比对结果分析
Table 2
| 编号 No | 过滤后的序列数 Clean reads | GC GC (%) | Q30 Q30 (%) | 原始数 Total reads | 比对到参考基因组的总数 Mapped reads | 比对到参考基因组唯一位置的数目 Uniq mapped reads | 比对到参考基因组多个位置的数目 Multiple mapped reads | 比对到正链数 Reads map to “+” | 比对到负链数 Reads map to “-“ |
|---|---|---|---|---|---|---|---|---|---|
| L01 | 6E+07 | 42.75 | 88.41 | 1.13E+08 | 100098347(88.79%) | 95615963 (84.82%) | 4482384 (3.98%) | 49356770 (43.78%) | 49542917 (43.95%) |
| L02 | 7E+07 | 42.89 | 87.89 | 1.49E+08 | 132564763(88.84%) | 126665191 (84.88%) | 5899572 (3.95%) | 65411403 (43.84%) | 65613688 (43.97%) |
| L03 | 6E+07 | 43.73 | 88.21 | 1.29E+08 | 114781600(89.23%) | 109876069 (85.42%) | 4905531 (3.81%) | 56731466 (44.10%) | 56901283 (44.24%) |
| L04 | 7E+07 | 43.32 | 87.93 | 1.31E+08 | 115706679(88.26%) | 109946001 (83.86%) | 5760678 (4.39%) | 57037850 (43.51%) | 57182987 (43.62%) |
| L05 | 6E+07 | 43.45 | 88.18 | 1.15E+08 | 101447417(88.47%) | 96871049 (84.47%) | 4576368 (3.99%) | 50115129 (43.70%) | 50235739 (43.81%) |
| L06 | 7E+07 | 43.65 | 88.41 | 1.36E+08 | 119977298(88.23%) | 114195412 (83.98%) | 5781886 (4.25%) | 59276583 (43.59%) | 59413894 (43.69%) |
| L07 | 9E+07 | 43.47 | 88.41 | 1.85E+08 | 164355512(88.86%) | 156168989 (84.43%) | 8186523 (4.43%) | 81092741 (43.84%) | 81289251 (43.95%) |
| L08 | 6E+07 | 43.21 | 87.12 | 1.24E+08 | 108363137(87.68%) | 102760244 (83.15%) | 5602893 (4.53%) | 53406006 (43.21%) | 53552268 (43.33%) |
| L09 | 6E+07 | 43.53 | 88.53 | 1.25E+08 | 110741117(88.61%) | 105077371 (84.08%) | 5663746 (4.53%) | 54559214 (43.66%) | 54711263 (43.78%) |
| L10 | 6E+07 | 43.93 | 88.53 | 1.12E+08 | 95982638(85.64%) | 89277314 (79.66%) | 6705324 (5.98%) | 47006407 (41.94%) | 47142478 (42.06%) |
| L11 | 6E+07 | 43.62 | 87.74 | 1.24E+08 | 106630817(85.76%) | 99506038 (80.03%) | 7124779 (5.73%) | 52250173 (42.03%) | 52415958 (42.16%) |
| L12 | 7E+07 | 43.65 | 88.90 | 1.45E+08 | 125095168(86.41%) | 116813022 (80.69%) | 8282146 (5.72%) | 61391019 (42.41%) | 61584013 (42.54%) |
| L13 | 6E+07 | 43.78 | 89.76 | 1.28E+08 | 109195486(85.27%) | 104512429 (81.62%) | 4683057 (3.66%) | 53869063 (42.07%) | 54027240 (42.19%) |
| L14 | 7E+07 | 43.40 | 90.20 | 1.31E+08 | 115537224(88.07%) | 110898150 (84.53%) | 4639074 (3.54%) | 57057026 (43.49%) | 57220578 (43.61%) |
| L15 | 8E+07 | 43.20 | 90.60 | 1.63E+08 | 144883755(88.62%) | 139117996 (85.09%) | 5765759 (3.53%) | 71628619 (43.81%) | 71848062 (43.94%) |
| L16 | 7E+07 | 44.19 | 89.70 | 1.44E+08 | 122786455(85.34%) | 116535579 (80.99%) | 6250876 (4.34%) | 60495477 (42.04%) | 60621983 (42.13%) |
| L17 | 8E+07 | 44.14 | 90.20 | 1.56E+08 | 130032853(83.13%) | 123871138 (79.19%) | 6161715 (3.94%) | 64115657 (40.99%) | 64295623 (41.10%) |
| L18 | 7E+07 | 43.38 | 90.49 | 1.44E+08 | 127237888(88.61%) | 121687992 (84.75%) | 5549896 (3.87%) | 62876344 (43.79%) | 63029624 (43.90%) |
| L19 | 6E+07 | 43.32 | 91.03 | 1.19E+08 | 106495539(89.28%) | 101489480 (85.08%) | 5006059 (4.20%) | 52555236 (44.06%) | 52649155 (44.14%) |
| L20 | 6E+07 | 43.38 | 91.29 | 1.16E+08 | 101384931(87.60%) | 96799488 (83.64%) | 4585443 (3.96%) | 50050156 (43.24%) | 50157927 (43.34%) |
| L21 | 6E+07 | 43.63 | 91.17 | 1.12E+08 | 98144438(87.78%) | 93630910 (83.74%) | 4513528 (4.04%) | 48444159 (43.33%) | 48541604 (43.42%) |
| L22 | 6E+07 | 44.24 | 87.94 | 1.22E+08 | 100479675(82.20%) | 92865206 (75.98%) | 7614469 (6.23%) | 49048631 (40.13%) | 49206041 (40.26%) |
| L23 | 6E+07 | 44.06 | 87.32 | 1.23E+08 | 102829882(83.46%) | 95769710 (77.73%) | 7060172 (5.73%) | 50222205 (40.76%) | 50359879 (40.87%) |
| L24 | 6E+07 | 44.39 | 86.86 | 1.14E+08 | 96749028(84.55%) | 90393952 (79.00%) | 6355076 (5.55%) | 47365502 (41.39%) | 47485322 (41.50%) |
新窗口打开|下载CSV
为了鉴定24个样品间的生物学重复关系, 对各样本间生物学重复数据进行皮尔逊相关系数(Pearson Correlation Coefficient, PCC)分析(图2), 结果发现, fz材料3个生物学重复间的皮尔逊相关系数均大于0.98 (图2-A), FZ 材料3个生物学重复间的皮尔逊相关系数均大于0.94 (图2-B), 说明这2个材料不同样本的3个生物学重复较好, 可进行差异基因表达分析。
图2
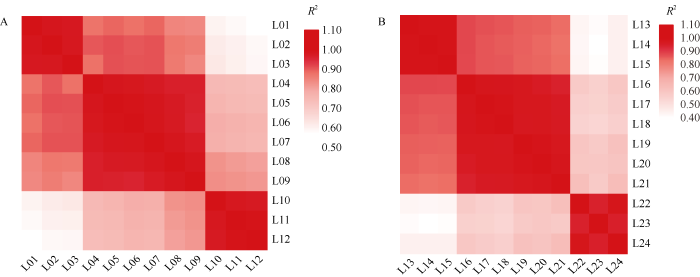 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2所有样本间皮尔逊相关系数分析
A和B分别代表fz和FZ
Fig. 2Pearson correlation coefficients of all samples
A and B indicate fz and FZ, respectively.
2.3 差异基因表达分析
计算FPKM值后, 选择P-value<0.05和|log2(ratio)| ≥1作为筛选差异表达基因的标准。通过比较突变体 FZ和野生型fz材料0、+3、+5、+8DPA的FPKM值, 共鉴定出3784个差异基因(图3-A)。其中在 fz vs. FZ 0DPA时期特异表达的基因有201个, fz vs. FZ +3DPA时期特异表达的基因有313个, fz vs. FZ +5DPA特异表达的基因有1108个, fz vs. FZ +8DPA时期特异表达的基因有1303个。突变体和野生型4个纤维发育时期发现91个共有的差异基因, 这些基因的差异表达情况见图3-B和表3, 52个基因明显上调, 39个基因明显的下调, 对差异基因功能分析发现一些未知蛋白、转录因子、乙烯受体和抗病基因都参与了短绒纤维的发育。图3
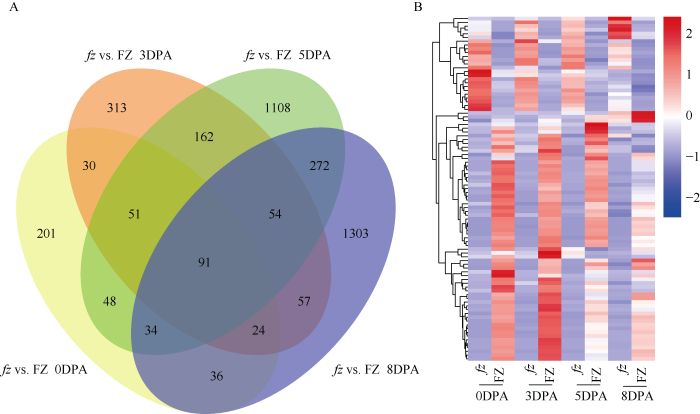 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3fz及其短绒突变体FZ不同纤维发育时期共有的差异基因
A: 在fz和FZ材料中不同纤维发育时期差异基因韦恩图; B: 在fz和FZ两个材料中4个纤维发育时期91个共有的差异基因热图。
Fig. 3Common DEG at different fiber development stages in fz and fuzz-mutant FZ
A: Venn diagram of significant DEGs found in fz and FZ across various fiber development stages; B: Heatmap showing 91 common significant DEGs in fz and FZ across fiber development stages.
