Bacterial Diversity of Soybean Rhizosphere Soil under Different Cropping Patterns
WANG Fang1, CHEN Jing-Sheng2, LIU Da-Wei3第一联系人:
收稿日期:2018-02-5接受日期:2018-07-20网络出版日期:2018-07-31
| 基金资助: |
Received:2018-02-5Accepted:2018-07-20Online:2018-07-31
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (2505KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王芳, 陈井生, 刘大伟. 不同种植方式大豆根际土壤细菌多样性分析[J]. 作物学报, 2018, 44(10): 1539-1547. doi:10.3724/SP.J.1006.2018.01539
WANG Fang, CHEN Jing-Sheng, LIU Da-Wei.
大豆胞囊线虫病是世界大豆种植区的主要土传病害之一, 中国大豆播种面积70%以上发现有该病的发生, 大豆胞囊线虫成为制约大豆生产的重要病原物[1]。大豆与小麦、玉米、谷子、亚麻、万寿菊等作物轮作, 大豆感病丰产品种-抗病品种-玉米之间的轮作是目前防治该病害的主要措施[2,3,4]。也有研究表明, 大豆长期连作也可以降低线虫的物种丰度, 连作14年后土壤中胞囊线虫数量极少[5,6]。
作物根际细菌是土壤微生物中的主要群体, 养分活化的驱动者, 对植物根际生物条件具有重要作用, 直接或间接影响病原物的定殖及存活, 甚至一些细菌是植物病原物的拮抗菌[7,8]。国内外****对不同种植方式下大豆根际细菌群落多样性研究表明, 种植制度对土壤中细菌的定殖、丰度及多样性都产生了影响[9,10,11,12,13]。因此, 鉴定大豆胞囊线虫病在建立及发展过程中不同种植方式对大豆根际细菌群落的影响, 研究这些变化对病害的控制及大豆健康生产具有重要意义。
本研究基于 Illumina MiSeq第二代高通量测序平台, 分析不同种植方式下大豆根际细菌菌群丰度及多样性的变化, 探索在大豆胞囊线虫侵染下, 大豆根际细菌菌群对不同种植方式的响应, 为促进大豆健康生长和稳定土壤生产力提供一定的理论依据。
1 材料与方法
1.1 试验小区概况及设计
试验样地设在黑龙江省农业科学院大庆分院与齐齐哈尔分院大豆胞囊线虫病圃试验区。土壤类型为黑钙土。本研究6个试验小区, 包括大豆4年连作(C4, 大庆)、10年连作(C10, 大庆)、20年连作(C20, 齐齐哈尔)、大豆轮作1 (茬口设计: RS, 感病品种-大麻-感病品种, 大庆)、大豆轮作2 (RR, 感病品种-大麻-抗病品种, 大庆)、大豆轮作3 (RQ, 玉米-谷子-感病品种, 齐齐哈尔)。1.2 土壤样品采集
于2016年7月28日至7月29日(大豆盛花期, R2)采集田间土壤样品。选用“Z”字型取样方法, 每个采样小区至少取10个采样点。采样时先将0~5 cm表土去除, 拔出大豆植株根部, 采集紧紧附着大豆根际表面~5 mm土层。将各采集点土壤充分混合均匀, 采用四分法选取土壤, 装于取样袋置冰盒中, 4℃冰箱保存备用。1.3 土壤细菌总DNA的提取及PCR扩增
取每个土壤样品0.5 g, 按照 Mo BIO Power Soil DNA Kit试剂盒说明书提取土壤微生物基因组DNA。经1.0%琼脂糖电泳检测DNA完整性, Nanodrop测定DNA浓度后, 将DNA样品置-20℃冰箱保存备用。以稀释后的基因组DNA为PCR模板, 采用细菌特异性引物341F/805R扩增16S rDNA-V4区, 同时在上游引物的5°端添加带6个碱基长度Barcode序列区分样品。PCR反应条件为95℃预变性2 min; 95℃变性30 s, 55℃退火60 s, 72℃延伸90 s, 30个循环; 72℃终止延伸10 min。取5 μL PCR扩增产物进行凝胶电泳检测。使用 Illumina Miseq高通量测序平台(派森诺生物, 上海)上机测序。1.4 土壤细菌16S rDNA基因测序及原始数据处理
对16S rDNA的V4进行双端测序, 将获得的原始数据进行质量过滤和双端序列的连接, 过滤和去除嵌合体, 统计样品优质序列。应用Qiime软件在97%相似水平下对得到的优质序列进行OUT (Operational Taxonomic Units)聚类和注释。1.5 物种丰度分析
从样本中抽取一定数量的序列, 统计这些序列代表的OTU数目, 根据序列数及OTU丰度从大到小的排序, 以各个丰度值取log2的对数做丰度分布曲线, 反映样品中物种的分布规律。根据OTU列表中的各样品丰度情况, 利用mothur软件计算4种常用的多样性指数。1.6 群落结构分析
在各分类水平上进行群落结构分析和物种丰度差异分析。在上述分析的基础上, 在属的水平下进一步对群落结构进行群落组成分析、主成分分析及聚类分析。1.7 Alpha数据分析
Alpha数据处理及统计分析在SPSS Statistics 17.0中进行, 使用Duncan’s法进行多重比较。2 结果与分析
2.1 土壤样本OTU稀释曲线分析
利用Illumina MiSeq高通量测序平台, 将获得的原始数据进一步过滤, 去除嵌合体, 共获得优质序列300 726条, 平均50 124条。从6个样本中随机抽取一定数量的序列, 与他们所能代表OTU数目构建稀释曲线, 所有样品平均覆盖率均超过90%, 各样品的稀释曲线趋于平坦, 表明该测序深度趋于饱和, 测序数据量已经基本覆盖样品中的所有物种, 可以反映样品中的物种丰度(图1)。图1
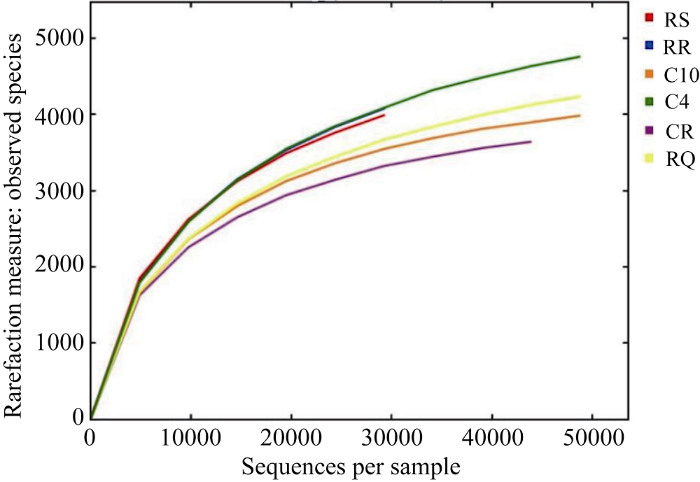 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1相似度为97%水平下各样品稀释性曲线
RR: 感病品种-大麻-抗病品种; RS: 感病品种-大麻-感病品种; RQ: 玉米-谷子-感病品种; C4: 连作4年; C10: 连作10年; C20: 连作20年。
Fig. 1Rarefaction curves for each sample OTUs at cutoff level of 97%
RR: susceptible variety-hemp-resistant variety; RS: susceptible variety-marijuana-susceptible variety; RQ: corn-millet-susceptible variety; C4: continuous cropping 4 years; C10: continuous cropping 10 years; C20: continuous cropping 20 years.
2.2 丰度分布曲线
对丰度值取log2的对数值得到样品丰度分布曲线图。横轴为OTU相对丰度含量等级降序排列, 纵轴是OTU所占相对丰度比例。由图2可知, 3种轮作土壤样品的曲线斜率较大, 表明轮作下土壤细菌物种多样性、均匀度及各物种的丰度高于连作。图2
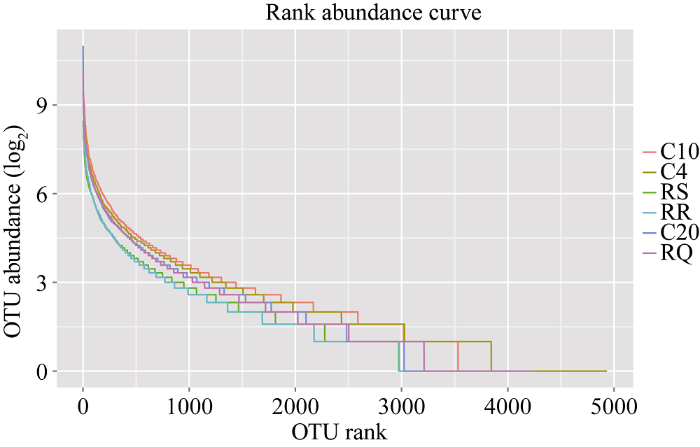 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图26个土壤样品丰度分布曲线
RR: 感病品种-大麻-抗病品种; RS: 感病品种-大麻-感病品种; RQ: 玉米-谷子-感病品种; C4: 连作4年; C10: 连作10年;C20: 连作20年。
Fig. 2Abundance distribution curve for six samples
RR: susceptible variety-hemp-resistant variety; RS: susceptible variety-marijuana-susceptible variety; RQ: corn-millet-susceptible variety; C4: continuous cropping 4 years; C10: continuous cropping 10 years; C20: continuous cropping 20 years.
