全文HTML
--> --> -->常温常压下, HfC和HfN均为B1型岩盐结构, 铪原子形成面心立方结构, 碳、氮原子处于[Hf6]八面体间隙. HfC和HfN可形成三元无限固溶体, 且实验和理论上发现三元Hf-C-N体系具有更好的力学性质[14-19]. 例如, Holleck[14]研究了HfC1–xNx的微观硬度与组分之间的关系, 发现HfN的含量x = N/(C + N) = 0.4时, 微观硬度最大. Yang等[15]研究得到x = 0.2—0.6时, HfC1–xNx的剪切模量、弹性模量、体模量最大, 而纳米硬度和显微硬度随着HfN含量的增大而减小. 理论上, Jhi等[16]采用第一性原理方法, 研究得到x = 0.4时, HfC1–xNx的剪切模量最大. Feng等[17]和Balasubramanian等[18]采用第一性原理方法, 计算了HfC1–xNx的力学性能和电子性质随组分变化的关系, 发现x = 0.25时, HfCxN1–x的弹性模量和硬度最大. Peng和Tikhonov[19]采用第一性原理和进化算法, 研究了HfC1–xNx的结构、力学性质等随组分的变化, 发现x = 0.25—0.3时, HfC1–xNx的剪切模量、弹性模量、维氏硬度等最大, 断裂韧性也得到了改善. 因此, 相比于二元化合物, 三元Hf-C-N体系的模量、硬度和韧性等都提高了; 合金化是改善力学性能的一种有效方法.
实际上, 二元及以上过渡金属碳/氮和碳氮化合物都存在一定浓度的空位, 被称为强非化学计量比化合物. Gusev等[20]发现在一定条件下, 空位会重新分布, 晶体结构发生改变, 即形成了空位有序结构. 强非化学计量比化合物的性质将受到空位浓度及其分布(有序无序性)等影响[11,14,20-23]. Rudy[22]研究发现Hf-C体系中, HfC0.94的熔点最高. Holleck[14]研究发现随着空位浓度的减小, TaNx, TaCx, NbNx和NbCx的微观硬度先增加后减小; 而TiCx, ZrCx, HfCx, TiNx和ZrNx的微观硬度随着空位浓度的减小而增大. Jhi等[11]采用第一性原理方法, 分别研究了Ti-N和Nb-C体系的剪切性质和电子性质, 发现空位对Ti-N和Nb-C体系的力学性质产生了完全不同的影响, 即空位硬化和软化, 且与Holleck[14]的实验结果一致. Gusev等[20]在著作中总结了二元强非化学计量比化合物的有序结构, 并分析了空位有序性对性质的影响.
近年来研究人员采用第一性原理结合晶体结构预测等方法, 对二元过渡金属碳/氮化合物的有序结构进行了大量研究[24-38]. 基于第一性原理和进化算法, Yu等[24-26]和Xie等[27,28]分别采用晶体结构预测软件USPEX[39-41], 对过渡金属碳化物(TM = Ti, Zr, Hf, V, Nb, Ta等)的结构进行了预测, 发现了一系列含阴离子空位的结构, 包括TM2C, TM3C2, TM4C3, TM5C4, TM6C5, TM7C6和TM8C7等, 并研究了空位对结构和力学性质的影响. Zhang等[29]和Gunda等[30]分别采用CE方法[31]预测了ZrCx和TiCx的结构, Weinberger和Thompson[32]基于有序参数函数方法[33]预测了第IVB, VB族过渡金属碳化物的稳定结构. 基于第一性原理和进化算法, Yu等[34,35]和Fan等[36]分别预测了TiNx, ZrNx和HfNx的结构, 除了含阴离子空位的化合物外, 还发现含过渡金属空位的结构如Zr15N16, Zr7N8, Zr4N5, Hf4N5和Hf5N6等, 并研究空位对力学性质等的影响. 采用USPEX[39-41], Zhao等[37]和Li等[38]分别预测了Nb-N和Ta-N体系的稳定结构.