Table 3
表3
表3在fz和FZ两个材料中4个纤维发育时期91个共有的差异基因及其表达情况
Table 3
| 基因编号 Gene ID | 基因注释Gene annotation | 0DPA | +3DPA | +5DPA | +8DPA |
|---|---|---|---|---|---|
| Ga11G2831 | 含有WD40结构的蛋白 WD40 repeat-like superfamily protein | 2.00 | 2.70 | 3.00 | 1.72 |
| Cotton_newGene_517935 | 假定蛋白 Hypothetical protein | 2.55 | 2.94 | 1.88 | 1.84 |
| Ga03G0324 | 谷胱甘肽S-转移酶 Glutathione S-transferase | 1.33 | 1.65 | 1.63 | 1.05 |
| Ga11G0115 | 假定蛋白 Hypothetical protein | -1.25 | -1.28 | -1.42 | -1.05 |
| Ga07G1809 | 硫代硫酸盐ATP结合蛋白 Sulfate/thiosulfate ATP-binding protein | 2.35 | 2.34 | 2.40 | 0.95 |
| Cotton_newGene_190500 | 未鉴定蛋白 Uncharacterized protein | 2.37 | 2.67 | 0.97 | 1.16 |
| Cotton_newGene_217366 | 未鉴定蛋白 Uncharacterized protein | 2.03 | 2.44 | 1.33 | 1.65 |
| Ga11G3311 | 葡萄糖基转移酶 UDP-glycosyltransferase 83A1 | 1.25 | 1.98 | 1.50 | 1.85 |
| Ga09G2202 | 乙烯响应因子 AP2/ERF | 1.95 | 1.31 | 2.13 | -1.42 |
| Ga09G0608 | 亮氨酸拉链蛋白 Leucine-rich repeat protein kinase | -1.90 | -1.49 | -0.70 | -0.20 |
| Cotton_newGene_190685 | 假定蛋白 Hypothetical protein | 1.39 | 1.60 | 0.94 | 0.89 |
| Cotton_newGene_184413 | 假定蛋白 Hypothetical protein | 0.86 | 0.82 | 0.68 | 0.20 |
| Cotton_newGene_216530 | 假定蛋白 Hypothetical protein | 1.42 | 1.92 | 0.70 | 1.48 |
| Ga06G0475 | 泛素连接酶 E3 ligase | -0.74 | -0.80 | -0.53 | -0.61 |
| Cotton_newGene_206595 | 假定蛋白 Hypothetical protein | 0.62 | 0.51 | 0.22 | 0.08 |
| Cotton_newGene_188033 | 假定蛋白 Hypothetical protein | -3.18 | -2.89 | -2.89 | -1.50 |
| Ga14G2119 | 谷氨酸受体 Glutamate receptor | 3.94 | 3.27 | 3.11 | 1.92 |
| Cotton_newGene_206588 | 假定蛋白 Hypothetical protein | 2.13 | 2.00 | 1.73 | 0.92 |
| Ga11G2832 | 自噬相关蛋白 Autophagy-related protein | 2.42 | 2.45 | 2.66 | 1.38 |
| Ga06G1674 | 锌依赖的包外多肽酶蛋白 Zn-dependent exopeptidases protein | -1.22 | -0.78 | -0.75 | -0.57 |
| Cotton_newGene_313214 | TMV抗性蛋白 TMV resistance protein | 0.42 | 1.14 | 1.27 | 0.73 |
| Ga04G1219 | 乙酰基葡萄糖转移酶 UDP-GlcNAc | 1.21 | 1.37 | 1.40 | 0.78 |
| Cotton_newGene_383086 | ATP合成酶 ATP synthase | 1.29 | 1.00 | 1.15 | 0.57 |
| Cotton_newGene_336363 | 核酸内切酶 Endonuclease | 1.47 | 1.00 | 1.30 | 0.46 |
| Cotton_newGene_521387 | 功能未知 Unknown | 1.87 | 1.81 | 1.73 | 1.36 |
| Ga04G0915 | 功能未知 Unknown | 1.43 | 1.30 | 1.23 | 0.32 |
| Cotton_newGene_190683 | 细胞色素氧化酶 P450 Cytochrome P450 | 2.78 | 3.25 | 2.09 | 1.73 |
| Cotton_newGene_94861 | 核酸酶 Putative nuclease | -1.00 | -0.86 | -0.93 | -0.65 |
| Cotton_newGene_195900 | 功能未知 Unknown | -3.47 | -3.50 | -3.45 | -2.85 |
| Ga10G0013 | 甲基转移酶 Histone-lysine N-methyltransferase | 1.78 | 1.81 | 2.10 | 1.35 |
| Ga11G0121 | 木糖梅 Alpha-xylosidase 1 | -1.64 | -0.77 | -0.92 | -1.26 |
| Ga11G0187 | 假定蛋白 Hypothetical protein | -0.85 | -0.74 | -0.81 | -0.48 |
| Ga11G0110 | 假定蛋白 Hypothetical protein | 0.82 | 1.21 | 0.71 | 0.84 |
| Ga06G1755 | 脯氨酸受体蛋白激酶Proline-rich receptor-like protein kinase | -1.00 | -0.52 | -0.98 | -0.95 |
| Ga05G2954 | 天冬氨酸酶 Asparticase | 0.65 | 0.58 | 1.25 | 0.24 |
| Cotton_newGene_572274 | 假定蛋白 Hypothetical protein | 2.24 | 2.62 | 1.48 | 1.69 |
| Cotton_newGene_353692 | 假定蛋白 Hypothetical protein | -0.56 | -0.63 | -0.71 | -0.49 |
| Cotton_newGene_483966 | 假定蛋白 Hypothetical protein | 2.77 | 2.77 | 2.71 | 2.30 |
| Ga11G0111 | 微丝蛋白 Fb17 | -0.31 | -0.75 | -1.21 | -2.11 |
| Cotton_newGene_99176 | 丝氨酸/苏氨酸蛋白磷酸酶 Serine/threonine protein phosphatase | 1.08 | 0.85 | 0.97 | 1.10 |
| Ga12G1137 | 乙烯受体蛋白 AP2/ERF | 1.25 | 1.53 | 1.81 | -1.25 |
| Ga11G2354 | 假定蛋白 Hypothetical protein | 1.73 | 1.58 | 1.68 | 0.31 |
| Cotton_newGene_517938 | 细胞色素氧化酶 P450 Cytochrome P450 | 4.11 | 4.65 | 3.60 | 3.14 |
| Cotton_newGene_300782 | 核酸内切酶亚基 Endonuclease subunit | 1.47 | 1.44 | 1.06 | 1.14 |
| Cotton_newGene_252500 | Copia型多蛋白 Copia-type polyprotein | 2.74 | 2.88 | 2.52 | 2.00 |
| Cotton_newGene_517934 | 功能未知 Unknown | 3.19 | 3.84 | 2.65 | 1.73 |
| Cotton_newGene_353671 | 缬氨酸-tRNA连接酶 Valine-tRNA ligase | -0.39 | -0.95 | -0.65 | -0.94 |
| Ga05G3965 | 功能未知 Unknown | -1.55 | -1.24 | -1.95 | -1.34 |
| Ga09G1652 | 丝氨酸/苏氨酸蛋白磷酸酶 Serine/threonine protein phosphatase | -1.61 | -1.34 | -1.01 | -1.25 |
| Ga04G1773 | 转录因子 NAC1 | 1.20 | 0.98 | 2.02 | -1.43 |
| Ga09G1364 | 乙酰辅酶A氧化酶 Acyl-coenzyme A oxidase | 2.66 | 2.52 | 2.53 | 2.06 |
| Cotton_newGene_213765 | 假定蛋白 Hypothetical protein | -1.28 | -1.29 | -1.89 | -1.22 |
| Ga04G0280 | 富含脯氨酸蛋白 Proline-rich protein | 0.71 | 0.77 | 1.46 | 1.21 |
| Cotton_newGene_217374 | 假定蛋白 Hypothetical protein | 1.56 | 1.89 | 0.72 | 1.26 |
| Ga10G0798 | 细胞壁受体激酶 Wall-associated receptor kinase | 0.44 | 0.41 | 0.30 | 0.13 |
| Ga03G0640 | 功能未知 Unknown | 1.88 | 1.62 | 1.14 | 1.16 |
| Cotton_newGene_577041 | 功能未知 Unknown | 3.08 | 1.66 | 3.58 | 0.40 |
| Ga07G1742 | 含有CBS结构域的蛋白 CBS domain-containing protein | -1.45 | -1.20 | -1.17 | -0.69 |
| Ga07G2703 | 丝氨酸/苏氨酸蛋白磷酸酶 Serine/threonine-protein kinase | -0.81 | -0.59 | -0.39 | -0.26 |