2.3 Alpha多样性分析
根据OTU 列表中各样品物种丰度情况, 应用Alpha多样性反映不同种植方式下大豆根际土壤群落丰富度(Chao指数)和多样性(Shannon指数)。6个土壤样品OTUs数量在3706~4928, 连作4年OTUs数量最高, 连作20年最低。连作4年细菌丰富度最高, 多样性也相对较高; 连作20年土壤丰富度及多样性均为最低。不同轮作方式土壤细菌丰富度差异不显著(P>0.05)(表1)。短期连作(4年)与长期连作(10年或20年)细菌丰富度及多样性差异显著(P<0.05) (表2)。Shannon指数显示, 大庆4份土壤样品细菌多样性都高于齐齐哈尔地区的2份土壤样品, 不同地区土壤细菌菌群多样性存在明显差异(P<0.05)。Table 1
表1
表1不同轮作下大豆根际土壤细菌序列统计及多样性指数
Table 1
| 样品 Sample | 有效序列数量 Effective sequence-number | OTUs数量 OTUs number (97%) | Chao指数 Chao index | Shannon指数 Shannon index |
|---|---|---|---|---|
| 感病品种-大麻-抗病品种(大庆) RR (Daqing) | 39530 | 4243 | 5247.7 a | 10.45 a |
| 感病品种-大麻-感病品种(大庆) RS (Daqing) | 37614 | 4140 | 5122.7 a | 10.57 a |
| 玉米-谷子-感病品种(齐齐哈尔) RQ (Qiqihar) | 54387 | 4229 | 4972.7 a | 10.20 b |
新窗口打开|下载CSV
Table 2
表2
表2不同连作下大豆根际土壤细菌序列统计及多样性指数
Table 2
| 样品 Sample | 有效序列数量 Effective sequence-number | OTUs数量 OTUs number (97%) | Chao指数 Chao index | Shannon指数 Shannon index |
|---|---|---|---|---|
| 连作4年(大庆) C4 (Daqing) | 67271 | 4928 | 5659.6 a | 10.47 a |
| 连作10年(大庆) C10 (Daqing) | 69356 | 4173 | 4583.8 b | 10.23 b |
| 连作20年(齐齐哈尔) C20 (Qiqihar ) | 52387 | 3706 | 4147.0 b | 10.09 c |
新窗口打开|下载CSV
2.4 土壤细菌群落结构分析
6个土壤样品细菌菌群在门的层次下, 所含物种基本相似, 鉴定到的OTUs归属于47个菌门(图3)。变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、芽单胞菌门(Gemmatimonadetes)、浮霉菌门(Planctomycetes)是供试土壤细菌的优势菌门, 占所有细菌群落总数的90%以上。变形菌门中的物种丰度最多, 变化范围为24.95%~36.00%, 均值31.08%; 其次为放线菌门, 变化范围在13.29%~26.96%, 均值18.78%; 酸杆菌门变化范围在12.99%~17.14%, 均值14.86%; 芽单胞菌门变化范围在5.53%~14.30%, 均值7.92%; 浮霉菌门的变化范围在5.40%~9.09%, 均值6.99%。拟杆菌门(Bacteroidetes)、疣微菌门 (Verrucomicrobia)、绿弯菌门(Chloroflexi)、硝化螺旋菌门(Nitrospirae)的物种丰度也相对较高, 均值均超过1%。图3
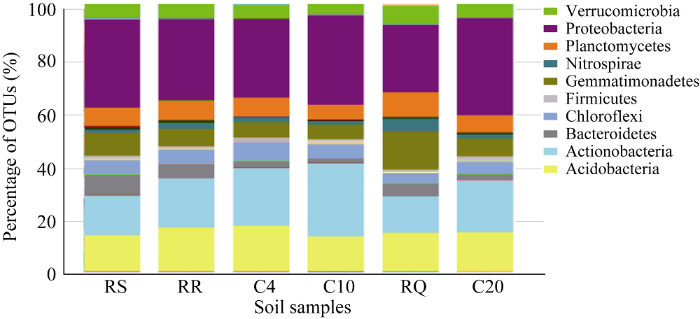 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3各样品在门水平的群落相对丰度
Fig. 3Relative abundance of soil bacterial communities on phylum level of six samples
不同轮作方式放线菌门的OTUs占数据的14.34%、18.07%、13.29%; 连作下放线菌门的OTUs都有上升的趋势, 分别为21.11%、26.96%、18.92%。轮作下芽单胞菌门OTUs所占比例(8.65%、6.59%、14.30%)高于同地区连作下(5.81%、5.53%、6.63%)的比例。轮作下拟杆菌门OTUs所占比例(7.49%、4.91%、4.71%)高于同地区连作下(2.30%、1.49%、2.19%)的比例。
6个土壤样品细菌类群分布在147个纲(图4)。α-变形菌纲(Alphaproteobacteria)物种数量最多, 占细菌群落占总OTUs数量的14.1%。其次为放线菌纲(Actinobacteria), 均值为9.5%, 酸杆菌纲-6 (Acidobacteria-6), 均值为9.0%。变形菌门下的Deltaproteobacteria、Gammaproteobacteria、Betaproteobacteria细菌丰度也相对较高, 均值分别为7.8%、4.8%和4.3%。Hermoleophilia (5.2%)、Gemmatimonadetes (5.1%)、Chloracidobacteria (3.4%)、Planctomycetia (3.1%)、Phycisphaerae (2.4%)、Pedosphaerae (2.3%)、Nitrospira (2.0%)、Acidimicrobiia (1.9%)、Rubrobacteria (1.6%)、Spartobacteria (1.4%)细菌群落占总OTUs数量也均超过1%。
图4
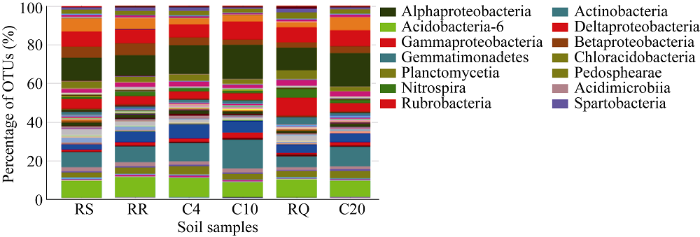 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4各样品在纲水平的群落相对丰度
Fig. 4Relative abundance of soil bacterial communities on class level of six samples
Actinobacteria和Alphaproteobacteria的细菌丰度在大豆连作之后都明显上升, 平均分别增加4.3%和5.2%。而Gemmatimonadetes的细菌丰度在轮作时高于连作下的菌群丰度, 平均增加2.4%。
2.5 群落聚类分析
6个土壤样品细菌类群分布在709个属。应用R (pheatmap)软件, 将所得数据在属的水平上对样品和样品所含菌属丰度相似性进行聚类, 根据聚类后的各样品中不同OTU所含序列的丰度绘制Heatmap图, 利用该图反映样品在菌属水平上细菌群落结构组成的相似性和差异性(图5)。6个土壤样品所含菌落可以分为3个大的分支, 其中最为优势的OTU单元为变形菌门, OTU包含序列最多。从总体上看, 轮作与连作土壤细菌群落结构差异明显。不同年限连作土壤细菌丰度相似性相对较小, 变化明显。连作4年, Streptomyces、Phenylobacterium、Aeromicrobium丰度较高。连作10年, Plesiocystis、Steptosporangium、Actinoplanes、Kibdelosporangium、Rubrobacter的丰度增加。连作20年土壤细菌丰度与轮作下的细菌菌群相似性更高, 但Lysobacter、Ramlibacter、Agrobacterium、Ochrobactrum、Bradyrhizobium、Pseudomonas、Erwinia丰度增加。图5
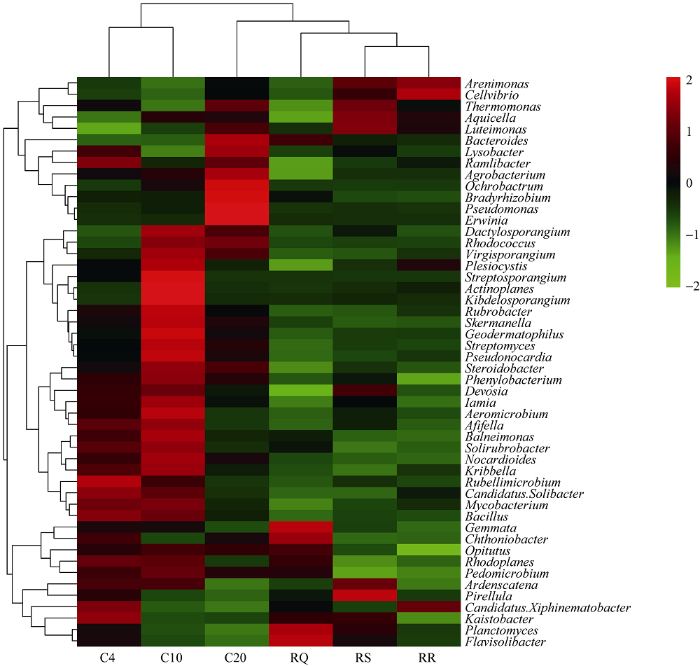 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5样品聚类分析热图
Fig. 5Heatmap tree for samples cluster analysis
2个地区不同轮作方式土壤细菌的丰度相似性较高, 属的丰度变化不明显。Arenimonas、Cellvibrio、Luteimonas、Thermomonas、Aquicella、Luteimonas、Bacteroides、Lysobacter、Planctomyces、Flavisolibacter 细菌丰度较高。
放线菌Rubrobacter、Geodermatophilus、Streptomyces、Pseudonocardia、Devosia 及变形菌Skermanella、 Steroidobacter、Phenylobacterium细菌的丰度差异在轮作及连作下变化明显。
2.6 主成分分析
应用R(vegan)软件绘制PCA图, 对各样品序列之间的内在关系属水平上的多样性进行主成分分析。由图6可知, 主成分1和主成分2分别占40.06%和28.04%。6个土壤样品群落组成可以分成3组, 地域和种植方式对大豆根际土壤细菌群落组成有重要影响, 地域对轮作下细菌多样性的影响高于连作。图6
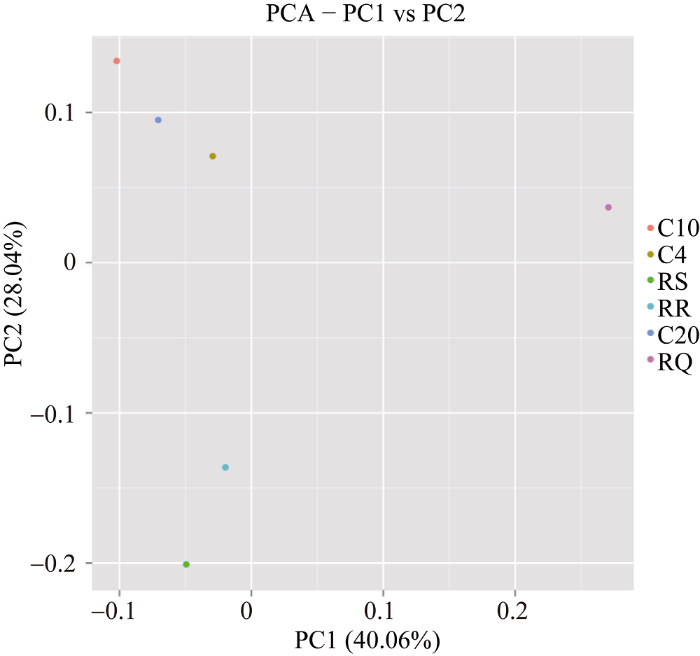 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图66个土壤样品群落主成分分析
Fig. 6Principal component analysis of six soil samples community