然而, 目前关于三元过渡金属碳/氮化合物空位有序结构及性质的报道还很少. Rudy[42]采用X射线粉末图谱法研究了Ta2VC2的结构, 空间群为




综上所述, 三元过渡金属碳氮化合物的性质除了受到化学组成的影响外, 还将受到空位的浓度及其分布等的影响. 为了研究三元Hf-C-N体系, 建立组分-结构-性质的关系, 必须考虑空位对结构及性质的影响. 然而, 目前实验和理论上, 很少关于三元Hf-C-N空位有序结构、力学性质及其关系的研究报道. 本文基于第一性原理和进化算法, 采用晶体结构预测软件USPEX[39-41]—已成功应用于二元过渡金属碳/氮化合物及许多新结构的预测[24-28,34-41], 搜索了三元Hf-C-N的空位有序结构, 研究了空位、化学组成等对力学性质的影响. 为该材料的实验合成、制备和应用等提供了理论指导和依据, 也为其他三元过渡金属碳氮化合物的研究提供了借鉴.
采用USPEX[39-41]分别搜索了Hf6C4N, Hf6C3N, Hf6C3N2, Hf4C2N, Hf3CN, Hf5C2N2, Hf4CN2, Hf6C2N3, HfCN3和Hf6CN4等组分的结构, 单胞中原子数分别为22, 20, 22, 14, 10/20, 22, 14, 22, 20和22. 初始结构(共60个)由USPEX[39-41]随机产生, 从第2代开始, 每代结构(共50个)分别由遗传(40%)、软模变异(20%)、晶格变异(10%)、原子位置交换(10%)、随机(20%)等进化操作产生. 对USPEX[39-41]产生的每个结构, 采用VASP软件[51]计算其能量, 筛选出能量最低的结构. 当连续20代能量最低的结构相同, 或搜索了30代结构时, 计算停止.
然后, 采用VASP软件[51]对Hf-C-N各组分能量最低结构进一步进行结构优化和性质计算. 结构优化时, 离子实与价电子之间的相互作用, 采用投影缀加波方法[52]进行描述, 截断能为600 eV, 电子与电子间交换关联能采用GGA-PBE[53]方法进行处理. 布里渊区高对称点间距为2π × 0.018 ?–1. 能量收敛判据为: 能量差为10–8 eV/atom, 压力差为10–3 eV/?. 基于“凸包结构”判断得到热力学稳定结构, 即某一结构分解为任意其他结构时, 分解能为正, 则该结构为热力学稳定结构. 然后, 采用VASP软件[51]计算弹性常数, 根据经验公式[54-58]计算得到体模量、剪切模量、弹性模量、泊松比、维氏硬度、Pugh比等. 最后, 基于密度泛函微扰理论, 采用Phonopy软件[59]计算Hf-C-N空位有序结构的声子谱曲线, 判断晶格动力学稳定性. 采用VESTA软件[60]画出其晶体结构和模拟X射线衍射图谱. 采用LOBSTER软件[61]计算Hf-C-N空位有序结构的晶体轨道哈密顿分布(–COHP).
3.1.晶体结构预测及Hf-C-N空位有序结构
由于Hf-C-N体系的性质受到空位浓度的影响, 首先采用空位调控的方法, 对三元Hf-C-N体系的组分进行设计, 采用晶体结构预测软件USPEX[39-41]结合VASP[51], 搜索了三元空位有序结构. 图1(a)为三元Hf-HfC-HfN体系的能量凸包图, 反应焓ΔH (eV/atom)的计算公式如下: 图 1 (a) 常压下, 三元Hf-HfC-HfN体系的能量凸包图, 黑色球表示热力学稳定结构, 其他为亚稳结构; (b) Hf-C-N空位有序结构的X射线衍射模拟图谱, 衍射源为Cu Kα射线
图 1 (a) 常压下, 三元Hf-HfC-HfN体系的能量凸包图, 黑色球表示热力学稳定结构, 其他为亚稳结构; (b) Hf-C-N空位有序结构的X射线衍射模拟图谱, 衍射源为Cu Kα射线Figure1. (a) Enthalpy convex-hull of ternary Hf-HfC-HfN system at ambient pressure. The black sphere indicates stable structure, and others are metastable structure. (b) The simulated X-ray diffractions of Hf-C-N vacancy ordered structures with a copper Kα X-ray source.