| Ga07G0566 | 转录因子 bHLH137 | 1.05 | 1.03 | 3.13 | -0.50 |
| Cotton_newGene_190686 | 假定蛋白 Hypothetical protein | 1.42 | 1.61 | 0.84 | 0.86 |
| Ga04G1342 | 未鉴定蛋白 Uncharacterized protein | 0.80 | 0.30 | 0.17 | 0.37 |
| Cotton_newGene_563611 | 未鉴定蛋白 Uncharacterized protein | 2.35 | 2.52 | 1.44 | 1.83 |
| Cotton_newGene_592029 | 丝裂原活化蛋白激酶 Mitogen-activated protein kinase MMK1 | -1.65 | -1.23 | -0.87 | -0.46 |
| Ga01G1687 | 富含半胱氨酸/组氨酸蛋白 Cysteine/histidine-rich protein | 0.74 | 0.58 | 0.54 | 0.48 |
| Cotton_newGene_54250 | 功能未知 Unknown | 2.88 | 2.87 | 2.67 | 1.88 |
| Cotton_newGene_353710 | 假定蛋白 Hypothetical protein | 1.50 | 1.25 | 1.10 | 1.44 |
| Cotton_newGene_474685 | 未鉴定蛋白 Uncharacterized protein | 2.45 | 2.75 | 1.40 | 1.79 |
| Cotton_newGene_67170 | 假定蛋白 Hypothetical protein | 3.06 | 2.61 | 2.59 | 1.44 |
| Cotton_newGene_353674 | 芳香类多肽 Pentafunctional arom polypeptide | 1.41 | 1.31 | 1.33 | 1.02 |
| Ga03G1055 | 木聚糖糖基化酶 Xyloglucan endotransglucosylase | 0.83 | 2.34 | -1.71 | 0.96 |
| Ga11G0123 | 甲硫氨酸tRNA转移酶 Methionine-tRNA ligase | -2.16 | -1.98 | -1.73 | -1.21 |
| Ga07G0272 | 咖啡酸盐酯酶 Caffeoylshikimate esterase | -1.59 | -1.52 | -1.82 | -1.20 |
| Cotton_newGene_57872 | 假定蛋白 Hypothetical protein | 0.51 | 0.71 | 0.36 | 0.36 |
| Ga03G2383 | 受体蛋白 Receptor | -0.81 | -1.43 | -2.23 | 1.21 |
| Ga02G0173 | 假定蛋白 Hypothetical protein | 1.44 | 1.10 | 1.19 | 1.04 |
| Cotton_newGene_509534 | 假定蛋白 Hypothetical protein | 1.82 | 2.28 | 1.03 | 1.35 |
| Ga11G0124 | 未鉴定蛋白 Uncharacterized protein | -2.92 | -4.04 | -3.64 | -3.84 |
| Ga13G1311 | 丝氨酸/苏氨酸蛋白磷酸酶 Serine/threonine-protein phosphatase | 1.80 | 1.60 | 1.61 | 1.42 |
| Ga10G1419 | NADH脱氢酶 NADH dehydrogenase | -0.99 | -1.20 | -1.37 | -1.31 |
| Cotton_newGene_190499 | 未鉴定蛋白 Uncharacterized protein | 1.30 | 1.60 | 0.80 | 0.91 |
| Cotton_newGene_217363 | 假定蛋白 Hypothetical protein | 1.69 | 2.16 | 0.97 | 1.75 |
| Ga07G2215 | 未鉴定蛋白 Uncharacterized protein | 1.38 | 2.23 | -2.23 | 0.98 |
| Ga11G2833 | 谷胱甘肽S-转移酶 Glutathione S-transferase | 4.06 | 5.20 | 5.45 | 1.92 |
| Cotton_newGene_473974 | 功能未知 Unknown | 1.76 | 1.56 | 1.46 | 0.69 |
| Ga07G0353 | 富含脯氨酸蛋白 Proline-rich protein | 1.48 | 1.61 | 1.06 | 0.36 |
| Cotton_newGene_99000 | 假定蛋白 Hypothetical protein | 0.73 | 0.61 | 0.46 | 0.92 |
| Ga07G1792 | CBL9 | -1.33 | -1.33 | -1.07 | -0.68 |
| Cotton_newGene_190497 | 假定蛋白 Hypothetical protein | 2.55 | 2.89 | 2.28 | 1.86 |
| Ga09G1540 | 蛋白YLS9 YLS9 protein | 0.88 | 1.12 | -0.82 | 0.66 |
| Ga09G1216 | 川芎嗪-3 Tetraspanin-3 | -1.02 | -1.53 | -0.94 | -2.15 |
新窗口打开|下载CSV
通过比较同一材料不同纤维发育时期的差异基因发现(图4-A)在fz材料中, 开花当天至+3DPA时共有2227个基因上调, 1113个基因下调; +3DPA至+5DPA时期, 601个基因上调, 194个基因下调; +5DPA至+8DPA时期2993个基因上调, 3703个基因下调。+3DPA至+5DPA时期差异基因数目较少。在FZ材料中, 0到+3DPA时期, 2420个基因发生了上调, 下调基因的数目是1201; +3DPA到+5DPA时期, 611个基因上调, 753个基因下调; +5DPA到+8DPA时期, 5465个基因上调, 6686个基因下调。
图4
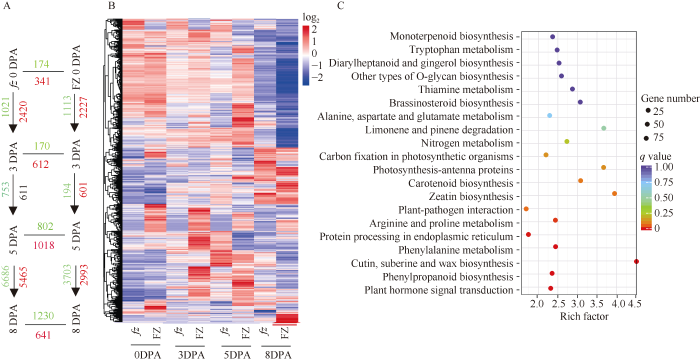 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4fz及其短绒突变体FZ不同纤维发育时期差异基因概况
A: 在fz和FZ材料中不同纤维发育时期差异基因数目; 红色代表上调的基因, 绿色代表下调基因。B: 1783个差异基因在fz和FZ材料4个短绒纤维发育时期的表达趋势。C: fz和FZ材料4个短绒纤维发育时期差异基因KEGG 富集通路。
Fig. 4Overview of DEGS at different fiber development stages in fz and fuzz-mutant FZ
A: number of genes differentially expressed during fiber development within and between fz and FZ; Red represent up-regulated, green represent down-regulated gene. B: the expression trends of 1783 DEGs in fz and FZ across fiber development stages; C: the DEGs KEGG pathway in fz and FZ across fiber development stages.
比较突变体FZ和野生型fz同一纤维时期的差异基因(图4-A, B)表明, 在开花当天与野生型材料相比, 突变体中341个基因上调, 174个基因下调; 在+3DPA时, 突变体中612个基因上调, 下调基因数目170; 在+5DPA时期, 突变体中发生了1018个基因上调, 下调基因数目为802个; 在+8DPA时期, 1230个基因下调, 641个基因上调; 突变体中在短绒起始期上调基因的数目多于下调基因的数目, 这些结果证明大部分基因负调控短绒纤维的起始。
2.4 与短绒起始相关的差异基因KEGG通路分析
通过对2个材料4个纤维发育时期的差异基因KEGG (Kyoto Encyclopedia of Genes and Genomes)通路富集分析(图4-C)发现, 40个基因参与到植物-病原菌互作过程; 20个基因参与到精氨酸和脯氨酸代谢过程; 52个基因参与到内质网上蛋白加工过程; 30个基因涉及到苯丙烷代谢过程; 17个基因涉及到角质、木质素和蜡质生物合成过程; 44个基因参与到苯丙烷生物合成过程; 92个基因汇集到植物激素信号传导过程。对KEGG途径富集因子较高的植物激素信号传导途径中的苯丙烷生物合成途径和角质木质素以及蜡质生物合成途径的差异基因进一步分析发现(表4和表5), 26个差异基因与生长素响应以及生物合成有关, 与野生型(fz)相比, 突变体(FZ)大部分基因在+5DPA时上调, 在+8DPA时下调, 在+3DPA时基因表达差异不明显, 表明生长素(IAA)对短绒的起始没有影响, 对短绒纤维的伸长起正调控作用; 12个基因参与脱落酸(ABA)和乙烯信号转导过程, 如乙烯响应因子和脱落酸受体, 这些基因在短绒纤维起始时期+5DPA突变体中显著上调, Ga11G2673基因差异倍数达到15以上, 而这些基因在纤维伸长时期+8DPA突变体中下调, 这些结果说明棉花体内过量的ABA和乙烯抑制纤维的发育, 而少量的乙烯能促进纤维伸长。参与角质、软木质、蜡质生物合成途径的基因有8个, 主要是细胞色素氧化酶P450和脂肪酸羟化酶(表4); 参与苯丙烷代谢和生物合成途径有关的基因有23个, 大部分为过氧化物酶以及2个苯丙氨酸裂合酶、3个甲基转移酶和1个花青素葡萄糖基转移酶, 说明这些基因与短绒纤维的起始和发育有关。Table 4
表4
表4角质、软木质、蜡质生物合成途径以及苯丙烷代谢途径的差异基因及其表达情况
Table 4
| 基因编号Gene ID | 基因功能 Gene function | 0DPA | +3DPA | +5DPA | +8DPA | KEGG_pathway_annotation |
|---|---|---|---|---|---|---|
| Ga01G2133 | ω-羟基棕榈酸酯-乙酰酰转移酶 Omega-hydroxypalmitate-feruloyl transferase | 0.05 | 0.52 | -0.11 | 1.07 | Cutin, suberine, and wax biosynthesis |
| Ga12G0500 | 细胞色素氧化酶 P450 Cytochrome P450 | -0.06 | 0.18 | -1.22 | -1.24 | Cutin, suberine, and wax biosynthesis |