3 讨论
本研究利用第二代高通量测序技术, 得到许多难以通过传统的分子生物学方法研究的细菌种群, 这些细菌基本可以代表大豆在不同种植方式下大部分的种类, 6个土壤样品的平均覆盖率均超过90%。由于目前还没有仅应用一种方法就能全面获得土壤微生物种群的信息, 依然有许多菌种未被发现。本研究发现, 轮作及连作大豆根际细菌优势菌群相似, 主要分布在变形菌门、放线菌门、酸杆菌门、芽单胞菌门、浮霉菌门、拟杆菌门、疣微菌门、绿弯菌门、硝化螺旋菌门、纤维杆菌门等。变形菌是土壤中最丰富的细菌类群, 其中α-变形杆菌是陆地土壤中的优势菌群[14]。本研究分类到变形菌门的物种数量在6个供试土壤样品中均最多, 并且α-变形菌亚群占优势, 与前人研究结果类似[15,16,17], 表明根际分泌物对其有吸引作用。朱英波等[18]对黑龙江富锦地区2个相邻的大豆土壤细菌多样性分析发现轮作土壤以变形菌为优势类群, 连作以酸杆菌为优势类群。厚壁菌门的物种数量在本实验所有样品中都较低, 平均占总数据的1.0%, 这与Zhu等[15]的研究结果不同, 表明不同门类细菌在不同生态地区, 根际和根围所占比例不同。
大豆连作障碍问题一直是研究的热点, 针对不同类型土壤和不同种植方式下大豆田土壤微生物的变化已有诸多报道。根据生态环境、栽培方式、研究对象以及连作年限的不同, 土壤微生物类群及丰度的变化结果也不尽相同。众多研究者认为大豆连作导致细菌数量减少, 真菌增多。曾有研究证明, 大豆持续连作后土壤细菌、放线菌的丰富度下降, 真菌丰富度增加, 主要原因是大豆根际沉积、根系和叶枯落物以及多年完全相同的田间管理措施导致了一种特殊的有利于病原真菌积累的土壤微生态环境[19,20]。本研究结果发现, 黑龙江西部地区大豆经过短期的连作(4年), 大豆根际细菌群落的多样性和丰富度呈增加现象。此结果与陈雪丽[21]的结果类似, 轮作、连作2年和连作4年处理的土壤多样性指数高于长期连作(7年以上)。前期也有研究发现, 大豆不同年限连作, 细菌仍是主要的微生物类群, 并且根际细菌16S rRNA基因的拷贝数随着连作年限的增加出现了先增加后降低再增加的现象, 短期连作细菌基因组DNA质量略高于20年连作和轮作, 此现象在其他作物连作时也显示出同样的趋势[22,23,24]。这些结果表明连作对微生物的影响是一个持续积累的过程。
在一种土壤病害爆发之后, 随着作物连作的延长, 病害开始降低, 长期连作导致健康土壤到致病土壤再到抑病土壤的转变[25,26]。连作使土壤出现抑制线虫的现象已在全世界发现[27]。这可能涉及到土壤根际微生物群落结构的变化, 不同功能生物类群结构的交换更替, 一方面有益真菌增加, 有害真菌和植食性线虫减少, 另一方面细菌优势菌群丰度的变更[28]。也有研究表明, 长期连作土壤较短期连作有更多特异性细菌类群存在, 如链霉菌属(Streptomyces)和根瘤菌(Bradyrhizobium)[16, 29]。本研究聚类分析结果显示, 长期连作(20年)的优势细菌丰度与轮作土壤相似性更高, 并且发现随着连作年限的增加, 一些有益菌的丰度高于轮作, 如与固氮有关的根瘤菌和与有机物矿化有关的链霉菌属的丰度。促进植物生长和病原菌有防控作用的芽孢杆菌属(Bacillus)的丰度在连作4年及10年都高于轮作。具有生物防控作用的溶杆菌属(Lysobacter)在连作4年和20年也表现出高丰度, 其中Lysobacter spp.已用在根结线虫的防治中[30,31]。在不同年限的连作下, 土微菌属(Pedomicrobium)相对丰度也高于轮作。土微菌属能够以烃类污染物为碳源来氧化滞留土壤环境中的铁和锰, 降低对植物的毒害作用[32]。这也表明土壤菌群结构随着连作年限增加处于不断的动态变化过程中, 优势菌群不断交替更新, 越来越接近于轮作土壤。本研究鉴定到一些促进植物生长且有抑制病原菌作用的细菌类群, 该结果对解释大豆长期连作对抑病土壤的形成具有一定价值。这些类群在大豆胞囊线虫病生态控制方面的具体作用待进一步研究。
Zhu等[15]发现放线菌门在健康的大豆植株根际比受到胞囊线虫侵染的土壤中物种数量高。而本研究发现轮作下放线菌门的OTUs占数据的比例、丰度及多样性低于连作下, 且连作时间越长放线菌门所占比例越高。芽单胞菌门及拟杆菌门物种数量所占比例在轮作下高于同地区连作下的比例, 不同年限连作导致这两种种群丰度和多样性都有不同程度的降低, 变化趋势明显。前人一部分发现随着大豆连作年限的增加, 放线菌数量减少, 也有一些研究发现呈增加趋势, 并且在其他作物上也发现类似现象[33,34,35,36,37,38]。但普遍认为与轮作相比, 连作1~6年土壤微生物三大类群变化的主要特征是细菌总量减少, 真菌总量增加和放线菌变化表现不规律[39]。基于以上研究结果可以说明不同种植方式及生态环境对各类微生物的影响并不一致, 不能一概而论。
4 结论
土壤是一个复杂的生态系统, 种植方式、作物品种甚至是根际范围均与土壤微生物类群密切相关。大豆轮作根际土壤细菌菌群多样性高于连作方式。短期连作, 根际土壤细菌菌群的丰度略有增加; 随着连作年限增加, 细菌菌群丰度及多样性都下降。不同轮作方式细菌菌属丰度差异不明显, 不同年限的连作细菌菌属丰度表现特异性, 土壤功能细菌根瘤菌、链霉菌属、芽孢杆菌属、溶杆菌属、土微菌属在连作下相对丰度较高, 长期连作细菌菌属相似性更接近轮作方式。放线菌及变形菌细菌丰度在不同种植方式下变化明显。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]

大豆胞囊线虫(Heterodera glycines,Ichinohe)病是大豆生产中的重要限制因子之一,其病原大豆胞囊线虫是一种典型定居型内寄生土传线虫,大豆连作会使土壤中大豆胞囊线虫不断积累,导致病害加重,因此大豆胞囊线虫也是引起大豆连作障碍的主要因素之一。然而,当大豆感病品种连续种植5年以上,会出现大豆胞囊线虫自然衰退现象,并将存在这种现象的土壤称为大豆胞囊线虫抑制性土壤。抑制性土壤
URL [本文引用: 1]
大豆胞囊线虫(Heterodera glycines,Ichinohe)病是大豆生产中的重要限制因子之一,其病原大豆胞囊线虫是一种典型定居型内寄生土传线虫,大豆连作会使土壤中大豆胞囊线虫不断积累,导致病害加重,因此大豆胞囊线虫也是引起大豆连作障碍的主要因素之一。然而,当大豆感病品种连续种植5年以上,会出现大豆胞囊线虫自然衰退现象,并将存在这种现象的土壤称为大豆胞囊线虫抑制性土壤。抑制性土壤
[本文引用: 1]
[本文引用: 1]
DOI:10.3321/j.issn:0577-7518.2007.04.003URL [本文引用: 1]
Soybean cyst nematode (SCN,Heterodera glycines) is a disastrous disease all over the world, which causing a lot of losses of soybean especially in USA, Canada and China. The number of SCN and soil nematode community structure were studied under continuous cropping and rotation soybean fields by root dying and traditional nematode classification in Wangjia, Shenyang City, Liaoning Province. The soil nematode community structure was surveyed and analysed using the mathematical statistics. The results would provide rightful tillage practices to control the soybean cyst nematode and other diseases of soybean, to keep the yield increasing sustainably in the northeast of China. The results showed that the number of SCN including the number of cysts and all juveniles of different stages in soil and roots of soybean was notably increased under continuous cropping soybean field against under rotation(95.6 in 2005 and 335.5 in 2006 vs. 10.3 and 31.8). The number of cysts in susceptible soybean was decided by the number of invaded J2, and it was the least under maize-maize-soybean rotation in the research. Total number of soil nematodes was much more in rotation than continuous cropping soybean field, because the relative aboundance of dominant genus Helicotylenchus which cp-scaling 3 and dominant tropic groups as plant parasitic feeders were higher in rotation than continuous cropping, but Heterodera glycines which cp-scaling 3 was more in continuous cropping than rotation. So H.glycines among of all kind of nematodes would be acted as key species that indicated the different degrees of SCN disease. Moreover dominant tropic groups and dominant genus would be more effective indicator to respond to soil health in soybean field in the northeast of China.