| Compound | Space group | Lattice constants/? | ΔH/(eV·atom–1) | CN | CV |
| Hf6C4N | $C2 $/m | a = 5.679, b = 9.799, c = 5.671, β = 70.6o | –0.0899 | 5 | 1/6 |
| Hf6C3N | $C2 $ | a = 5.658, b = 9.763, c = 9.262, β = 144.8o | –0.0980 | 4 | 1/3 |
| Hf6C3N2 | $C2 $m | a = 5.660, b = 9.783, c = 5.619, β = 109.6o | –0.1038 | 5 | 1/6 |
| Hf3CN | $C2 $ | a = 5.632, b = 9.705, c = 5.625, β = 109.8o | –0.1107 | 4 | 1/3 |
| Hf6C2N3 | $C2 $ | a = 5.624, b = 9.725, c = 5.602, β = 109.6o | –0.1047 | 5 | 1/6 |
| Hf4CN2 | Cmmm | a = 6.427, b = 9.147, c = 3.235 | –0.1082 | 4/5 | 1/4 |
| Hf6CN3 | $C2 $/m | a = 5.592, b = 9.658, c = 6.455, β = 125.3o | –0.0894 | 4 | 1/3 |
| Hf6CN4 | $C2 $/m | a = 5.580, b = 9.681, c = 5.587, β = 70.3o | –0.0815 | 5 | 1/6 |
表1Hf-C-N空位有序结构的空间群、晶格常数、反应焓ΔH (eV/atom)、Hf原子的配位数(CN) 和空位浓度(CV)
Table1.Space group, lattice constants, the enthalpy of reaction ΔH (eV/atom), coordination number (CN) of Hf and the concentration of vacancy (CV) of Hf-C-N vacancy ordered structures.
图1(b)为Hf-C-N空位有序结构的X射线衍射模拟图谱, 三元化合物的衍射峰形状与HfC, HfN的相同, 且衍射角位于HfC, HfN的衍射角之间; 即结构与HfC和HfN的结构相同, 都具有岩盐结构. 此结果与Binder等[47]和Buinevich等[49]发现的Hf-C-N无序固溶体的结构类型一致. 本文的理论预测结果证明了Hf-C-N空位化合物能够以有序结构的形式存在.
图2为Hf-C-N空位有序结构某一晶面上的空位分布, 黑色方框表示空位, 取代的是部分C和N原子的位置; 即对于Hf-C-N化合物, 原子数比Hf/(C + N)小于化学计量比1∶1时, 空位占据剩余C和N原子的晶格位置. 则C和N原子的配位数均为6, 而Hf的配位数小于6, Hf原子的配位数如表1所列. 与二元HfCx等[24-28]的结构相似, 过渡金属原子形成[Hf6]八面体, 八面体共棱连接, C, N原子和空位都处于八面体间隙位置. 图3为Hf-C-N空位有序结构的声子谱曲线, 在布里渊区(高对称点路径采用SeeK-path软件[62]得到), 都不存在虚频, 证明这些结构都是晶格动力学稳定的.