| Ga07G2444 | 细胞色素氧化酶 P450 Cytochrome P450 | 0.06 | 1.06 | -0.18 | -0.40 | Cutin, suberine, and wax biosynthesis |
| Ga08G1573 | 细胞色素氧化酶 P450 Cytochrome P450 | -0.81 | -0.68 | -0.63 | 1.02 | Cutin, suberine, and wax biosynthesis |
| Ga08G2220 | 细胞色素氧化酶 P450 Cytochrome P450 | -1.95 | -2.11 | -2.22 | 0.14 | Cutin, suberine, and wax biosynthesis |
| Ga10G2228 | ω-羟基棕榈酸酯-乙酰酰转移酶 Omega-hydroxypalmitate-feruloyl transferase | 0.59 | -1.55 | 0.32 | 0.59 | Cutin, suberine, and wax biosynthesis |
| Ga04G1297 | 脂肪酸羟化酶 Fatty acid hydroxylase | -0.95 | -0.15 | 0.31 | 2.10 | Cutin, suberine, and wax biosynthesis |
| Ga07G2443 | 细胞色素氧化酶 P450 Cytochrome P450 | 0.05 | 1.16 | -0.09 | -0.37 | Cutin, suberine, and wax biosynthesis |
| Ga05G2018 | 过氧化物酶Peroxidase | 0.34 | 0.88 | 1.20 | -0.25 | Phenylalanine metabolism |
| Ga14G0143 | 肉桂醇脱氢酶 Cinnamyl alcohol dehydrogenase | -0.06 | 0.47 | 0.77 | -1.41 | Phenylpropanoid biosynthesis |
| Ga10G0561 | 甲基转移酶 Caffeic acid-methyltransferase | 0.08 | -0.02 | -0.19 | -1.30 | Phenylalanine metabolism |
| Ga11G3625 | 过氧化物酶 Peroxidase | 0.65 | 2.86 | 1.37 | 1.11 | Phenylalanine metabolism |
| Ga12G1737 | 甲基转移酶 Caffeic acid-methyltransferase | -1.18 | -0.54 | -0.96 | 1.15 | Phenylpropanoid biosynthesis |
| Ga05G1817 | 过氧化物酶 Peroxidase | -0.05 | 0.33 | -0.17 | -2.71 | Phenylalanine metabolism |
| Ga12G2843 | 过氧化物酶 Peroxidase | -1.71 | -1.17 | -0.29 | -0.02 | Phenylalanine metabolism |
| Ga12G0336 | 甲基转移酶 Caffeic acid-methyltransferase | 0.35 | 0.44 | -0.05 | -1.12 | Phenylpropanoid biosynthesis |
| Ga03G0366 | 花青素葡萄糖基转移酶 Anthocyanidin-glucosyltransferase | 0.25 | 0.72 | -0.22 | 1.45 | Phenylpropanoid biosynthesis |
| Ga05G0239 | 乙烯脱氢酶 Aldehyde dehydrogenase | -0.01 | 0.02 | 0.37 | -3.41 | Phenylpropanoid biosynthesis |
| Ga09G0681 | 葡萄糖苷酶 Glucosidase | -0.18 | 0.30 | -0.15 | -1.20 | Phenylpropanoid biosynthesis |
| Ga04G1983 | 过氧化物酶 Peroxidase | 0.08 | 0.33 | 0.23 | -1.16 | Phenylalanine metabolism |
| Ga09G1700 | 苯丙氨酸氨裂合酶 Phenylalanine ammonia-lyase | -0.19 | 0.37 | -1.21 | -0.84 | Phenylalanine metabolism |
| Ga01G2475 | 过氧化物酶 Peroxidase 68 | 0.42 | 2.69 | 0.60 | -0.49 | Phenylalanine metabolism |
| Ga10G0968 | 肉桂酸4-羟化酶 Cinnamate 4-hydroxylase | 0.24 | -0.26 | -1.28 | -0.01 | Phenylalanine metabolism |
| Ga11G1671 | 过氧化物酶 Peroxidase 17 | 0.13 | 0.46 | 0.05 | -2.06 | Phenylalanine metabolism |
| Ga10G1786 | 过氧化物酶 Cationic peroxidase 1 | 0.46 | 1.38 | 0.50 | -0.25 | Phenylalanine metabolism |
| Ga10G1772 | 莽草酸O-羟基肉桂酰基转移酶 Shikimate O-hydroxycinnamoyltransferase | 0.04 | 1.14 | 1.78 | 1.38 | Phenylpropanoid biosynthesis |
| Ga11G2088 | 过氧化物酶 Peroxidase 72 | 0.42 | 0.22 | 0.79 | -2.38 | Phenylalanine metabolism |
| Ga13G0640 | 细胞色素氧化酶 P450 Cytochrome P450 | -0.09 | 0.15 | 0.12 | -1.35 | Phenylpropanoid biosynthesis |
| Ga12G1739 | 氨基甲酸N-甲基转移酶 Anthranilate N-methyltransferase | -1.30 | -0.86 | -0.68 | 0.34 | Phenylpropanoid biosynthesis |
| Ga04G0842 | 苯丙氨酸裂解酶 Phenylalanine ammonia-lyase | -0.12 | 0.06 | -1.08 | -0.88 | Phenylalanine metabolism |
| Ga04G1242 | 过氧化物酶 Peroxidase 51 | 0.44 | 1.08 | -0.02 | 0.17 | Phenylalanine metabolism |
新窗口打开|下载CSV
Table 5
表5
表5植物激素转导途径中的差异基因及其表达情况
Table 5
| 基因编号 Gene ID | 基因注释 Gene annotation | 基因名称Gene name | 激素Hormone | 0DPA | +3DPA | +5DPA | +8DPA |
|---|---|---|---|---|---|---|---|
| Ga06G1125 | 乙烯响应因子 EIN3-binding F-box protein 1 | EIN3 | ETH | 1.1 | 1.1 | 4.6 | 0.4 |
| Ga01G1781 | 生长素响应因子Auxin response factor 9 | ARF9 | IAA | 1.0 | 1.2 | 2.4 | 0.6 |
| Ga09G2536 | 生长素诱导蛋白Auxin-induced protein 22D | AUX22D | IAA | 1.6 | 1.0 | 2.6 | 1.6 |
| Ga02G1198 | 脱落酸不敏感蛋白Abscisic acid-insensitive protein | ABI3 | ABA | 0.7 | 0.8 | 2.1 | 0.4 |
| Ga01G2735 | 转录因子MYC4 Transcription factor MYC4 | MYC4 | JA | 1.7 | 1.4 | 4.1 | 0.5 |
| Ga13G1098 | 生长素诱导蛋白Auxin-induced protein X10A | AIP10A | IAA | 1.0 | 1.0 | 0.6 | 2.2 |
| Ga03G2153 | 吲哚乙酸合成酶Indole-3-acetic acid-amido synthetase | IAA | IAA | 0.9 | 5.0 | 2.7 | 0.6 |
| Ga05G2728 | 生长素诱导蛋白Auxin-induced protein 6B | AIP6B | IAA | 1.2 | 0.9 | 3.2 | 1.0 |
| Ga11G1087 | 生长素响应蛋白Auxin-responsive protein IAA16 | IAA16 | IAA | 0.8 | 1.1 | 4.2 | 0.4 |
| Ga07G0242 | 脱落酸受体Abscisic acid receptor | PYL2 | ABA | 1.2 | 1.7 | 1.5 | 0.7 |
| Ga01G0771 | 吲哚乙酸合成酶 Indole-3-acetic acid-amido synthetase | IAA | IAA | 1.4 | 1.3 | 3.0 | 0.5 |
| Ga08G2804 | 乙烯响应因子 Ein3-binding F-box protein 4 | EIN3 | ETR | 1.4 | 1.2 | 4.6 | 0.7 |
| Ga06G2183 | 磷酸转移蛋白Histidine-containing phosphotransfer protein 4 | AHP4 | CK | 1.0 | 1.0 | 0.6 | 0.4 |
| Ga10G2395 | 生长素响应蛋白 Auxin-responsive protein IAA26 | SRK2I | IAA | 0.8 | 1.4 | 2.3 | 0.7 |
| Ga10G3077 | 生长素诱导蛋白Auxin-induced protein X15 | AIPX15 | IAA | 0.3 | 0.4 | 0.7 | 1.0 |
| Ga08G2252 | 生长素响应蛋白Auxin-responsive protein IAA11 | IAA11 | IAA | 1.2 | 0.9 | 2.5 | 0.6 |
| Ga03G2608 | 生长素诱导蛋白Auxin-induced protein X10A | AIP10A | IAA | 2.2 | 0.8 | 1.3 | 1.0 |