DOI:10.3321/j.issn:0577-7518.2007.04.003URL [本文引用: 1]
Soybean cyst nematode (SCN,Heterodera glycines) is a disastrous disease all over the world, which causing a lot of losses of soybean especially in USA, Canada and China. The number of SCN and soil nematode community structure were studied under continuous cropping and rotation soybean fields by root dying and traditional nematode classification in Wangjia, Shenyang City, Liaoning Province. The soil nematode community structure was surveyed and analysed using the mathematical statistics. The results would provide rightful tillage practices to control the soybean cyst nematode and other diseases of soybean, to keep the yield increasing sustainably in the northeast of China. The results showed that the number of SCN including the number of cysts and all juveniles of different stages in soil and roots of soybean was notably increased under continuous cropping soybean field against under rotation(95.6 in 2005 and 335.5 in 2006 vs. 10.3 and 31.8). The number of cysts in susceptible soybean was decided by the number of invaded J2, and it was the least under maize-maize-soybean rotation in the research. Total number of soil nematodes was much more in rotation than continuous cropping soybean field, because the relative aboundance of dominant genus Helicotylenchus which cp-scaling 3 and dominant tropic groups as plant parasitic feeders were higher in rotation than continuous cropping, but Heterodera glycines which cp-scaling 3 was more in continuous cropping than rotation. So H.glycines among of all kind of nematodes would be acted as key species that indicated the different degrees of SCN disease. Moreover dominant tropic groups and dominant genus would be more effective indicator to respond to soil health in soybean field in the northeast of China.
[本文引用: 1]
[本文引用: 1]
DOI:10.3321/j.issn:1007-9084.2006.02.017URL [本文引用: 1]
在白浆土设计长期轮作和连作试验区,采取麦麦豆油玉豆6区轮作和麦玉豆连作,18a内连续调 查土壤中大豆胞囊线虫(SCN)的胞囊数量,观察轮作或连作对土壤中SCN胞囊数量的影响。试验结果表明,长期轮作使土壤中胞囊数量有减少的趋势,各茬口 间胞囊数量变化幅度减小,轮作12a后土壤中胞囊数量达到动态平衡;大豆连作的前2a土壤中胞囊数量急速增加,以后缓慢增加,7a后有下降趋势,14a后 土壤中的胞囊数量在较高水平上趋于平衡;小麦或玉米连作前3a土壤中的胞囊数量呈快速下降,4a后缓慢减少,14a后土壤中胞囊数量极少。
DOI:10.3321/j.issn:1007-9084.2006.02.017URL [本文引用: 1]
在白浆土设计长期轮作和连作试验区,采取麦麦豆油玉豆6区轮作和麦玉豆连作,18a内连续调 查土壤中大豆胞囊线虫(SCN)的胞囊数量,观察轮作或连作对土壤中SCN胞囊数量的影响。试验结果表明,长期轮作使土壤中胞囊数量有减少的趋势,各茬口 间胞囊数量变化幅度减小,轮作12a后土壤中胞囊数量达到动态平衡;大豆连作的前2a土壤中胞囊数量急速增加,以后缓慢增加,7a后有下降趋势,14a后 土壤中的胞囊数量在较高水平上趋于平衡;小麦或玉米连作前3a土壤中的胞囊数量呈快速下降,4a后缓慢减少,14a后土壤中胞囊数量极少。
[本文引用: 1]
DOI:10.1146/annurev.phyto.40.030402.110010URL [本文引用: 1]
DOI:10.1126/science.1203980URLPMID:21551032 [本文引用: 1]
Disease-suppressive soils are exceptional ecosystems in which crop plants suffer less from specific soil-borne pathogens than expected owing to the activities of other soil microorganisms. For most disease-suppressive soils, the microbes and mechanisms involved in pathogen control are unknown. By coupling PhyloChip-based metagenomics of the rhizosphere microbiome with culture-dependent functional analyses, we identified key bacterial taxa and genes involved in suppression of a fungal root pathogen. More than 33,000 bacterial and archaeal species were detected, with Proteobacteria, Firmicutes, and Actinobacteria consistently associated with disease suppression. Members of the 驴-Proteobacteria were shown to have disease-suppressive activity governed by nonribosomal peptide synthetases. Our data indicate that upon attack by a fungal root pathogen, plants can exploit microbial consortia from soil for protection against infections.