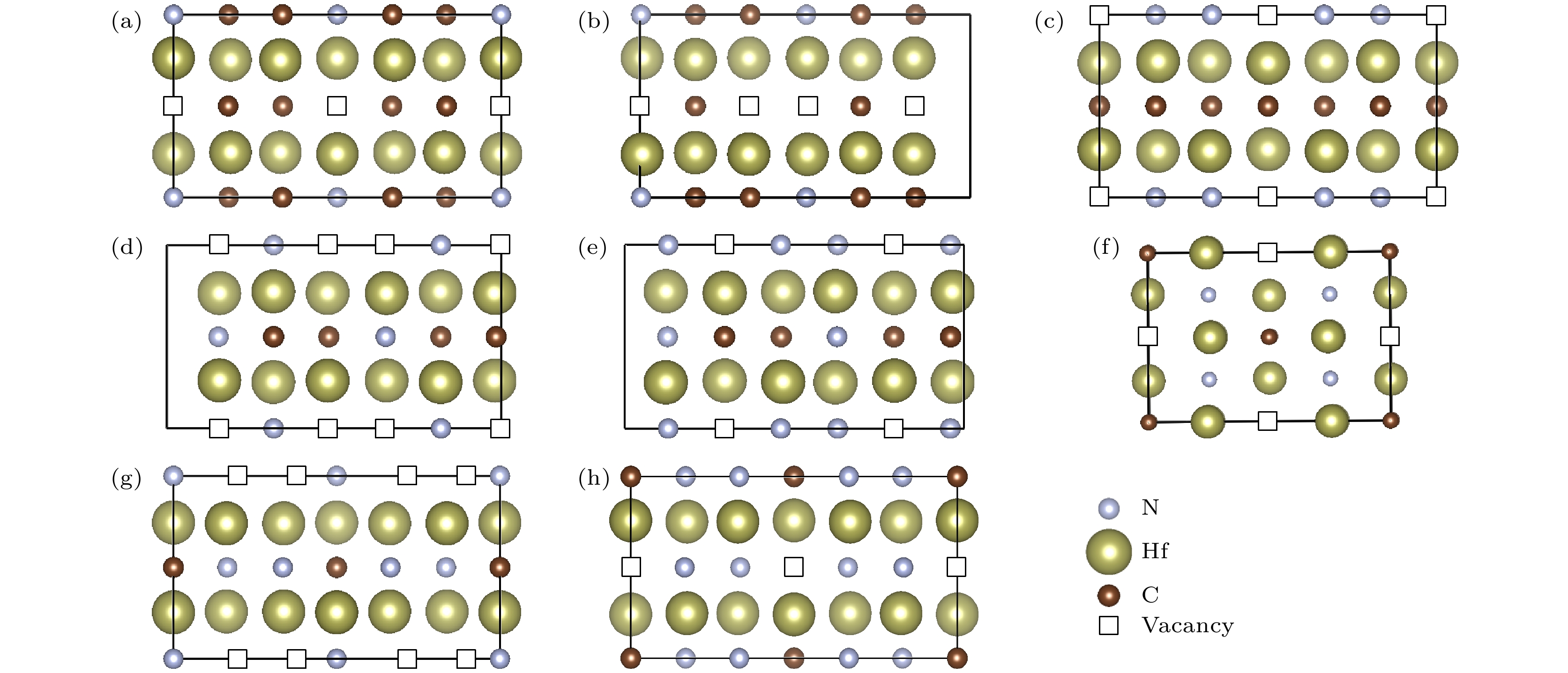 图 2 Hf-C-N空位有序结构在某一晶面上的空位分布 (a) Hf6C4N-
图 2 Hf-C-N空位有序结构在某一晶面上的空位分布 (a) Hf6C4N-






Figure2. Vacancies on the crystallographic plane: (a) Hf6C4N-







 图 3 Hf-C-N空位有序结构的声子谱曲线 (a) Hf6C4N-
图 3 Hf-C-N空位有序结构的声子谱曲线 (a) Hf6C4N-






Figure3. Phonon dispersion curves of (a) Hf6C4N-







2
3.2.Hf-C-N空位有序结构的力学性质
首先采用VASP软件[51]计算了Hf-C-N空位有序结构的弹性常数, 如表2所列, 这些结构的弹性常数都满足Born判据[63], 即都是力学稳定的. 再根据计算的弹性常数, 基于Voigt-Reuss-Hill近似[54-56], 得到体模量B、剪切模量G、弹性模量E、泊松比等. 采用Chen-Niu模型[57], 计算Hf-C-N空位有序结构的维氏硬度HV, 计算公式如下:| Compounds | C11 | C22 | C33 | C44 | C55 | C66 | C12 | C13 | C23 |
| Hf6C4N-$C2 $/m | 414.3 | 406.6 | 415.6 | 158.0 | 170.6 | 148.7 | 94.1 | 116.1 | 104.5 |
| Hf6C3N-$C2 $ | 358.5 | 362.8 | 352.2 | 100.0 | 114.3 | 132.3 | 87.5 | 98.3 | 91.6 |
| Hf6C3N2-$C2 $/m | 414.6 | 417.4 | 407.6 | 152.2 | 157.6 | 147.8 | 111.9 | 115.0 | 116.3 |
| Hf3CN-$C2 $ | 354.5 | 363.5 | 348.7 | 90.6 | 103.6 | 128.7 | 102.1 | 109.5 | 101.3 |
| Hf6C2N3-$C2 $ | 409.7 | 418.7 | 418.1 | 149.6 | 160.2 | 148.9 | 123.4 | 122.9 | 126.5 |
| Hf4CN2-Cmmm | 373.4 | 368.8 | 406.8 | 142.2 | 133.1 | 135.8 | 146.4 | 112.0 | 124.4 |
| Hf6CN3-$C2 $/m | 361.1 | 358.4 | 351.7 | 84.9 | 99.8 | 124.9 | 108.1 | 121.7 | 114.2 |
| Hf6CN4-$C2 $/m | 401.2 | 414.1 | 403.5 | 146.5 | 157.2 | 139.8 | 134.0 | 139.9 | 147.8 |
表2Hf-C-N空位有序结构的弹性常数Cij (单位: GPa)
Table2.Calculated elastic constants Cij (in GPa) of Hf-C-N vacancy ordered structures.