| Ga03G0428 | 乙烯受体Ethylene receptor 2 | ETR2 | ETH | 1.3 | 0.9 | 2.6 | 0.7 |
| Ga11G1046 | 乙烯受体Ethylene receptor | EIN4 | ETH | 1.3 | 1.2 | 2.2 | 0.8 |
| Ga07G0142 | 蛋白磷酸酶 Protein phosphatase 2C | PP2C | ABA | 1.5 | 1.6 | 4.0 | 0.8 |
| Ga05G0163 | 乙烯响应蛋白EIN3-binding F-box protein 1 | EIN3 | ETH | 0.9 | 1.2 | 3.6 | 0.4 |
| Ga09G2537 | 生长素响应蛋白 Auxin-responsive protein IAA14 | IAA14 | IAA | 1.0 | 0.5 | 0.4 | 1.2 |
| Ga12G2846 | 赤霉素受体 Gibberellin receptor GID1B | GID1B | GA | 1.2 | 1.8 | 2.1 | 0.4 |
| Ga08G2885 | 蛋白磷酸酶 Protein phosphatase 2C | PP2C77 | ABA | 1.4 | 1.3 | 3.9 | 0.4 |
| Ga07G0455 | 生长素响应蛋白 Auxin-responsive protein IAA9 | IAA9 | IAA | 1.6 | 0.8 | 2.6 | 0.3 |
| Ga03G2632 | 生长素转运蛋白 Auxin transporter-like protein 5 | ATP5 | IAA | 0.9 | 1.1 | 2.3 | 0.8 |
| Ga11G3045 | 脱落酸受体 Abscisic acid receptor | PYL9 | ABA | 1.6 | 1.2 | 4.2 | 0.4 |
| Ga11G1086 | 生长素诱导蛋白 Auxin-induced protein 22D | AUX22D | IAA | 1.3 | 1.8 | 6.4 | 0.5 |
| Ga03G2592 | 生长素诱导蛋白 Auxin-induced protein 15A | AUX15A | IAA | 0.9 | 0.6 | 0.5 | 2.2 |
| Ga05G1344 | 生长素响应蛋白 Auxin-responsive protein IAA16 | IAA16 | IAA | 1.5 | 1.0 | 2.3 | 0.6 |
| Ga05G2981 | 细胞分裂素响应蛋白 Cytokinin ARR17 | ARR17 | IAA | 1.0 | 1.2 | 1.0 | 0.5 |
| Ga11G2673 | 乙烯响应转录因子 Ethylene-responsive transcription factor | ERF10 | ETH | 1.8 | 1.0 | 15.7 | 0.1 |
| Ga12G2331 | ABA相关蛋白激酶 ABA-related protein kinase | SNF1 | ABA | 1.2 | 1.3 | 2.1 | 0.9 |
| Ga05G0157 | 生长素响应蛋白IAA29 Auxin-responsive protein IAA29 | IAA29 | IAA | 1.3 | 0.8 | 0.5 | 0.5 |
| Ga02G1269 | 水杨酸相关蛋白 Salicylic acid-related protein | PRP1 | SA | 0.8 | 0.9 | 0.4 | 0.5 |
| Ga06G2185 | 磷酸转移蛋白 Histidine phosphotransfer protein | AHP3 | CK | 1.0 | 0.9 | 0.7 | 0.5 |
| Ga13G2114 | 生长素诱导蛋白Auxin-induced protein 22B | AUX15A | IAA | 1.4 | 1.2 | 2.1 | 1.9 |
| Ga05G3500 | 生长素诱导蛋白 Auxin-induced protein X15 | AUX15 | IAA | 3.6 | 0.7 | 0.9 | 0.6 |
| Ga07G1535 | 吲哚乙酸合成酶 Indole-3-acetic acid-amido synthetase | IAA | IAA | 0.8 | 1.3 | 1.5 | 0.4 |
| Ga09G2560 | 乙烯受体 Ethylene receptor 2 | ETR2 | ETH | 1.3 | 1.4 | 6.3 | 0.3 |
| Ga03G2582 | 生长素诱导蛋白 Auxin-induced protein 15A | AUX15A | IAA | 1.0 | 0.9 | 2.1 | 0.8 |
| Ga05G1383 | 生长素响应蛋白 Auxin-responsive protein IAA27 | IAA27 | IAA | 1.1 | 1.1 | 1.3 | 0.2 |
| Ga11G1865 | 生长素响应蛋白 Auxin-responsive protein | IAA | IAA | 1.3 | 0.9 | 2.0 | 0.4 |
| Ga05G1988 | 生长素诱导蛋白 Auxin-induced protein 6B | AUX6B | IAA | 1.1 | 0.8 | 2.9 | 0.3 |
新窗口打开|下载CSV
2.5 差异基因共表达趋势分析
为了研究野生型(fz)和短绒突变体(FZ)的差异基因在胚珠和纤维中的表达情况, 对2个材料4个纤维发育时期的3784个差异基因绘制时空表达模式图(图5-A), 结果发现3784个基因在2个材料4个纤维发育时期可以划分为9组(cluster), 同一组的基因有相同的表达趋势。clusters 1和2的基因在突变体(FZ)的开花当天表达高, 主要参与应答胁迫反应以及能量传递过程; clusters 4、5和7的基因在突变体的+3DPA表达高, 主要参与转录调控和钙离子, 铁离子, 铜离子的结合; clusters 6 和8的基因在突变体材料开花后5 d表达高, 也主要涉及转录调控, 钙离子, 铁离子和铜离子的结合过程; clusters 3和9的基因主要在突变体+8DPA表达高, 主要参与水解酶活性, 转运活性, 核酸激活, 脂肪酸生物合成途径和ATP结合过程(图5-B)。图5
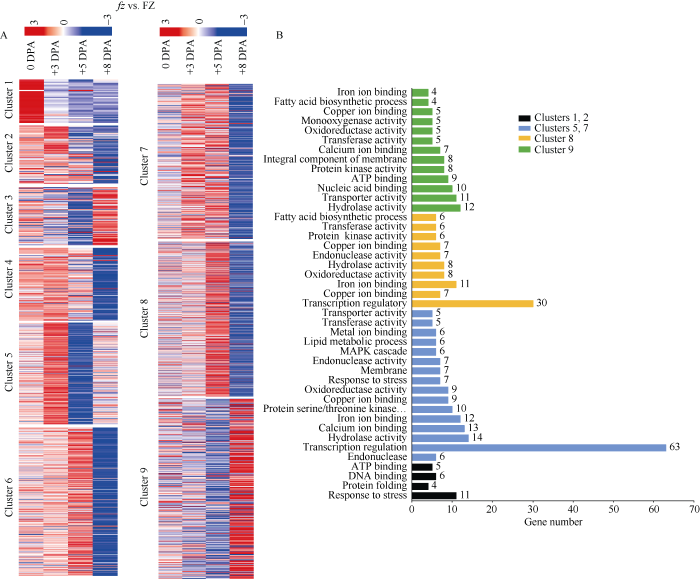 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5差异基因共表达趋势
A: 在fz 和FZ两个材料中4个纤维发育时期所有差异表达基因的热图分析。B: 差异基因功能注释; 黑色代
Fig. 5Coexpression analysis of all significant DEGs
A: heatmap analysis of the expression of all significant DEGs between fz and FZ at four time points. B: functional annotations of significant DEGs; Black bars indicate genes highly expressed at 0DPA, blue bars indicate genes highly expressed at +3DPA, orange bars indicate genes highly expressed at +5DPA, and green bars indicate genes highly expressed at +8DPA.
2.6 陆地棉控制短绒基因在亚洲棉短绒突变体FZ及其野生型fz的表达分析
前人通过图位克隆, 在四倍体陆地棉中分别定位出了一个控制短绒发育和长纤维发育的基因, 即MYBMIXTA-like转录因子GhMML3_A12和GhMML4_D12; GhMML3_A12在TM-1短绒起始期表达量明显高于其在短绒突变体N1中的表达量; GhMML4_D12在仅有长纤维的短绒突变体n2NSM纤维起始期的表达量明显高于其在陆无絮XZ142FLM中的表达量[17,18]。为了验证GhMML3_A12和GhMML4_D12在亚洲棉短绒突变体FZ和其野生型fz中的表达量, 将这2个基因的蛋白序列在亚洲棉中进行同源性比对(图6)表明, GhMML3_A12和GhMML4_D12在亚洲棉中为同一个基因Ga12G1199, 基因功能注释显示该基因属于MYB类转录因子, 其相似性达到97%。转录组数据分析表明, Ga12G1199在FZ材料短绒起始期表达量高于其在fz材料中的表达量(图7), 说明Ga12G1199可能负调控短绒发育。图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6MYBMIXTA-like基因在亚洲棉中的同源序列比对
Fig. 6Homologous sequence alignment of MYBMIXTA-like in G. arboreum
图7
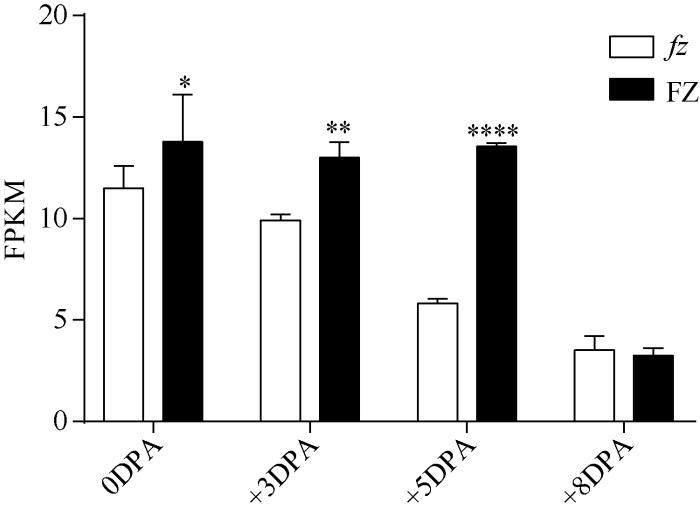 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7Ga12G1199在FZ和fz材料中的表达差异
* P < 0.05, ** P < 0.01, **** P < 0.0001.
Fig. 7Expression of Ga12G1199 in FZ and fz materials
2.7 差异基因实时荧光定量PCR验证
为了验证转录组数据的可靠性, 随机选择22个差异基因在2个材料+3DPA胚珠中进行荧光定量PCR验证。结果显示, RNA-seq结果与荧光定量PCR中的基因表达变化一致。通过单样本t检验, 野生型fz +3DPA胚珠中22个差异基因表达量与突变体FZ胚珠中表达量有显著差异。将qRT-PCR和RNA-seq中差异基因表达量经 log2 转化后做相关性分析, 所得的皮尔逊相关系数为0.89 (图8), 证明RNA-Seq结果可信。图8
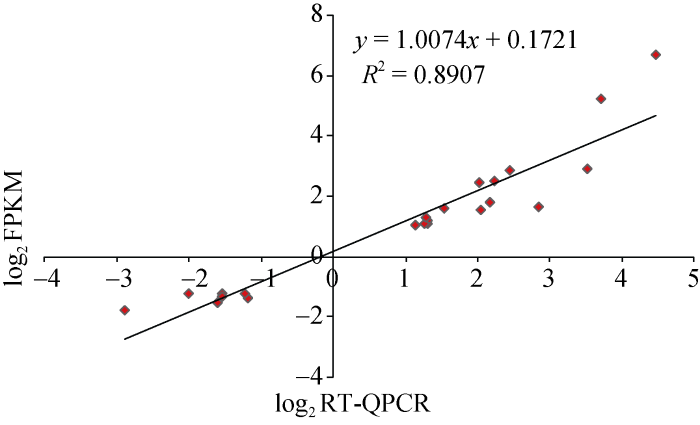 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8RT-QPCR (X-轴)结果与RNA-seq (Y-轴)差异基因相关性分析
散点图代表RT-QPCR与RNA-seq的差异基因相对表达量的log2值。
Fig. 8Comparison of expression ratios from RT-QPCR (Y-axis) and transcriptomic (X-axis) profiling
Scatter plots represent log2 expression ratios calculated from RT- QPCR and RNA-seq.