DOI:10.3724/SP.J.1006.2013.02016URL [本文引用: 1]
在大豆开花期分别对3个施氮水平下(0、50和100 kg hm 2)大豆连作(大豆-大豆-大豆)、玉米-大豆轮作I(大豆-玉米-大豆)及玉米-大豆轮作II(玉米-玉米-大豆),应用PCR-DGGE技术研究玉米-大豆轮作及施氮对土壤细菌群落结构变化的影响。结果表明,随着施氮水平的提高,3种种植方式土壤中细菌群落多样性、丰富度均呈减少趋势。高氮处理(100 kg hm 2)明显降低了大豆连作、玉米-大豆轮作II根际土壤细菌群落多样性及丰富度,玉米-大豆轮作I根际土壤细菌群落多样性及丰富度略有降低。玉米-大豆轮作I种植方式可减轻氮肥对其根际细菌群落多样性和丰富度的影响,但施氮明显改变了其细菌群落结构。玉米-大豆轮作II中大豆根际土壤细菌群落结构较为稳定,受氮肥影响较小。在3种种植方式的土壤中,分布着酸杆菌门、变形菌门及厚壁菌门细菌,其中前两门菌群占主导地位。
DOI:10.3724/SP.J.1006.2013.02016URL [本文引用: 1]
在大豆开花期分别对3个施氮水平下(0、50和100 kg hm 2)大豆连作(大豆-大豆-大豆)、玉米-大豆轮作I(大豆-玉米-大豆)及玉米-大豆轮作II(玉米-玉米-大豆),应用PCR-DGGE技术研究玉米-大豆轮作及施氮对土壤细菌群落结构变化的影响。结果表明,随着施氮水平的提高,3种种植方式土壤中细菌群落多样性、丰富度均呈减少趋势。高氮处理(100 kg hm 2)明显降低了大豆连作、玉米-大豆轮作II根际土壤细菌群落多样性及丰富度,玉米-大豆轮作I根际土壤细菌群落多样性及丰富度略有降低。玉米-大豆轮作I种植方式可减轻氮肥对其根际细菌群落多样性和丰富度的影响,但施氮明显改变了其细菌群落结构。玉米-大豆轮作II中大豆根际土壤细菌群落结构较为稳定,受氮肥影响较小。在3种种植方式的土壤中,分布着酸杆菌门、变形菌门及厚壁菌门细菌,其中前两门菌群占主导地位。
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s11248-013-9691-xURL [本文引用: 1]
DOI:10.3839/jksabcURL [本文引用: 1]
DOI:10.1007/s11104-013-1796-8URL [本文引用: 1]
DOI:10.1023/A:1006853600205URLPMID:9421915 [本文引用: 1]
Abstract Genomic DNA was isolated from the active layer of tundra soil collected from the Kolyma lowland, Northeast Eurasia, near the Arctic Ocean coast. The SSU (small subunit) rRNA genes were amplified with eubacterial primers from the bulk genomic community DNA and cloned into plasmid vectors. Forty-three SSU rDNA clones were obtained, and all of them had different RFLP patterns. Phylogenetic analysis based on partial sequences (about 300 bp) established with the maximum likelihood method revealed the presence of three major and several minor groups that fell into 11 of the established lines of bacteria, and one sequence that could not be assigned to any of the described groups. Most of the clones belonged to the alpha (20.9%) and delta (25.6%) subdivisions of the Proteobacteria, with lesser proportions in the beta (9.3%) and gamma (4.7%) subdivisions, groups typically isolated from soil by culture methods. Fewer than 12% of the clones belonged to Gram-positive bacteria, and 16% of the clones were related to Fibrobacter. The majority of the clones (70%) had sequences that were 5-15% different from those in the current databases, and 7% of the clones had sequences that differed by more than 20% from those in the database. The results suggest that these tundra-derived clones are very diverse in phylogeny, and that many probably reflect new genera or families. Hence, most of the tundra soil bacterial community has never been isolated and thus the physiology and function of its dominant members appears to be unknown.
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
DOI:10.7666/d.Y2887846URL [本文引用: 1]
中国农业生产中,豆禾间作历史悠久,不仅可以消除大豆农田中的“氮阻遏”问题,提高根瘤菌的固氮结瘤能力,还能够实现双增产。为此,本文将豆禾间作与接种根瘤菌WGF03及其缺失突变体△exoD相结合,研究二因素对大豆生长及细菌群落结构的影响。大豆植株生理指标方面结果表明:(1)甘蔗间作大豆与单作大豆相比,在大豆株高、瘤数、瘤重、根瘤酶活方面均有增加,间作方式能够促进大豆生长和结瘤固氮能力。(2)接种野生型根瘤菌WGF03促进效果最明显,间作及接种因素的叠加作用,共同促进了大豆的生长:株高在花期和成熟期分别增加了7.29%和5.3%,瘤数、瘤重方面增加了44.95%~70.89%不等,根瘤固氮酶活在4个处理中最高,达到28.99μ mol·g-1·h-1。接种突变菌△exoD后,增加趋势不明显,甚至在花期瘤数、花期瘤重以及根瘤固氮酶活方面出现抑制作用,减少了34.23%~51.02%不等。说明此突变菌的影响力超过问作的影响,起到负面抑制作用,推测根瘤菌的exoD基因会影响大豆植株生长,特别是在根瘤方面表现明显。对根瘤内生细菌群落结构分析发现:(1)根瘤内生细菌在花期数量较多,处理组之间相似性指数在84.3~92.3之间,差异较大;成熟期数量较少,处理组之间相似性指数在90.3~95之间,差异逐渐减小。(2)单作和间作方式下,多样性和相似性无明显差别,这是由于间作处理时,甘蔗根系距离大豆根瘤较远,影响不到根瘤内细菌群落结构。(3)在花期,接菌处理使得根瘤内生细菌的种类和数量明显减少,花期占优势的蓝藻门细菌协助外来根瘤菌进行生物固氮。在成熟期则由于接种根瘤菌竞争力减弱,土著根瘤菌最终占优势,使得四个处理组之间细菌群落结构差异不大,并且蓝藻门细菌数量也急剧减少。大豆根际、根围土壤细菌群落分析表明:(1)土壤细菌比对应的根瘤内生细菌更为丰富,且根际土壤中大多数细菌为PGPR菌,能够促进植物生长和营养的吸收。(2)两种分析方法均得出,根围细菌群落种类和数量的多样性大于根际细菌的。(3)高通量测序方法展示的相似性数据表现为根围土壤不间作处理组W4、W5、W6相似性极高,而由于间作及接菌处理,W1、W2、W3各有不同。表明间作体系中,甘蔗根系伸向大豆的根围区,根围群落结构受其影响。(4)接种的外源根瘤菌WGF03在根际土壤中,比不接种处理组的多样性要大,而突变菌则在根际、根围土壤中均没有表现出差异。推测野生型菌株WGF03的适应能力强,对周围土壤细菌的影响较大,分泌的大量胞外多糖或其他物质吸引着细菌往根际部分聚集。
DOI:10.7666/d.Y2887846URL [本文引用: 1]
