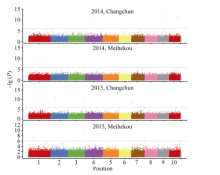
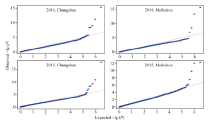
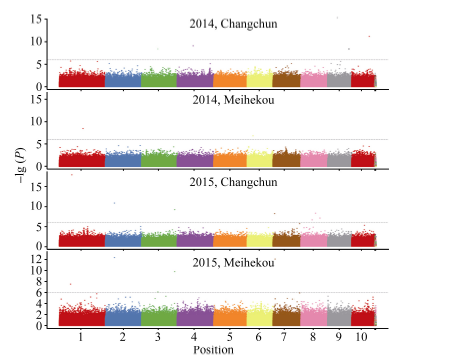
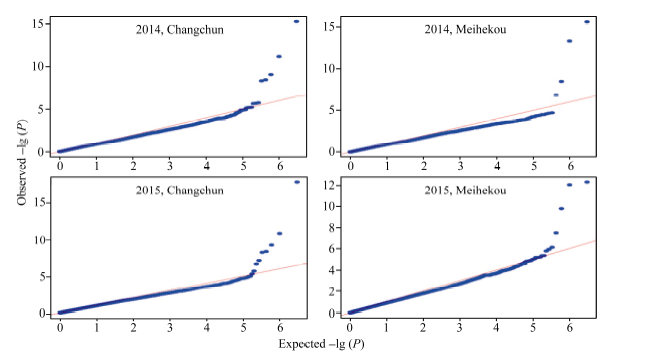
关键词:玉米; 行粒数; 单核苷酸多态性; 关联分析 Genome-wide Association Analysis of Kernel Number per Row in Maize WU Lyu, DAI Li-Qiang, DONG Qing-Song, SHI Ting-Ting, WANG Pi-Wu* Jilin Agricultural University, Changchun 130000, China Fund:This study was supported by grants from the Special Fund for Modern Crop Seed Industry Development of Jilin Province and the 948 Project of the Ministry of Agriculture (2013-Z47). AbstractKernel number per row in maize is a significant trait in determining yield components and it has great significance to study its genetic mechanism. This report studied 80 Jilin maize inbred lines in field experiments at Jilin Changchun and Jilin Meihekou, and measured kernel number per row in 2014 and 2015. At the same time, whole-genome resequencing was performed for the association population using second generation sequencing technology, and the obtained single nucleotide polymorphisms (SNPs) markers were used for subsequent analysis. The results revealed that the range of phenotypic traits of kernel number per row was from 12.0 to 41.6 and the broad-sensed heritability was 70.5% in four environments. A total of 19 SNP markers significantly associated with kernel number per row were detected by a genome-wide association study. Of these, two markers located at bins 2.04 and 3.08 of chromosome frame were detected in the experiments at Changchun and Meihekou in 2015, respectively, and 14 SNP markers located within the quantitative trait loci had been previously mapped. Four candidate genes, such as the genes encoding the receptor for ubiquitination targets protein, metal dependent phosphohydrolase, heavy metal transport/detoxification protein and putative protein with no characteristic function, were identified from the range of linkage disequilibrium of the significant SNP makers and predicted that they were closely associated to the development of the kernel number per row.
Keyword:Maize; Kernel number per row; Single nucleotide polymorphism; Association analysis Show Figures Show Figures
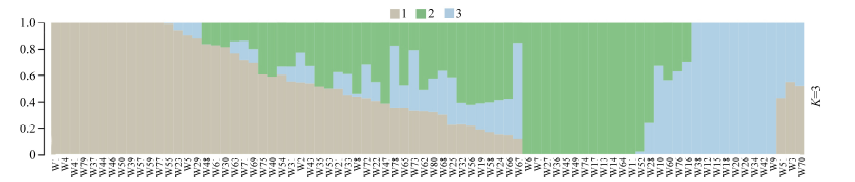
图1 群体结构图不同颜色片段的长度表示该个体基因组中某个祖先所占的比例。Fig. 1 Group structure plot The length of the different color segments represents the proportion of an ancestor in the individual genome.