经qRT-PCR验证, 细胞色素氧化酶P450 (Ga04G0014)、甲基转移酶(Ga07G0568)、转录因子WD40(Ga07G1530)、蛋白受体激酶(Ga08G0121)、二氢黄酮醇-4-还原酶(Ga09G0661)和谷胱甘肽S-转移酶(Ga11G2833)在突变体FZ中上调; 分子伴侣(Ga05G3965)、钙联蛋白(Ga07G1742)、转录因子MYB108(Ga07G2088)、网状蛋白(Ga07G2611)和NADH脱氢酶(Ga11G3670)在突变体材料中下调。
3 讨论
棉纤维起始是决定纤维产量的关键过程。在过去的10年中, 许多参与棉纤维发育的基因已经被鉴定出来。相比之下, 对于野生型种皮上的短绒发育了解比较少。长纤维在生产上主要用于纺织, 短纤维在生产上主要用于火药、造纸和医药用品, 由于短纤维一些特有性质和价值, 吸引了一些科学家研究其发育机制[19]。植物激素例如赤霉素(GA)、生长素、细胞分裂素、油菜素内酯(BR)、脱落酸(ABA)、乙烯、茉莉酸甲酯(JA)和水杨酸是植物内源小信号分子[20]。许多研究证明生长素在纤维发育过程中起重要作用。外源补充吲哚乙酸(IAA)能弥补纤维伸长的缺陷性, 并且能显著增加纤维的总体积[21]。生长素响应蛋白GhARF2和GhARF18, 在纤维起始的细胞中表达高, 当把这2个生长素响应蛋白在拟南芥中超表达后, 能显著促进拟南芥表皮毛起始, 说明GhARF2和GhARF18正调控棉花纤维细胞起始[22]。与GhARF2和GhARF18作用相反, INDOLEACETIC ACIDINDUCED PROTEIN 16 (GhIAA16)负调控纤维的起始和伸长。GhIAA16在野生型胚珠中表达非常低, 然而在无短绒无长纤维突变体中转录水平在开花后急剧升高[23], 这与本研究结果一致。GaIAA16在短绒突变体+5DPA的表达量比野生型的高4倍。前人研究显示向体外离体培养的胚珠中加入ABA, 纤维细胞的起始受到抑制, 而且Ligon-lintless 1突变体0DPA的胚珠中积累ABA的量比野生型的高[24]。这些结果说明ABA负调控棉花纤维起始。本研究结果显示, 与ABA信号传递相关的基因表达量在突变体中明显升高, 与纤维起始呈负相关的植物激素ABA响应因子(Ga11G3045、Ga12G2331)在突变体短绒起始期也显著的上调, 进而抑制了短绒纤维的起始。这些转录组分析结果需要进一步的试验验证。
最近研究显示, 植物体内信号分子, 如钙离子(Ca2+)和活性氧(ROS), 特别是超氧阴离子(O2-)和过氧化氢(H2O2)参与棉花纤维发育。当用H2O2处理光子突变体(N1)和短绒突变体(XinFLM)时, 突变体纤维在0DPA时开始起始, 说明ROS对纤维起始具有促进作用[25]。本研究显示参与活性氧清除的谷胱甘肽转移酶(GST)基因在突变体FZ短绒起始期明显升高。Ca2+也参与棉纤维细胞的起始, 与-1DPA的胚珠相比, 0DPA的胚珠中钙离子的积累明显升高, 基因芯片分析结果显示, 参与钙离子信号转导的基因钙结合蛋白在+1DPA胚珠中表达量明显上调[26]。钙调蛋白(CBL)在Ca2+信号转导过程中起重要作用, 其表达量在短绒突变体胚珠中下调了2倍, 与前人结果一致。Ca2+正调控纤维起始[27,28]。
前人研究结果显示, GhMML3 (GhMYB25-like)基因正调控陆地棉短绒纤维起始, 而GhMML4基因正调控陆地棉长纤维起始[17,18]。而这2个基因在亚洲棉中的同源基因Ga12G1199在短绒突变体中的表达量明显高于其在野生型中的表达量, 该基因可能负调控短绒的发育。造成这种现象的原因可能是陆地棉和亚洲棉短绒发育机制不一样。
本文通过短绒突变体及其野生型4个纤维发育时期的RNA-Seq测序数据分析筛选到4988个差异表达的基因, 其中上调表达的2612个, 下调表达的2376个, 但仅21.5%的基因在棉花基因组数据库中找到注释信息, 有大量未知基因信息存在。参与KEGG排名前5富集通路的基因, 如参与角质、木质素和蜡质合成过程的基因(Ga08G2220, 细胞色素氧化酶)在突变体(FZ)中下调, 说明这些基因可能正调控短绒的发育; 参与苯丙烷代谢和生物合成过程的基因(Ga11G3625、Ga05G2018, 过氧化物酶)在FZ中明显上调, 说明这些基因可能负调控短绒发育; 参与植物激素信号转导途径中的乙烯响应因子相关基因(Ga06G1125、Ga08G2804、Ga05G0163和Ga11G2673)以及ABA合成和信号转导相关基因(Ga02G1198、Ga07G0242、Ga07G0142、Ga08G2885和Ga11G3045)在FZ短绒起始期+5DPA明显上调, 说明这些基因可能负调控短绒发育。在91个共有的差异基因中, 与转座现象相关的基因Cotton_newGene_252500 copia型多蛋白, 转录因子NAC1(Ga04G1773), 以及氧化还原相关基因谷胱甘肽S-转移酶(Ga11G2833)在FZ中明显上调, 说明这些基因可能负调控短绒发育; 而NADPH脱氢酶(Ga10G1419)基因, 与钙信号转导相关基因(Ga07G1792)在FZ中下调, 说明这些基因正调控短绒发育。
4 结论
短绒突变体(FZ)及其野生型(fz)的表型差异为阐明调控棉纤维发育的机制和品系间的形态差异提供了信息。短绒的起始在+3DPA。造成短绒纤维细胞在突变体中未起始的原因是, 一些促进纤维伸长的基因下调, 而另外一些抑制纤维起始的基因上调。例如参与蜡质合成的基因以及Ca2+信号转导的基因在突变体中都下调; 清除活性氧的基因以及参与ABA信号转导的基因在突变体中上调。该研究对于理解亚洲棉短绒纤维起始的机制具有一定的理论意义, 对于提高棉纤维产量和品质具有重要的实践意义。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1093/aob/mcm232URLPMID:17905721 [本文引用: 1]

Cotton is the dominant source of natural textile fibre and a significant oil crop. Cotton fibres, produced by certain species in the genus Gossypium, are seed trichomes derived from individual cells of the epidermal layer of the seed coat. Cotton fibre development is delineated into four distinct and overlapping developmental stages: fibre initiation, elongation, secondary wall biosynthesis and maturation.
URLPMID:11743074 [本文引用: 1]
DOI:10.1093/jhered/93.5.359URL [本文引用: 1]
DOI:10.1007/s11103-004-0392-yURL [本文引用: 1]
Cotton fibers are single-celled seed trichomes of major economic importance. Factors that regulate the rate and duration of cell expansion control fiber morphology and important agronomic traits. For genetic characterization of rapid cell elongation in cotton fibers, ∼ 14,000 unique genes were assembled from 46,603 expressed sequence tags (ESTs) from developmentally staged fiber cDNAs of a cultivated diploid species (Gossypium arboreumL.). Conservatively, the fiber transcriptome represents 35–40% of the genes in the cotton genome. In silico expression analysis revealed that rapidly elongating fiber cells exhibit significant metabolic activity, with the bulk of gene transcripts, represented by three major functional groups – cell wall structure and biogenesis, the cytoskeleton and energy/carbohydrate metabolism. Oligonucleotide microarrays revealed dynamic changes in gene expression between primary and secondary cell wall biogenesis showing that fiber genes in the dbEST are highly stage-specific for cell expansion – a conclusion supported by the absence of known secondary cell wall-specific genes from our fiber dbEST. During the developmental switch from primary to secondary cell wall syntheses, 2553 “expansion-associated” fiber genes are significantly down regulated. Genes (81) significantly up-regulated during secondary cell wall synthesis are involved in cell wall biogenesis and energy/carbohydrate metabolism, which is consistent with the stage of cellulose synthesis during secondary cell wall modification in developing fibers. This work provides the first in-depth view of the genetic complexity of the transcriptome of an expanding cell, and lays the groundwork for studying fundamental biological processes in plant biology with applications in agricultural biotechnology.
DOI:10.1105/tpc.105.040303URLPMID:16461577 [本文引用: 1]
Upland cotton (Gossypium hirsutum) produces the most widely used natural fibers, yet the regulatory mechanisms governing fiber cell elongation are not well understood. Through sequencing of a cotton fiber cDNA library and subsequent microarray analysis, we found that ethylene biosynthesis is one of the most significantly upregulated biochemical pathways during fiber elongation. The 1-Aminocyclopropane-1-Carboxylic Acid Oxidase1-3 (ACO1-3) genes responsible for ethylene production were expressed at significantly higher levels during this growth stage. The amount of ethylene released from cultured ovules correlated with ACO expression and the rate of fiber growth. Exogenously applied ethylene promoted robust fiber cell expansion, whereas its biosynthetic inhibitor l-(2-aminoethoxyvinyl)-glycine (AVG) specifically suppressed fiber growth. The brassinosteroid (BR) biosynthetic pathway was modestly upregulated during this growth stage, and treatment with BR or its biosynthetic inhibitor brassinazole (BRZ) also promoted or inhibited, respectively, fiber growth. However, the effect of ethylene treatment was much stronger than that of BR, and the inhibitory effect of BRZ on fiber cells could be overcome by ethylene, but the AVG effect was much less reversed by BR. These results indicate that ethylene plays a major role in promoting cotton fiber elongation. Furthermore, ethylene may promote cell elongation by increasing the expression of sucrose synthase, tubulin, and expansin genes.
DOI:10.1111/j.1365-313X.2006.02829.xURLPMID:16889650 [本文引用: 1]
Gene expression during the early stages of fiber cell development and in allopolyploid crops is poorly understood. Here we report computational and expression analyses of 32 789 high-quality ESTs derived from Gossypium hirsutum L. Texas Marker-1 (TM-1) immature ovules (GH_TMO). The ESTs were assembled into 8540 unique sequences including 4036 tentative consensus sequences (TCs) and 4504 singletons, representing approximately 15% of the unique sequences in the cotton EST collection. Compared with approximately 178 000 existing ESTs derived from elongating fibers and non-fiber tissues, GH_TMO ESTs showed a significant increase in the percentage of genes encoding putative transcription factors such as MYB and WRKY and genes encoding predicted proteins involved in auxin, brassinosteroid (BR), gibberellic acid (GA), abscisic acid (ABA) and ethylene signaling pathways. Cotton homologs related to MIXTA, MYB5, GL2 and eight genes in the auxin, BR, GA and ethylene pathways were induced during fiber cell initiation but repressed in the naked seed mutant (N1N1) that is impaired in fiber formation. The data agree with the known roles of MYB and WRKY transcription factors in Arabidopsis leaf trichome development and the well-documented phytohormonal effects on fiber cell development in immature cotton ovules cultured in vitro. Moreover, the phytohormonal pathway-related genes were induced prior to the activation of MYB-like genes, suggesting an important role of phytohormones in cell fate determination. Significantly, AA sub-genome ESTs of all functional classifications including cell-cycle control and transcription factor activity were selectively enriched in G. hirsutum L., an allotetraploid derived from polyploidization between AA and DD genome species, a result consistent with the production of long lint fibers in AA genome species. These results suggest general roles for genome-specific, phytohormonal and transcriptional gene regulation during the early stages of fiber cell development in cotton allopolyploids.
DOI:10.1038/s41588-018-0282-xURLPMID:30510239 [本文引用: 1]
Allotetraploid cotton species (Gossypium hirsutum and Gossypium barbadense) have long been cultivated worldwide for natural renewable textile fibers. The draft genome sequences of both species are available but they are highly fragmented and incomplete1-4. Here we report reference-grade genome assemblies and annotations for G. hirsutum accession Texas Marker-1 (TM-1) and G. barbadense accession 3-79 by integrating single-molecule real-time sequencing, BioNano optical mapping and high-throughput chromosome conformation capture techniques. Compared with previous assembled draft genomes1,3, these genome sequences show considerable improvements in contiguity and completeness for regions with high content of repeats such as centromeres. Comparative genomics analyses identify extensive structural variations that probably occurred after polyploidization, highlighted by large paracentric/pericentric inversions in 14 chromosomes. We constructed an introgression line population to introduce favorable chromosome segments from G. barbadense to G. hirsutum, allowing us to identify 13 quantitative trait loci associated with superior fiber quality. These resources will accelerate evolutionary and functional genomic studies in cotton and inform future breeding programs for fiber improvement.