中国农业生产中,豆禾间作历史悠久,不仅可以消除大豆农田中的“氮阻遏”问题,提高根瘤菌的固氮结瘤能力,还能够实现双增产。为此,本文将豆禾间作与接种根瘤菌WGF03及其缺失突变体△exoD相结合,研究二因素对大豆生长及细菌群落结构的影响。大豆植株生理指标方面结果表明:(1)甘蔗间作大豆与单作大豆相比,在大豆株高、瘤数、瘤重、根瘤酶活方面均有增加,间作方式能够促进大豆生长和结瘤固氮能力。(2)接种野生型根瘤菌WGF03促进效果最明显,间作及接种因素的叠加作用,共同促进了大豆的生长:株高在花期和成熟期分别增加了7.29%和5.3%,瘤数、瘤重方面增加了44.95%~70.89%不等,根瘤固氮酶活在4个处理中最高,达到28.99μ mol·g-1·h-1。接种突变菌△exoD后,增加趋势不明显,甚至在花期瘤数、花期瘤重以及根瘤固氮酶活方面出现抑制作用,减少了34.23%~51.02%不等。说明此突变菌的影响力超过问作的影响,起到负面抑制作用,推测根瘤菌的exoD基因会影响大豆植株生长,特别是在根瘤方面表现明显。对根瘤内生细菌群落结构分析发现:(1)根瘤内生细菌在花期数量较多,处理组之间相似性指数在84.3~92.3之间,差异较大;成熟期数量较少,处理组之间相似性指数在90.3~95之间,差异逐渐减小。(2)单作和间作方式下,多样性和相似性无明显差别,这是由于间作处理时,甘蔗根系距离大豆根瘤较远,影响不到根瘤内细菌群落结构。(3)在花期,接菌处理使得根瘤内生细菌的种类和数量明显减少,花期占优势的蓝藻门细菌协助外来根瘤菌进行生物固氮。在成熟期则由于接种根瘤菌竞争力减弱,土著根瘤菌最终占优势,使得四个处理组之间细菌群落结构差异不大,并且蓝藻门细菌数量也急剧减少。大豆根际、根围土壤细菌群落分析表明:(1)土壤细菌比对应的根瘤内生细菌更为丰富,且根际土壤中大多数细菌为PGPR菌,能够促进植物生长和营养的吸收。(2)两种分析方法均得出,根围细菌群落种类和数量的多样性大于根际细菌的。(3)高通量测序方法展示的相似性数据表现为根围土壤不间作处理组W4、W5、W6相似性极高,而由于间作及接菌处理,W1、W2、W3各有不同。表明间作体系中,甘蔗根系伸向大豆的根围区,根围群落结构受其影响。(4)接种的外源根瘤菌WGF03在根际土壤中,比不接种处理组的多样性要大,而突变菌则在根际、根围土壤中均没有表现出差异。推测野生型菌株WGF03的适应能力强,对周围土壤细菌的影响较大,分泌的大量胞外多糖或其他物质吸引着细菌往根际部分聚集。
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.soilbio.2014.01.019URL [本文引用: 1]
DOI:10.1111/nph.1993.124.issue-3URL [本文引用: 1]
URL [本文引用: 1]
在农田生态系统中,不同作物耐连作程度大不相同,有些农作物对连作不敏感,连续种植对产量和品质影响不大,如玉米和水稻;有些作物不耐连作,大豆是典型的对连作敏感的作物。大豆连作产量降低,品质变差,生长发育受阻,土传病虫害加重。而土传病害发生,主要是土壤中的病原菌多样性和丰度增加,生长发育受阻的另一个原因是氮代谢受阻,氮代谢受阻来源于土壤中的氮素转化,而影响氮素转化就是土壤中的一些功能菌。系统研究大豆连作和这些微生物关系,对解析连作大豆根部病害加重和氮素循环受阻机制和为解决连作障碍提供技术原理具有重大的理论意义和广泛的应用价值。本文利用黑龙江省农业科学院土壤肥料与环境资源研究所的一组大豆长期连作实验为平台,采用了高通量测序等方法,研究结果如下:1、在肥沃的黑土上,大豆连续种植17年,并没有导致土壤有机质、总氮、总碳、p H等指标的显著变化。对土壤酶活性的测定结果表明,连作4年和连作7年处理的土壤酶活性变化较大,尤其脲酶和蔗糖酶活性。其中连作4年处理的脲酶活性最低为0.88 NH2-N mg/(g 24h),比轮作处理降低28.5%,随后随着连作年限的增加而增加。而蔗糖酶活性在连作7年以前随着连作年限的增加而增加,到连作7年蔗糖酶活性与轮作处理相似。连作11年蔗糖酶活性最高,随后随着连作年限的增加而降低,连作17年处理的蔗糖酶活性为73.27 G mg/(g·24h)。由此可以看出,连作4年到7年是连作大豆土壤生物活性变化最大的时间段,之后又趋于平稳。土壤酶活性与土壤微生物的群落数量和活性密切相关,因此认为土壤酶活性的变化是大豆连作土壤被认为是抑制性土壤的一种表现。2、应用PCR-DGGE技术,对不同连作年限大豆根际细菌和真菌群落结构多样性分析。其中细菌群落结构组成和多样性在连作7年以后明显低于轮作和连作4年以前,由此可以看出,大豆连续种植导致土壤中细菌群落组成的变化,但是变化过程是缓慢的,直到连作7年才足以被检测出来。对连作大豆根际真菌群落结构多样性分析结果表明,大豆连作2年和连作4根际真菌群落组成丰富,多样性较高。大豆连续种植7年以上,根际真菌群落组成和多样性与轮作相似,由此也可以看出大豆连作土壤被称作抑制性土壤的机制所在。3、通过PCR-DGGE,q PCR,克隆测序等方法研究连作大豆根际土壤氨氧化细菌群落结构组成和多样性。结果表明,连作7年处理的amo A基因数量为4.71×10-5 amo A gene copies/g dry soil,低于其他处理。通过测序结果表明该研究中amo A基因属于7 Clusters,其中,连作7年处理土壤中氨氧化细菌种类最为丰富,6个簇的氨氧化细菌均有出现;其次是连作2年处理除Cluster 4以外均有出现;而连作17年处理土壤中氨氧化细菌种类最少4个簇。以往的研究表明氨氧化细菌群落结构与土壤p H、有机质含量以及空间地理分布等因素密切相关,而本研究基于相同的施肥量和环境条件下,氨氧化细菌群落结构组成与环境因子相关性分析认为大豆连作年限的长短是影响氨氧化细菌群落组成的主要因子。4、通过PCR-DGGE,q PCR,Illumina Miseq测序等方法研究连作大豆根际氨氧化古菌群落组成和数量,结果表明,连作2年和连作17年处理的根际氨氧化古菌数量低于其他处理;轮作和连作17年处理根际氨氧化古菌群落组成不同于其他处理。连作大豆根际氨氧化古菌种类来自泉古菌门(Crenarchaeota)、和奇古菌门两大类(Thaumarchaeota),仍有42%为未分类的古菌种类。5、土壤的潜在硝化速率是表征土壤氮转化能力的重要指标,与氨氧化微生物活性密切相关。本研究中连作大豆根际土壤的潜在硝化速率在0.24μg NO3-g-1soil h-1到0.58μg NO3-g-1 soil h-1之间。其中连作4年和连作7年处理的土壤潜在硝化速率显著低于其他处理。与轮作处理相比,连作4年和连作7年处理的土壤潜在硝化速率分别降低51.79%和31.58%。而连作2年处理硝化速率最高,比轮作处理高17.5%。土壤潜在硝化速率与氨氧化细菌和氨氧化古菌的群落结构组成和丰度相关性分析结果表明,氨氧化细菌在黑土上连作大豆根际的氨氧化过程中起主要作用。
URL [本文引用: 1]
在农田生态系统中,不同作物耐连作程度大不相同,有些农作物对连作不敏感,连续种植对产量和品质影响不大,如玉米和水稻;有些作物不耐连作,大豆是典型的对连作敏感的作物。大豆连作产量降低,品质变差,生长发育受阻,土传病虫害加重。而土传病害发生,主要是土壤中的病原菌多样性和丰度增加,生长发育受阻的另一个原因是氮代谢受阻,氮代谢受阻来源于土壤中的氮素转化,而影响氮素转化就是土壤中的一些功能菌。系统研究大豆连作和这些微生物关系,对解析连作大豆根部病害加重和氮素循环受阻机制和为解决连作障碍提供技术原理具有重大的理论意义和广泛的应用价值。本文利用黑龙江省农业科学院土壤肥料与环境资源研究所的一组大豆长期连作实验为平台,采用了高通量测序等方法,研究结果如下:1、在肥沃的黑土上,大豆连续种植17年,并没有导致土壤有机质、总氮、总碳、p H等指标的显著变化。对土壤酶活性的测定结果表明,连作4年和连作7年处理的土壤酶活性变化较大,尤其脲酶和蔗糖酶活性。其中连作4年处理的脲酶活性最低为0.88 NH2-N mg/(g 24h),比轮作处理降低28.5%,随后随着连作年限的增加而增加。而蔗糖酶活性在连作7年以前随着连作年限的增加而增加,到连作7年蔗糖酶活性与轮作处理相似。连作11年蔗糖酶活性最高,随后随着连作年限的增加而降低,连作17年处理的蔗糖酶活性为73.27 G mg/(g·24h)。由此可以看出,连作4年到7年是连作大豆土壤生物活性变化最大的时间段,之后又趋于平稳。土壤酶活性与土壤微生物的群落数量和活性密切相关,因此认为土壤酶活性的变化是大豆连作土壤被认为是抑制性土壤的一种表现。2、应用PCR-DGGE技术,对不同连作年限大豆根际细菌和真菌群落结构多样性分析。其中细菌群落结构组成和多样性在连作7年以后明显低于轮作和连作4年以前,由此可以看出,大豆连续种植导致土壤中细菌群落组成的变化,但是变化过程是缓慢的,直到连作7年才足以被检测出来。对连作大豆根际真菌群落结构多样性分析结果表明,大豆连作2年和连作4根际真菌群落组成丰富,多样性较高。大豆连续种植7年以上,根际真菌群落组成和多样性与轮作相似,由此也可以看出大豆连作土壤被称作抑制性土壤的机制所在。3、通过PCR-DGGE,q PCR,克隆测序等方法研究连作大豆根际土壤氨氧化细菌群落结构组成和多样性。结果表明,连作7年处理的amo A基因数量为4.71×10-5 amo A gene copies/g dry soil,低于其他处理。