张怀胜, 陈士林, 王铁固. 玉米行粒数主基因+多基因混合遗传模型分析. 河南农业科学, 2013, 42(2): 30-33Zhang HS, Chen SL, Wang TG. Genetic analysis on kernel number per row by mixed inheritance model of major gene and polygene in maize. J Henan Agric Sci, 2013, 42(2): 30-33 (in Chinese with English abstract)[本文引用:2]
兰进好, 李新海, 高树仁, 张宝石, 张世煌. 不同生态环境下玉米产量性状QTL分析. 作物学报, 2005, 31: 1253-1259Lan JH, Li XH, Gao SR, Zhang BS, Zhang SH. QTL analysis of yield components in maize under different environments. Acta Agron Sin, 2005, 31: 1253-1259 (in Chinese with English abstract)[本文引用:2]
[4]
LiM, Guo XH, ZhangM, Wang XP, Zhang GD, Tian YC, Wang ZL. Mapping QTLs for grain yield and yield components under high and low phosphorus treatments in maize ( Zea mays L. ). Plant Sci, 2010, 178: 454-462[本文引用:1]
[5]
HuoD, NingQ, ShenX, LiuL, ZhangZ. QTL mapping of kernel number-related traits and validation of one major QTL for ear length in maize. PLoS One, 2016, 11: e0155506[本文引用:3]
[6]
ChenJ, ZhangL, LiuS, LiZ, HuangR, LiY, ChengH, LiX, ZhouB, WuS, ChenW, WuJ, DingJ. The genetic basis of natural variation in kernel size and related traits using a four-way cross population in maize. PLoS One, 2016, 11: e0153428[本文引用:1]
[7]
Knapp SJ, Stroup WW, Ross WM. Exact confidence intervals for heritability on a progeny mean basis. Crop Sci, 1985, 25: 192-194[本文引用:1]
[8]
LiH, DurbinR. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics, 2009, 25: 1754-1760[本文引用:1]
[9]
LiH, Hand sakerB, WysokerA, FennellT, RuanJ, HomerN, MarthG, AbecasisG, DurbinR. The sequence alignment/map format and SAMtools. Bioinformatics, 2009, 25: 2078-2079[本文引用:1]
[10]
PurcellS, NealeB, Todd-BrownK, ThomasL, Ferreira MA, BenderD, MallerJ, Sklar P, de Bakker P I, Daly M J, Sham P C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet, 2007, 81: 559-575[本文引用:1]
[11]
Lipka AE, TianF, WangQ, PeifferJ, LiM, Bradbury PJ, Gore MA, Buckler ES, ZhangZ. GAPIT: genome association and prediction integrated tool. Bioinformatics, 2012, 28: 2397-2399[本文引用:1]
[12]
LiuX, HuangM, FanB, Buckler ES, ZhangZ. Iterative usage of fixed and rand om effect models for powerful and efficient genome-wide association studies. PLoS Genet, 2016, 12: e1005767[本文引用:1]
[13]
TuberosaR, SalviS, Sanguineti MC, Land iP, MaccaferriM, ContiS. Mapping QTL regulating morpho-physiological traits and yield: case studies, shortcomings and perspectives in drought-stressed maize. Ann Bot, 2002, 89: 941-963[本文引用:1]
[14]
Tenaillon MI, Sawkins MC, Long AD, Gaut RL, Doebley JF, Gaut BS. Patterns of DNA sequence polymorphism along chromosome 1 of maize ( Zea mays ssp. mays L. ). Proc Natl Acad Sci USA, 2001, 98: 9161-9166[本文引用:1]
[15]