DOI:10.1038/s41588-018-0116-xURLPMID:29736014 [本文引用: 3]
The ancestors of Gossypium arboreum and Gossypium herbaceum provided the A subgenome for the modern cultivated allotetraploid cotton. Here, we upgraded the G. arboreum genome assembly by integrating different technologies. We resequenced 243?G. arboreum and G. herbaceum accessions to generate a map of genome variations and found that they are equally diverged from Gossypium raimondii. Independent analysis suggested that Chinese G. arboreum originated in South China and was subsequently introduced to the Yangtze and Yellow River regions. Most accessions with domestication-related traits experienced geographic isolation. Genome-wide association study (GWAS) identified 98 significant peak associations for 11 agronomically important traits in G. arboreum. A nonsynonymous substitution (cysteine-to-arginine substitution) of GaKASIII seems to confer substantial fatty acid composition (C16:0 and C16:1) changes in cotton seeds. Resistance to fusarium wilt disease is associated with activation of GaGSTF9 expression. Our work represents a major step toward understanding the evolution of the A genome of cotton.
DOI:10.1038/nbt.3207URLPMID:25893781 [本文引用: 1]
Upland cotton is a model for polyploid crop domestication and transgenic improvement. Here we sequenced the allotetraploid Gossypium hirsutum L. acc. TM-1 genome by integrating whole-genome shotgun reads, bacterial artificial chromosome (BAC)-end sequences and genotype-by-sequencing genetic maps. We assembled and annotated 32,032 A-subgenome genes and 34,402 D-subgenome genes. Structural rearrangements, gene loss, disrupted genes and sequence divergence were more common in the A subgenome than in the D subgenome, suggesting asymmetric evolution. However, no genome-wide expression dominance was found between the subgenomes. Genomic signatures of selection and domestication are associated with positively selected genes (PSGs) for fiber improvement in the A subgenome and for stress tolerance in the D subgenome. This draft genome sequence provides a resource for engineering superior cotton lines.
DOI:10.1186/s12870-015-0700-5URLPMID:26728271 [本文引用: 1]
The in vivo determination of the cell-specific chromosome number provides a valuable tool in several aspects of plant research. However, current techniques to determine the endosystemic ploidy level do not allow non-destructive, cell-specific chromosome quantification. Particularly in the gametophytic cell lineages, which are physically encapsulated in the reproductive organ structures, direct in vivo ploidy determination has been proven very challenging. Using Arabidopsis thaliana as a model, we here assess the applicability of recombinant CENH3-GFP reporters for the labeling of the cell's chromocenters and for the monitoring of the gametophytic and somatic chromosome number in vivo.
DOI:10.1186/s12864-017-4066-yURLPMID:28859611 [本文引用: 1]
Cotton fibre is a single cell and it is one of the best platforms for unraveling the genes express during various stages of fibre development. There are reports devoted to comparative transcriptome study on fiber cell initiation and elongation in tetraploid cultivated cotton. However, in the present investigation, comparative transcriptome study was made in diploid cultivated cotton using isogenic fuzzy-lintless (Fl) and normal fuzzy linted (FL) lines belong to Gossypium arboreum, diploid species at two stages, 0 and 10 dpa (days post anthesis), using Affymetrix cotton GeneChip genome array.
DOI:10.1093/bioinformatics/btp120URLPMID:19289445 [本文引用: 1]
A new protocol for sequencing the messenger RNA in a cell, known as RNA-Seq, generates millions of short sequence fragments in a single run. These fragments, or 'reads', can be used to measure levels of gene expression and to identify novel splice variants of genes. However, current software for aligning RNA-Seq data to a genome relies on known splice junctions and cannot identify novel ones. TopHat is an efficient read-mapping algorithm designed to align reads from an RNA-Seq experiment to a reference genome without relying on known splice sites.
DOI:10.1038/nprot.2012.016URLPMID:22383036 [本文引用: 2]
Recent advances in high-throughput cDNA sequencing (RNA-seq) can reveal new genes and splice variants and quantify expression genome-wide in a single assay. The volume and complexity of data from RNA-seq experiments necessitate scalable, fast and mathematically principled analysis software. TopHat and Cufflinks are free, open-source software tools for gene discovery and comprehensive expression analysis of high-throughput mRNA sequencing (RNA-seq) data. Together, they allow biologists to identify new genes and new splice variants of known ones, as well as compare gene and transcript expression under two or more conditions. This protocol describes in detail how to use TopHat and Cufflinks to perform such analyses. It also covers several accessory tools and utilities that aid in managing data, including CummeRbund, a tool for visualizing RNA-seq analysis results. Although the procedure assumes basic informatics skills, these tools assume little to no background with RNA-seq analysis and are meant for novices and experts alike. The protocol begins with raw sequencing reads and produces a transcriptome assembly, lists of differentially expressed and regulated genes and transcripts, and publication-quality visualizations of analysis results. The protocol's execution time depends on the volume of transcriptome sequencing data and available computing resources but takes less than 1 d of computer time for typical experiments and ~1 h of hands-on time.
DOI:10.1093/nar/27.1.29URLPMID:9847135 [本文引用: 1]
Kyoto Encyclopedia of Genes and Genomes (KEGG) is a knowledge base for systematic analysis of gene functions in terms of the networks of genes and molecules. The major component of KEGG is the PATHWAY database that consists of graphical diagrams of biochemical pathways including most of the known metabolic pathways and some of the known regulatory pathways. The pathway information is also represented by the ortholog group tables summarizing orthologous and paralogous gene groups among different organisms. KEGG maintains the GENES database for the gene catalogs of all organisms with complete genomes and selected organisms with partial genomes, which are continuously re-annotated, as well as the LIGAND database for chemical compounds and enzymes. Each gene catalog is associated with the graphical genome map for chromosomal locations that is represented by Java applet. In addition to the data collection efforts, KEGG develops and provides various computational tools, such as for reconstructing biochemical pathways from the complete genome sequence and for predicting gene regulatory networks from the gene expression profiles. The KEGG databases are daily updated and made freely available (http://www.genome.ad.jp/kegg/).
DOI:10.1093/nar/gkt1076URLPMID:24214961 [本文引用: 1]
In the hierarchy of data, information and knowledge, computational methods play a major role in the initial processing of data to extract information, but they alone become less effective to compile knowledge from information. The Kyoto Encyclopedia of Genes and Genomes (KEGG) resource (http://www.kegg.jp/ or http://www.genome.jp/kegg/) has been developed as a reference knowledge base to assist this latter process. In particular, the KEGG pathway maps are widely used for biological interpretation of genome sequences and other high-throughput data. The link from genomes to pathways is made through the KEGG Orthology system, a collection of manually defined ortholog groups identified by K numbers. To better automate this interpretation process the KEGG modules defined by Boolean expressions of K numbers have been expanded and improved. Once genes in a genome are annotated with K numbers, the KEGG modules can be computationally evaluated revealing metabolic capacities and other phenotypic features. The reaction modules, which represent chemical units of reactions, have been used to analyze design principles of metabolic networks and also to improve the definition of K numbers and associated annotations. For translational bioinformatics, the KEGG MEDICUS resource has been developed by integrating drug labels (package inserts) used in society.
DOI:10.1006/meth.2001.1262URLPMID:11846609 [本文引用: 1]
The two most commonly used methods to analyze data from real-time, quantitative PCR experiments are absolute quantification and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control. The 2(-Delta Delta C(T)) method is a convenient way to analyze the relative changes in gene expression from real-time quantitative PCR experiments. The purpose of this report is to present the derivation, assumptions, and applications of the 2(-Delta Delta C(T)) method. In addition, we present the derivation and applications of two variations of the 2(-Delta Delta C(T)) method that may be useful in the analysis of real-time, quantitative PCR data.
DOI:10.1111/nph.13860URLPMID:26832840 [本文引用: 2]
Natural antisense transcripts (NATs) are commonly observed in eukaryotic genomes, but only a limited number of such genes have been identified as being involved in gene regulation in plants. In this research, we investigated the function of small RNA derived from a NAT in fiber cell development. Using a map-based cloning strategy for the first time in tetraploid cotton, we cloned a naked seed mutant gene (N1 ) encoding a MYBMIXTA-like transcription factor 3 (MML3)/GhMYB25-like in chromosome A12, GhMML3_A12, that is associated with fuzz fiber development. The extremely low expression of GhMML3_A12 in N1 is associated with NAT production, driven by its 3' antisense promoter, as indicated by the promoter-driven histochemical staining assay. In addition, small RNA deep sequencing analysis suggested that the bidirectional transcriptions of GhMML3_A12 form double-stranded RNAs and generate 21-22?nt small RNAs. Therefore, in a fiber-specific manner, small RNA derived from the GhMML3_A12 locus can mediate GhMML3_A12 mRNA self-cleavage and result in the production of naked seeds followed by lint fiber inhibition in N1 plants. The present research reports the first observation of gene-mediated NATs and siRNA directly controlling fiber development in cotton.
DOI:10.1111/nph.14844URLPMID:29034968 [本文引用: 2]
Cotton, with cellulose-enriched mature fibers, is the largest source of natural textiles. Through a map-based cloning strategy, we isolated an industrially important lint fiber development gene (Li3 ) that encodes an MYB-MIXTA-like transcription factor (MML) on chromosome D12 (GhMML4_D12). Virus-induced gene silencing or decreasing the expression of the GhMML4_D12 gene in n2 NSM plants resulted in a significant reduction in epidermal cell prominence and lint fiber production. GhMML4_D12 is arranged in tandem with GhMML3, another MIXTA gene responsible for fuzz fiber development. These two very closely related MIXTA genes direct fiber initiation production in two specialized cell forms: lint and fuzz fibers. They may control the same metabolic pathways in different cell types. The MIXTAs expanded in Malvaceae during their evolution and produced a Malvaceae-specific family that regulates epidermal cell differentiation, different from the gene family that regulates leaf hair trichome development. Cotton has developed a unique transcriptional regulatory network for fiber development. Characterization of target genes regulating fiber production has provided insights into the molecular mechanisms underlying cotton fiber development and has allowed the use of genetic engineering to increase lint yield by inducing more epidermal cells to develop into lint rather than fuzz fibers.