通过测序结果表明该研究中amo A基因属于7 Clusters,其中,连作7年处理土壤中氨氧化细菌种类最为丰富,6个簇的氨氧化细菌均有出现;其次是连作2年处理除Cluster 4以外均有出现;而连作17年处理土壤中氨氧化细菌种类最少4个簇。以往的研究表明氨氧化细菌群落结构与土壤p H、有机质含量以及空间地理分布等因素密切相关,而本研究基于相同的施肥量和环境条件下,氨氧化细菌群落结构组成与环境因子相关性分析认为大豆连作年限的长短是影响氨氧化细菌群落组成的主要因子。4、通过PCR-DGGE,q PCR,Illumina Miseq测序等方法研究连作大豆根际氨氧化古菌群落组成和数量,结果表明,连作2年和连作17年处理的根际氨氧化古菌数量低于其他处理;轮作和连作17年处理根际氨氧化古菌群落组成不同于其他处理。连作大豆根际氨氧化古菌种类来自泉古菌门(Crenarchaeota)、和奇古菌门两大类(Thaumarchaeota),仍有42%为未分类的古菌种类。5、土壤的潜在硝化速率是表征土壤氮转化能力的重要指标,与氨氧化微生物活性密切相关。本研究中连作大豆根际土壤的潜在硝化速率在0.24μg NO3-g-1soil h-1到0.58μg NO3-g-1 soil h-1之间。其中连作4年和连作7年处理的土壤潜在硝化速率显著低于其他处理。与轮作处理相比,连作4年和连作7年处理的土壤潜在硝化速率分别降低51.79%和31.58%。而连作2年处理硝化速率最高,比轮作处理高17.5%。土壤潜在硝化速率与氨氧化细菌和氨氧化古菌的群落结构组成和丰度相关性分析结果表明,氨氧化细菌在黑土上连作大豆根际的氨氧化过程中起主要作用。
URL [本文引用: 1]
在大豆长期连作定位试验地,利用T-RFLP和qPCR方法研究了连作1-13年土壤中细菌及氨氧化微生物群落结构和丰度的动态变化,同时通过构建微宇宙系统,研究染料木因和大豆苷元对细菌和氨氧化微生物的影响,并结合田间和盆栽13C02标记试验,研究大豆根系淀积物对土壤细菌群落的影响,结果如下: (1)不同连作年限土壤中微生物量碳变化不显著,但是微生物量氮发生了显著的变化,并且细菌仍是主要的微生物类群。根际土壤中细菌16S rRNA基因的基因拷贝数随着连作年限的增加,出现了先增加后降低再增加的现象。不同连作年限土壤中与碳、氮循环相关的细菌类群的相对丰度发生变化,且根系分泌物与细菌群落结构的变化具有相关性,此外还与全氮、微生物量碳等因子有关。 (2) DNA-SIP在田间和盆栽条件下都没有找到利用13C的根际微生物,而盆栽RNA-SIP的结果找到158bp、438bp所代表的细菌是根际中活跃的微生物。在研究植物-微生物相互作用方面,RNA-SIP比DNA-SIP可能更为有效。 (3)大豆连作对氨氧化细菌的数量没有产生显著影响,但是改变了其群落结构,NMDS分析显示连作1年与其余年限分开,群落结构与染料木因、土壤全氮含量相关;氨氧化古菌的群落结构和数量均随着连作年限发生了显著的变化,连作1-4年土壤中氨氧化古菌amoA基因的拷贝数显著低于连作5-13年,而染料木因和土壤有机质含量与氨氧化古菌群落结构的变化有相关性。 (4)与对照相比,只有在培养3天时,添加较高浓度的大豆苷元和染料木因的混合液改变了细菌的群落结构,但是在培养7天和14天后细菌的群落结构却没有发生变化。培养时间为3天和7天时,大豆苷元对土壤中细菌群落的丰度产生了抑制作用,而培养14天时抑制作用减弱。染料木因对细菌群落丰度的作用表现在较低浓度时产生抑制作用,中等浓度时抑制作用消失,而高浓度时,培养3天时对细菌群落有抑制作用,而在培养7天和14天时抑制作用消失。 (5)添加异黄酮对氨氧化细菌和氨氧化古菌的群落结构均没有产生影响。在培养的7天和14天后,较高浓度的大豆苷元和染料木因的混合液显著增加了氨氧化细菌的数量。添加较高浓度的大豆苷元和染料木因的混合液对氧化古菌数量也有促进作用,其他处理在3天、7天时对氨氧化古菌的数量没有影响,培养14天时氨氧化古菌的数量显著性增加。 通过上述试验结果得出以下结论:(1)大豆连作后,主要对参与碳、氮循环的细菌类群产生影响,改变了其相对丰度;(2)大豆连作会影响土壤中氨氧化细菌和氨氧化古菌的群落结构和数量;(3)异黄酮在培养时间内对氨氧化细菌和古菌的群落结构没有产生影响,但是改变了其数量;(4)异黄酮对土壤细菌16S rRNA基因的基因拷贝数有影响,较高浓度的大豆苷元和染料木因混合液会改变细菌的群落结构;(5)在研究植物-微生物相互作用方面,RNA-SIP比DNA-SIP可能更为有效。
URL [本文引用: 1]
在大豆长期连作定位试验地,利用T-RFLP和qPCR方法研究了连作1-13年土壤中细菌及氨氧化微生物群落结构和丰度的动态变化,同时通过构建微宇宙系统,研究染料木因和大豆苷元对细菌和氨氧化微生物的影响,并结合田间和盆栽13C02标记试验,研究大豆根系淀积物对土壤细菌群落的影响,结果如下: (1)不同连作年限土壤中微生物量碳变化不显著,但是微生物量氮发生了显著的变化,并且细菌仍是主要的微生物类群。根际土壤中细菌16S rRNA基因的基因拷贝数随着连作年限的增加,出现了先增加后降低再增加的现象。不同连作年限土壤中与碳、氮循环相关的细菌类群的相对丰度发生变化,且根系分泌物与细菌群落结构的变化具有相关性,此外还与全氮、微生物量碳等因子有关。 (2) DNA-SIP在田间和盆栽条件下都没有找到利用13C的根际微生物,而盆栽RNA-SIP的结果找到158bp、438bp所代表的细菌是根际中活跃的微生物。在研究植物-微生物相互作用方面,RNA-SIP比DNA-SIP可能更为有效。 (3)大豆连作对氨氧化细菌的数量没有产生显著影响,但是改变了其群落结构,NMDS分析显示连作1年与其余年限分开,群落结构与染料木因、土壤全氮含量相关;氨氧化古菌的群落结构和数量均随着连作年限发生了显著的变化,连作1-4年土壤中氨氧化古菌amoA基因的拷贝数显著低于连作5-13年,而染料木因和土壤有机质含量与氨氧化古菌群落结构的变化有相关性。 (4)与对照相比,只有在培养3天时,添加较高浓度的大豆苷元和染料木因的混合液改变了细菌的群落结构,但是在培养7天和14天后细菌的群落结构却没有发生变化。培养时间为3天和7天时,大豆苷元对土壤中细菌群落的丰度产生了抑制作用,而培养14天时抑制作用减弱。染料木因对细菌群落丰度的作用表现在较低浓度时产生抑制作用,中等浓度时抑制作用消失,而高浓度时,培养3天时对细菌群落有抑制作用,而在培养7天和14天时抑制作用消失。 (5)添加异黄酮对氨氧化细菌和氨氧化古菌的群落结构均没有产生影响。在培养的7天和14天后,较高浓度的大豆苷元和染料木因的混合液显著增加了氨氧化细菌的数量。添加较高浓度的大豆苷元和染料木因的混合液对氧化古菌数量也有促进作用,其他处理在3天、7天时对氨氧化古菌的数量没有影响,培养14天时氨氧化古菌的数量显著性增加。 通过上述试验结果得出以下结论:(1)大豆连作后,主要对参与碳、氮循环的细菌类群产生影响,改变了其相对丰度;(2)大豆连作会影响土壤中氨氧化细菌和氨氧化古菌的群落结构和数量;(3)异黄酮在培养时间内对氨氧化细菌和古菌的群落结构没有产生影响,但是改变了其数量;(4)异黄酮对土壤细菌16S rRNA基因的基因拷贝数有影响,较高浓度的大豆苷元和染料木因混合液会改变细菌的群落结构;(5)在研究植物-微生物相互作用方面,RNA-SIP比DNA-SIP可能更为有效。
URL [本文引用: 1]
根腐病作为大豆连作后主要发生的根部病害,是东北黑土区大豆连作 障碍的主要原因之一。然而近年发现经过长期连作,大豆根腐病发病情况得到明显的控制,因此提出了长期连作可能形成根腐病抑制性土壤的假设。为了验证这一假 设,应用中国科学院海伦农业生态试验站大豆连作定位试验区,以大豆长期连作—大豆根腐病病原微生物—病原抑制性微生物三者关系为研究对象,结合传统分离计 数、形态学鉴定、致病性检测、核酸序列分析、实时荧光定量PCR(Real-timePCR)和变性梯度凝胶电泳(DGGE)等方法,研究了大豆连作方式 下根腐病原菌种类,以及其在3年连作、20年连作和20年...
URL [本文引用: 1]
根腐病作为大豆连作后主要发生的根部病害,是东北黑土区大豆连作 障碍的主要原因之一。然而近年发现经过长期连作,大豆根腐病发病情况得到明显的控制,因此提出了长期连作可能形成根腐病抑制性土壤的假设。为了验证这一假 设,应用中国科学院海伦农业生态试验站大豆连作定位试验区,以大豆长期连作—大豆根腐病病原微生物—病原抑制性微生物三者关系为研究对象,结合传统分离计 数、形态学鉴定、致病性检测、核酸序列分析、实时荧光定量PCR(Real-timePCR)和变性梯度凝胶电泳(DGGE)等方法,研究了大豆连作方式 下根腐病原菌种类,以及其在3年连作、20年连作和20年...
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/nph.2009.184.issue-3URL [本文引用: 1]
DOI:10.1128/AEM.00481-10URL [本文引用: 1]
In: Manzanilla-Lopez R H, Marban-Mendoza N, eds.