Lu GH, Tang JH, Yan JB, Ma XQ, Li JS, Chen SJ, Ma JC, Liu ZX, ZhuL, Zhang YR, Dai JR. Quantitative trait loci mapping of maize yield and its components under different water treatments at flowering time. J Integr Plant Biol, 2006, 48: 1233-1243[本文引用:1]
[16]
刘宗华, 汤继华, 卫晓轶, 王春丽, 田国伟, 胡彦民, 陈伟程. 氮胁迫和正常条件下玉米穗部性状的QTL分析. 中国农业科学, 2007, 40: 2409-2417Liu ZH, Tang JH, Wei XY, Wang CL, Tian GW, Hu YM, Chen WC. QTL mapping of ear traits under low and high nitrogen conditions in maize. Sci Agric Sin, 2007, 40: 2409-2417 (in Chinese with English abstract)[本文引用:1]
[17]
代国丽, 蔡一林, 徐德林, 吕学高, 王国强, 王久光, 孙海艳. 玉米穗部性状的QTL定位. 西南师范大学学报(自然科学版), 2009, 34(5): 133-138Dai GL, Cai YL, Xu DL, Lyu XG, Wang GQ, Wang JG, Sun HY. QTL mapping for ear traits in maize( Zea mays L. ). J Southwest China Norm Univ, 34(5): 133-138 (in Chinese with English abstract)[本文引用:1]
杨国虎. 玉米两个相关RILs群体遗传图谱构建及主要性状QTL分析. 河南农业大学博士学位论文, 河南郑州, 2011Yang GH. Construction of Genetic Map and QTL Analysis for Main Traits Using Two Connected RIL Populations in Maize. PhD Dissertation of Henan Agricultural University, Zhengzhou, China, 2011 (in Chinese with English abstract)[本文引用:1]
[20]
Веденеев ГИ (王富德译). 玉米数量性状的遗传控制: III. 穗行数和行粒数. 国外农学——杂粮作物, 1988, (3): 10-15Веденеев ГИ (Wang F DTrans). Genetic control of maize quantitative traits: III. Row number per ear and kernel number per row. Foreign Agron: Minor Cereals, 1988, (3): 10-15 (in Chinese)[本文引用:1]
[21]
王秀燕, 孙莉萍, 张建锋, 李辉, 吕文清, 张其清. F-box蛋白家族及其功能. 生命科学, 2008, 20: 807-811Wang XY, Sun LP, Zhang JF, LiH, Lyu WQ, Zhang QQ. F-box proteins and their functions. Chin Bull Life Sci, 2008, 20: 807-811 (in Chinese with English abstract)[本文引用:1]
[22]
AravindL, Koonin EV. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem Sci, 1998, 23: 469-472[本文引用:1]
[23]
Yakunin AF, ProudfootM, KuznetsovaE, SavchenkoA, BrownG, Arrowsmith CH, Edwards AM. The HD domain of the Escherichia coli tRNA nucleotidyltransferase has 2’, 3’-cyclic phosphodiesterase, 2’-nucleotidase, and phosphatase activities. J Biol Chem, 2004, 279: 36819-36827[本文引用:1]
[24]
Palmgren MG, Axelsen KB. Evolution of P-type ATPases. Biochim Biophys Acta, 1998, 1365: 37-45[本文引用:1]
[25]
金枫, 王翠, 林海建, 沈亚欧, 张志明, 赵茂俊, 潘光堂. 植物重金属转运蛋白研究进展. 应用生态学报, 2010, 21: 1875-1882JinF, WangC, Lin HJ, Shen YO, Zhang ZM, Zhao MJ, Pan GT. Heavy metal-transportproteins in plants: a review. Chin J Appl Ecol, 2010, 21: 1875-1882 (in Chinese with English abstract)[本文引用:1]
[26]
Seigneurin-BernyD, GravotA, AuroyP, MazardC, KrautA, FinazziG, GrunwaldD, RappaportF, VavasseurA, JoyardJ, RichaudP, Rolland N. HMA1, a new Cu-ATPase of the chloroplast envelope, is essential for growth under adverse light conditions. J Biol Chem, 2006, 281: 2882-2892[本文引用:1]
 , 代力强, 董青松, 施婷婷, 王丕武
, 代力强, 董青松, 施婷婷, 王丕武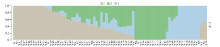

{kind=link}
{kind=link}
{kind=link}