Hf-C-N空位有序结构的力学性质如表3所列, 可以看到这些结构都具有非常高的体模量、剪切模量、弹性模量和维氏硬度等. 为了对比, HfC1–xNx的力学性质也在表3中列出, 尽管这些结构的力学性质已经在文献[19]中被报道了.
| Compound | B /GPa | G /GPa | E /GPa | μ | G/B | HV /GPa |
| Hf6C4N | 229.0 | 140.8 | 350.6 | 0.2449 | 0.6148 | 17.5 |
| Hf5C4N[19] | 260.6 | 201.3 | 480.3 | 0.1928 | 0.7727 | 29.9 |
| Hf6C3N | 180.9 | 121.5 | 297.9 | 0.2256 | 0.6717 | 17.8 |
| Hf4C3N[19] | 262.2 | 202.1 | 482.4 | 0.1934 | 0.7707 | 29.9 |
| Hf6C3N2 | 214.0 | 151.1 | 366.9 | 0.2143 | 0.7059 | 22.1 |
| Hf3CN | 188.0 | 113.4 | 283.3 | 0.2489 | 0.6031 | 14.6 |
| Hf2CN[19] | 268.1 | 198.5 | 477.6 | 0.2031 | 0.7403 | 28.1 |
| Hf6C2N3 | 221.3 | 149.7 | 366.6 | 0.2239 | 0.6766 | 20.7 |
| Hf4CN2 | 212.7 | 132.8 | 329.7 | 0.2417 | 0.6242 | 17.1 |
| Hf3CN2[19] | 272.8 | 185.1 | 452.8 | 0.2233 | 0.6786 | 23.9 |
| Hf6CN3 | 195.4 | 108.9 | 275.6 | 0.2650 | 0.5574 | 12.7 |
| Hf4CN3[19] | 276.2 | 179.6 | 442.8 | 0.2328 | 0.6504 | 22.2 |
| Hf6CN4 | 207.2 | 156.1 | 374.4 | 0.1989 | 0.7535 | 24.6 |
| Hf5CN4[19] | 279.0 | 171.5 | 427.0 | 0.2449 | 0.6147 | 20.0 |
表3Hf-C-N空位有序结构和HfC1–xNx[19]的力学性质—体模量(B)、剪切模量(G )、弹性模量(E )、泊松比(μ)、Pugh比(G/B)、维氏硬度(HV)等
Table3.Mechanical properties—bulk modulus (B), shear modulus (G ), elastic modulus (E ), Poisson’s ratio (μ), Pugh’s ratio (G/B), Vickers hardness (HV) of Hf-C-N vacancy ordered structures and HfC1–xNx[19].
图4为三元Hf-HfC-HfN体系的力学性质-组分相图, 其中包括三元空位有序结构, Hf-C, Hf-N体系和HfC1–xNx等的性质[19,25,36]. 从图4可以看到: 相同C/N比下, 随着空位浓度的增大, 体模量、剪切模量、弹性模量等减小; 如图5和图6所示, 空位浓度增大, Hf-C-N化合物的有效价电子浓度及总体键强减弱, 材料抵抗外力的能力减小, 则Hf-C-N化合物的模量减小. 然而, Hf6CN4 (空位浓度为1/6)的维氏硬度和Pugh比大于Hf5CN4(无空位)的维氏硬度和Pugh比, 表现出空位硬化现象; 而其他组分下, C/N比相同时, 维氏硬度和Pugh比等随空位浓度的增大而减小. 如图4(f)所示, 泊松比的变化规律与Pugh比的正好相反.