[本文引用: 1]
URL [本文引用: 1]
为了加深理解大豆连作的生物学障碍机理,本文采用高通量测序和末端限制性片段多态性分析(T-RFLP)、聚丙烯酰胺梯度凝胶电泳(DGGE)和定量(qPCR)方法,研究了连作13年的大豆根际细菌、真菌和线虫,以及镰刀菌、大豆疫霉菌的群落结构和丰度的动态。同时,通过与环境因子进行相关性分析,阐明驱动根际生物类群动态变化的关键因子。另外,通过建立微宇宙系统,利用DGGE和qPCR的方法,研究了大豆异黄酮对镰刀菌和大豆疫霉菌的影响,探讨了大豆异黄酮对主要病原菌的生态功能。结果如下: (1)连作1-3年根际真菌群落的丰度显著高于连作6-13年,而真菌的香农指数和均匀度在6-13年显著高于1-3年。在连作2-3年内,疾病真菌(Boeremia和Clavariopsis)的相对丰度更高,而有益菌(Bionectria, Melanospora和Hypocrea)的相对丰度较低。另外,根的分泌物如类黄酮、染料木因和pH是调节真菌群落的重要因子。细菌丰度从1-3年迅速减少,之后保持相对稳定。细菌群落组分受可利用性磷(AP)、硝态氮(N03-)和pH的影响。这些结果说明从起初的健康土壤到致病土壤的转化可能主要是由于致病真菌的相对丰度较高,而且真菌群落与细菌比例增加有关。之后,从致病土壤到抑病土壤的转化和土壤抑制特性的可持续性可能与土壤中有益菌的比例提高有关。根系分泌物和土壤性质是驱动微生物群落变化的因子。 (2)在1到13年,镰刀菌的群落结构和丰度都没有清楚的时间趋势。土壤的异黄酮物质和镰刀菌群落的变化具有明显相关性。另外,大豆疫霉菌的丰度从第1到第3年增加,之后开始减少。大豆疫霉菌与类黄酮的浓度呈负相关。物种少的镰刀菌群落是物种丰富的镰刀菌群落的子集,说明在种库中连续的演替是巢氏和物种的丧失。镰刀菌群落可能由随机和环境过程影响,镰刀菌群落可能不是导致大豆产量降低的主要因子,根际中大豆疫霉菌的密度或许更重要。 (3)线虫群落结构在连作期间发生了明显变化。线虫的物种丰富度随着连作年限的增加呈逐渐降低的趋势。第1年物种丰富度最高,第3年的丰富度显著低于第1年,之后逐渐降低。大豆根际土中共检测到16个线虫群落T-RF(末端限制性片段),且大多数T-RF能从克隆文库中鉴定。在大豆根际土中,食细菌线虫(Acrobeloides)是最丰富的有机体。在连作2-3年,植物寄生线虫相对丰度增加,而在连作后期,植物寄生线虫相对丰度减少。NMDS分析显示,第1年线虫群落与其余年限分开,而第2和第3年聚集较近,后面9、11和13年聚集较近。另外,线虫群落结构与pH、土壤有机质(SOM)、速效磷(AP)、细菌数量和真菌数量相关。线虫丰度呈先增后降的趋势,最高值出现在第6年。线虫的基因拷贝数与土壤NH4+和染料木因(genisten)浓度呈显著正相关,而与N03-和细菌的基因拷贝数呈显著负相关。说明多个因素驱动着连作过程中线虫群落变化,不仅受到土壤性质的影响,而且受到微生物群落影响。线虫群落的变化,特别是植食性线虫的变化,可能是导致连作障碍的因素之一。 (4)外源添加黄酮物质改变了镰刀菌群落结构。培养前期(第3天),黄酮溶液对镰刀菌丰度影响最大,随着培养时间的延长,黄酮溶液对镰刀菌丰度的影响较小。可是,在培养第3天,黄酮溶液没有改变大豆疫霉菌的丰度,当培养7天后,高浓度的大豆苷元和染料木因以及混合液对大豆疫霉菌产生了显著的抑制作用。培养14天,只有最高浓度的混合液促进了大豆疫霉菌的数量。大豆苷元和染料木因对镰刀菌和大豆疫霉菌能产生促进或抑制作用,主要取决于培养时间和黄酮物质的浓度。 通过上述研究得出以下结论(1)长期大豆连作使土壤细菌、真菌、线虫和镰刀菌群落的物种丰富度降低;(2)连作使土壤生物群落的基因拷贝数减少。
URL [本文引用: 1]
为了加深理解大豆连作的生物学障碍机理,本文采用高通量测序和末端限制性片段多态性分析(T-RFLP)、聚丙烯酰胺梯度凝胶电泳(DGGE)和定量(qPCR)方法,研究了连作13年的大豆根际细菌、真菌和线虫,以及镰刀菌、大豆疫霉菌的群落结构和丰度的动态。同时,通过与环境因子进行相关性分析,阐明驱动根际生物类群动态变化的关键因子。另外,通过建立微宇宙系统,利用DGGE和qPCR的方法,研究了大豆异黄酮对镰刀菌和大豆疫霉菌的影响,探讨了大豆异黄酮对主要病原菌的生态功能。结果如下: (1)连作1-3年根际真菌群落的丰度显著高于连作6-13年,而真菌的香农指数和均匀度在6-13年显著高于1-3年。在连作2-3年内,疾病真菌(Boeremia和Clavariopsis)的相对丰度更高,而有益菌(Bionectria, Melanospora和Hypocrea)的相对丰度较低。另外,根的分泌物如类黄酮、染料木因和pH是调节真菌群落的重要因子。细菌丰度从1-3年迅速减少,之后保持相对稳定。细菌群落组分受可利用性磷(AP)、硝态氮(N03-)和pH的影响。这些结果说明从起初的健康土壤到致病土壤的转化可能主要是由于致病真菌的相对丰度较高,而且真菌群落与细菌比例增加有关。之后,从致病土壤到抑病土壤的转化和土壤抑制特性的可持续性可能与土壤中有益菌的比例提高有关。根系分泌物和土壤性质是驱动微生物群落变化的因子。 (2)在1到13年,镰刀菌的群落结构和丰度都没有清楚的时间趋势。土壤的异黄酮物质和镰刀菌群落的变化具有明显相关性。另外,大豆疫霉菌的丰度从第1到第3年增加,之后开始减少。大豆疫霉菌与类黄酮的浓度呈负相关。物种少的镰刀菌群落是物种丰富的镰刀菌群落的子集,说明在种库中连续的演替是巢氏和物种的丧失。镰刀菌群落可能由随机和环境过程影响,镰刀菌群落可能不是导致大豆产量降低的主要因子,根际中大豆疫霉菌的密度或许更重要。 (3)线虫群落结构在连作期间发生了明显变化。线虫的物种丰富度随着连作年限的增加呈逐渐降低的趋势。第1年物种丰富度最高,第3年的丰富度显著低于第1年,之后逐渐降低。大豆根际土中共检测到16个线虫群落T-RF(末端限制性片段),且大多数T-RF能从克隆文库中鉴定。在大豆根际土中,食细菌线虫(Acrobeloides)是最丰富的有机体。在连作2-3年,植物寄生线虫相对丰度增加,而在连作后期,植物寄生线虫相对丰度减少。NMDS分析显示,第1年线虫群落与其余年限分开,而第2和第3年聚集较近,后面9、11和13年聚集较近。另外,线虫群落结构与pH、土壤有机质(SOM)、速效磷(AP)、细菌数量和真菌数量相关。线虫丰度呈先增后降的趋势,最高值出现在第6年。线虫的基因拷贝数与土壤NH4+和染料木因(genisten)浓度呈显著正相关,而与N03-和细菌的基因拷贝数呈显著负相关。说明多个因素驱动着连作过程中线虫群落变化,不仅受到土壤性质的影响,而且受到微生物群落影响。线虫群落的变化,特别是植食性线虫的变化,可能是导致连作障碍的因素之一。 (4)外源添加黄酮物质改变了镰刀菌群落结构。培养前期(第3天),黄酮溶液对镰刀菌丰度影响最大,随着培养时间的延长,黄酮溶液对镰刀菌丰度的影响较小。可是,在培养第3天,黄酮溶液没有改变大豆疫霉菌的丰度,当培养7天后,高浓度的大豆苷元和染料木因以及混合液对大豆疫霉菌产生了显著的抑制作用。培养14天,只有最高浓度的混合液促进了大豆疫霉菌的数量。大豆苷元和染料木因对镰刀菌和大豆疫霉菌能产生促进或抑制作用,主要取决于培养时间和黄酮物质的浓度。 通过上述研究得出以下结论(1)长期大豆连作使土壤细菌、真菌、线虫和镰刀菌群落的物种丰富度降低;(2)连作使土壤生物群落的基因拷贝数减少。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1128/MMBR.00039-10URLPMID:3122624 [本文引用: 1]
The majority of life on Earth--notably, microbial life--occurs in places that do not receive sunlight, with the habitats of the oceans being the largest of these reservoirs. Sunlight penetrates only a few tens to hundreds of meters into the ocean, resulting in large-scale microbial ecosystems that function in the dark. Our knowledge of microbial processes in the dark ocean-the aphotic pelagic ocean, sediments, oceanic crust, hydrothermal vents, etc.-has increased substantially in recent decades. Studies that try to decipher the activity of microorganisms in the dark ocean, where we cannot easily observe them, are yielding paradigm-shifting discoveries that are fundamentally changing our understanding of the role of the dark ocean in the global Earth system and its biogeochemical cycles. New generations of researchers and experimental tools have emerged, in the last decade in particular, owing to dedicated research programs to explore the dark ocean biosphere. This review focuses on our current understanding of microbiology in the dark ocean, outlining salient features of various habitats and discussing known and still unexplored types of microbial metabolism and their consequences in global biogeochemical cycling. We also focus on patterns of microbial diversity in the dark ocean and on processes and communities that are characteristic of the different habitats.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}