DOI:10.1016/j.jprot.2013.02.020URLPMID:23474080 [本文引用: 1]
In this study, a comparative proteomic analysis was employed to identify fuzz fiber initiation-related proteins in wild-type diploid cotton (Gossypium arboreum L.) and its fuzzless mutant. Temporal changes in global proteomes were examined using 2-DE at five developmental time points for fuzz fiber initiation, and 71 differentially expressed protein species were identified by MS, 45 of which were preferentially accumulated in the wild-type. These proteins were assigned to several functional categories, mainly in cell response/signal transduction, redox homeostasis, protein metabolism and energy/carbohydrate metabolism. It was remarkable that more than ten key proteins with high-abundance were involved in gibberellic acid (GA) signaling and ROS scavenging, and increasing concentrations of active GAs and H2O2 were also detected approximately 5dpa in wild type ovules. Furthermore, in vivo GA and H2O2 treatments of ovules inside young bolls showed that these compounds can synergistically promote fuzz fiber initiation. Our findings not only described a dynamic protein network supporting fuzz initiation in diploid cotton fiber ovules, but also deepened our understanding of the molecular basis of cotton fiber initiation.
DOI:10.1016/j.molp.2015.09.011URLPMID:26415696 [本文引用: 1]
Plant phenotypic plasticity is controlled by diverse hormone pathways, which integrate and convey information from multiple developmental and environmental signals. Moreover, in plants many processes such as growth, development, and defense are regulated in similar ways by multiple hormones. Among them, gibberellins (GAs) are phytohormones with pleiotropic actions, regulating various growth processes throughout the plant life cycle. Previous work has revealed extensive interplay between GAs and other hormones, but the molecular mechanism became apparent only recently. Molecular and physiological studies have demonstrated that DELLA proteins, considered as master negative regulators of GA signaling, integrate multiple hormone signaling pathways through physical interactions with transcription factors or regulatory proteins from different families. In this review, we summarize the latest progress in GA signaling and its direct crosstalk with the main phytohormone signaling, emphasizing the multifaceted role of DELLA proteins with key components of major hormone signaling pathways.
DOI:10.1038/nbt.1858URLPMID:21552241 [本文引用: 1]
DOI:10.1093/jxb/ery219URLPMID:29897556 [本文引用: 1]
Auxin signalling plays an essential role in regulating plant development. Auxin response factors (ARFs), which are critical components of auxin signalling, modulate the expression of early auxin-responsive genes by binding to auxin response factor elements (AuxREs). However, there has been no comprehensive characterization of this gene family in cotton. Here, we identified 56 GhARF genes in the assembled Gossypium hirsutum genome. This gene family was divided into 17 subfamilies, and 44 members of them were distributed across 21 chromosomes. GhARF6 and GhARF11 subfamily genes were predominantly expressed in vegetative tissues, whereas GhARF2 and GhARF18 subfamily genes were highly expressed during seed fibre cell initiation. GhARF2-1 and GhARF18-1 were exclusively expressed in trichomes, organs similar to cotton seed fibre cells, and overexpression of these genes in Arabidopsis enhances trichome initiation. Comparative transcriptome analysis combined with AuxRE prediction revealed 11 transcription factors as potential target genes of GhARF2 and GhARF18. Six of these genes were significantly expressed during seed fibre cell initiation and were bound by GhARF2-1 and GhARF18-1 in yeast one-hybrid assays. Our results suggest that GhARF2 and GhARF18 genes may be key regulators of cotton seed fibre initiation by regulating the expression of several transcription factor genes. This study deepens our understanding of auxin-mediated initiation of cotton seed fibre cells and helps us in breeding better cotton varieties in the future.
DOI:10.1016/j.gene.2012.03.069URLPMID:22575728 [本文引用: 1]
Members of the Aux/IAA gene family encode proteins that mediate the responses of auxin-regulated gene expression and regulate various aspects of plant morphological development. Here, we provide the first identification and characterization of nine cDNAs encoding the complete open reading frame (ORF) of the Aux/IAA family in cotton. These were designated GhAux1 to GhAux9 (Gossypiumhirsutum Aux/IAA). The proteins encoded by these nine genes had either whole or partially conserved domains of the Aux/IAA superfamily, with sequence identity ranging from 14% to 69%. A pair of homeologs exists for each Aux/IAA in G. hirsutum acc. TM-1 with high identity both in ORF sequences and amino acid level. Tissue- and organ-specific analysis showed that transcripts of GhAux1, GhAux2, and GhAux3 were abundant in vegetative organs, whereas GhAux4, GhAux5, GhAux6, and GhAux7 were preferentially expressed in ovules on the day of anthesis. GhAux8 and GhIAA16 (previously reported) were also preferentially expressed during fiber developmental stages, especially GhAux8 in fiber early elongation stages, and GhIAA16 in fiber initiation and secondary cell wall thickening stage. GhAux9 was specifically expressed in developing fibers. During the fiber initiation stage, except for GhAux3 and GhAux6, the expression of the other eight GhAuxs in various lintless-fuzzless and linted-fuzzless mutants demonstrated that they were significantly up-regulated compared with linted-fuzzy TM-1.
[本文引用: 1]
DOI:10.1016/j.jplph.2009.10.005URLPMID:19931935 [本文引用: 1]
Single-celled fibers initiate at anthesis from cotton seed epidermal cells of normal developmental cotton cultivars; however, fiber initiation is retarded in some cotton fiber mutants. In this study, the relationship between genes associated with fiber initiation retardation and fiber initiation development was investigated using three cotton fiber developmental mutants: recessive naked seed n2; dominant naked seed N1; and Xinxiangxiaoji Linted-Fuzzless Mutant (XinFLM); with genetic standard line TM-1 (TM-1) as control. Retardation during fiber initiation development was observed in N1 and XinFLM by scanning electron microscope (SEM) analysis. Reverse transcription-polymerase chain reaction (RT-PCR) analysis of genes related to the fiber initiation development showed that the expression of GhEXP1 and GhMYB25 was lower in N1 and XinFLM than in TM-1 and n2, however, the expression of GhTTG1 and GhTTG3 in XinFLM and n2 was higher than in TM-1 and N1. In vivo and in vitro treatments on ovules demonstrated that 30% hydrogen peroxide (H2O2) could prevent fiber initiation retardation in XinFLM, but no evident effect on N1. To further confirm the relationship between gene expression and the effects of H2O2 in XinFLM, qRT-PCR analysis of four differentially expressed genes was performed using -1d post-anthesis (DPA) ovules of XinFLM treated for 24 and 48h with 30% H2O2 and H2O, respectively, with 0 and 1 DPA untreated ovules from XinFLM and TM-1 as control. The results showed that the expression of GhMYB25 and GhEXP1 showed significant difference in XinFLM after -1 DPA ovule treated for 24h relative to the untreated or H2O-treated ovules, with the expression of GhMYB25 increased significantly and that of GhEXP1 decreased. This implied that H2O2 might be one of the upstream signal molecules affecting the expression of GhMYB25 and GhEXP1 genes. The fiber initiation retardation in XinFLM might be related to the production of reactive oxygen species (ROS).
DOI:10.1186/1471-2229-7-22URLPMID:17506877 [本文引用: 1]
Cotton (Gossypium hirsutum L.) fibers are trichomes that initiate from the ovule epidermis. Little is known about the developmental pathway causing fiber to differentiate from ovular epidermal cells even though limits on the number of cells that differentiate into fiber will limit yield.
DOI:10.1105/tpc.109.072686URLPMID:20354197 [本文引用: 1]
Ca(2+) signals are core transducers and regulators in many adaptation and developmental processes of plants. Ca(2+) signals are represented by stimulus-specific signatures that result from the concerted action of channels, pumps, and carriers that shape temporally and spatially defined Ca(2+) elevations. Cellular Ca(2+) signals are decoded and transmitted by a toolkit of Ca(2+) binding proteins that relay this information into downstream responses. Major transduction routes of Ca(2+) signaling involve Ca(2+)-regulated kinases mediating phosphorylation events that orchestrate downstream responses or comprise regulation of gene expression via Ca(2+)-regulated transcription factors and Ca(2+)-responsive promoter elements. Here, we review some of the remarkable progress that has been made in recent years, especially in identifying critical components functioning in Ca(2+) signal transduction, both at the single-cell and multicellular level. Despite impressive progress in our understanding of the processing of Ca(2+) signals during the past years, the elucidation of the exact mechanistic principles that underlie the specific recognition and conversion of the cellular Ca(2+) currency into defined changes in protein-protein interaction, protein phosphorylation, and gene expression and thereby establish the specificity in stimulus response coupling remain to be explored.
DOI:10.1016/j.cub.2015.03.053URLPMID:25936548 [本文引用: 1]
Polarized tip growth is a fundamental process of specialized eukaryotic cells like neuronal axons, fungal hyphae, and plant root hairs and pollen tubes. In pollen tubes, a tip-focused oscillating Ca(2+) gradient governs ions fluxes, vesicle transport, and cytoskeleton dynamics to ensure proper polarized cell growth [1, 2]. While a crucial role of vacuolar Ca(2+) signaling is established for cellular movements like guard cell dynamics [3-5], its contribution to polarized growth remains to be defined. Here we identified the two closely related tonoplast-localized Ca(2+)-sensor proteins CBL2 and CBL3 as crucial regulators of vacuolar dynamics and polarized pollen tube growth. Overexpression of CBL2 or CBL3 in Arabidopsis and tobacco pollen tubes affected vacuolar morphology, pollen germination, and tube growth, but did not alter actin organization, PI(4,5)P2 distribution, or?tip-focused Ca(2+) oscillations. Similarly, loss of function of each single Ca(2+) sensor and cbl2/cbl3 double mutants exhibited impaired pollen tube growth in?vitro and in?vivo. Both Ca(2+) sensors interacted with the kinase CIPK12, which translocated from the cytoplasm to the vacuolar membrane upon this interaction. Also, overexpression of CIPK12 induced severe vacuolar phenotypes, and loss of function of CIPK12 lead to impairment of polar growth. Remarkably, co-expression of CBL2 or CBL3 with CIPK12 resulted in a phosphorylation-dependent, massively enhanced vacuolar inflation and further disruption of polar growth. Together, these findings identify an essential role of the vacuole and vacuolar Ca(2+) signaling for polarized tip growth. We propose that a faithfully balanced activity of Ca(2+)-activated CBL2/3-CIPK12 complexes fulfills fundamental functions to enable the fast growth of pollen tubes in higher plants.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}