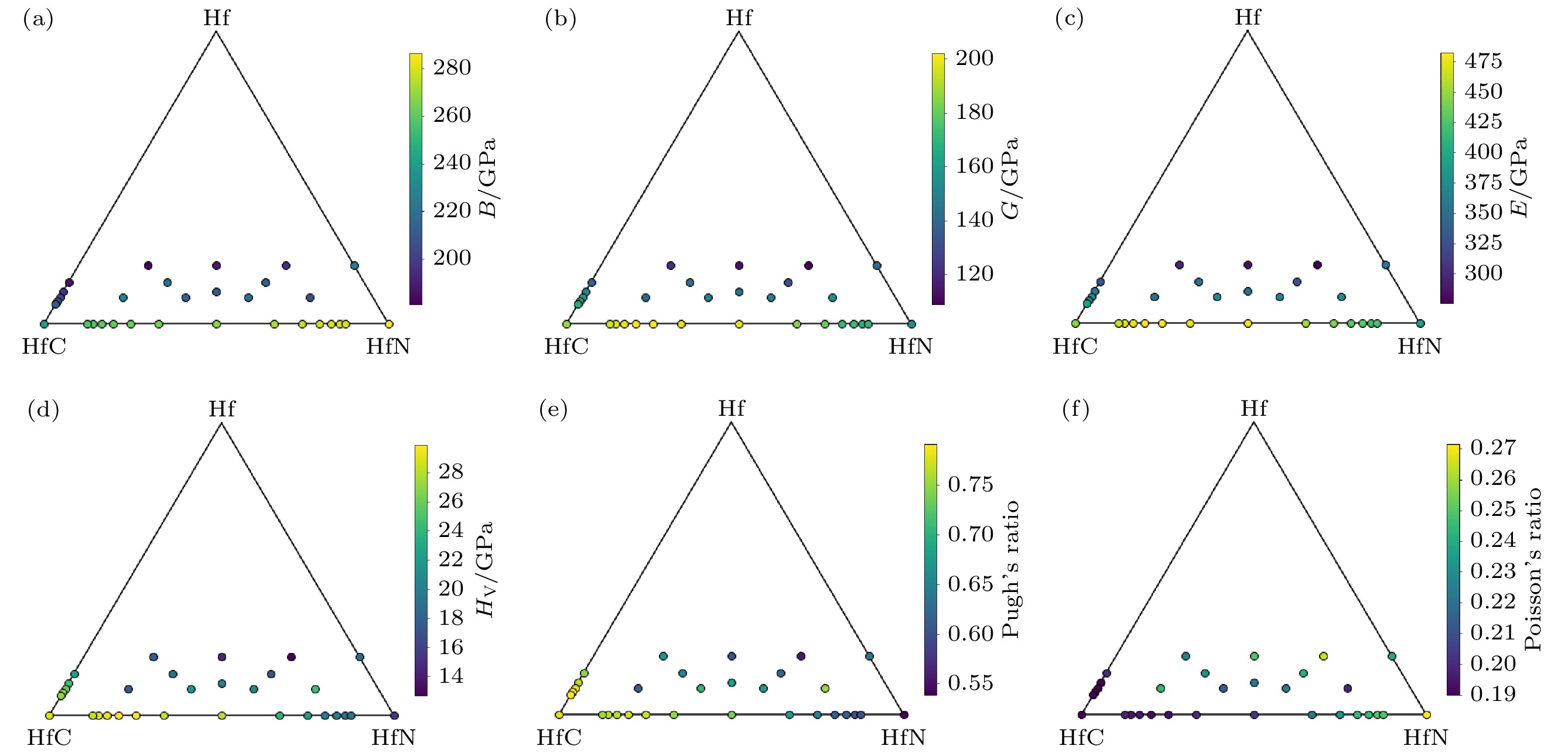 图 4 三元Hf-HfC-HfN体系的力学性质-组分相图 (a) 体模量(B); (b) 剪切模量(G ); (c) 弹性模量(E ); (d) 维氏硬度(HV); (e) Pugh比(G/B); (f) 泊松比(μ)
图 4 三元Hf-HfC-HfN体系的力学性质-组分相图 (a) 体模量(B); (b) 剪切模量(G ); (c) 弹性模量(E ); (d) 维氏硬度(HV); (e) Pugh比(G/B); (f) 泊松比(μ)Figure4. Mechanical properties-composition diagrams of ternary Hf-HfC-HfN system: (a) Bulk modulus (B); (b) shear modulus (G ); (c) elastic modulus (E ); (d) Vickers hardness (HV); (e) Pugh’s ratio (G/B); (f) Poisson’s ratio (μ).
 图 5 (a) Hf6C4N-
图 5 (a) Hf6C4N-






Figure5. Density of state (DOS) and partial density of state (PDOS) normalized by per HfCxNy of (a) Hf6C4N-







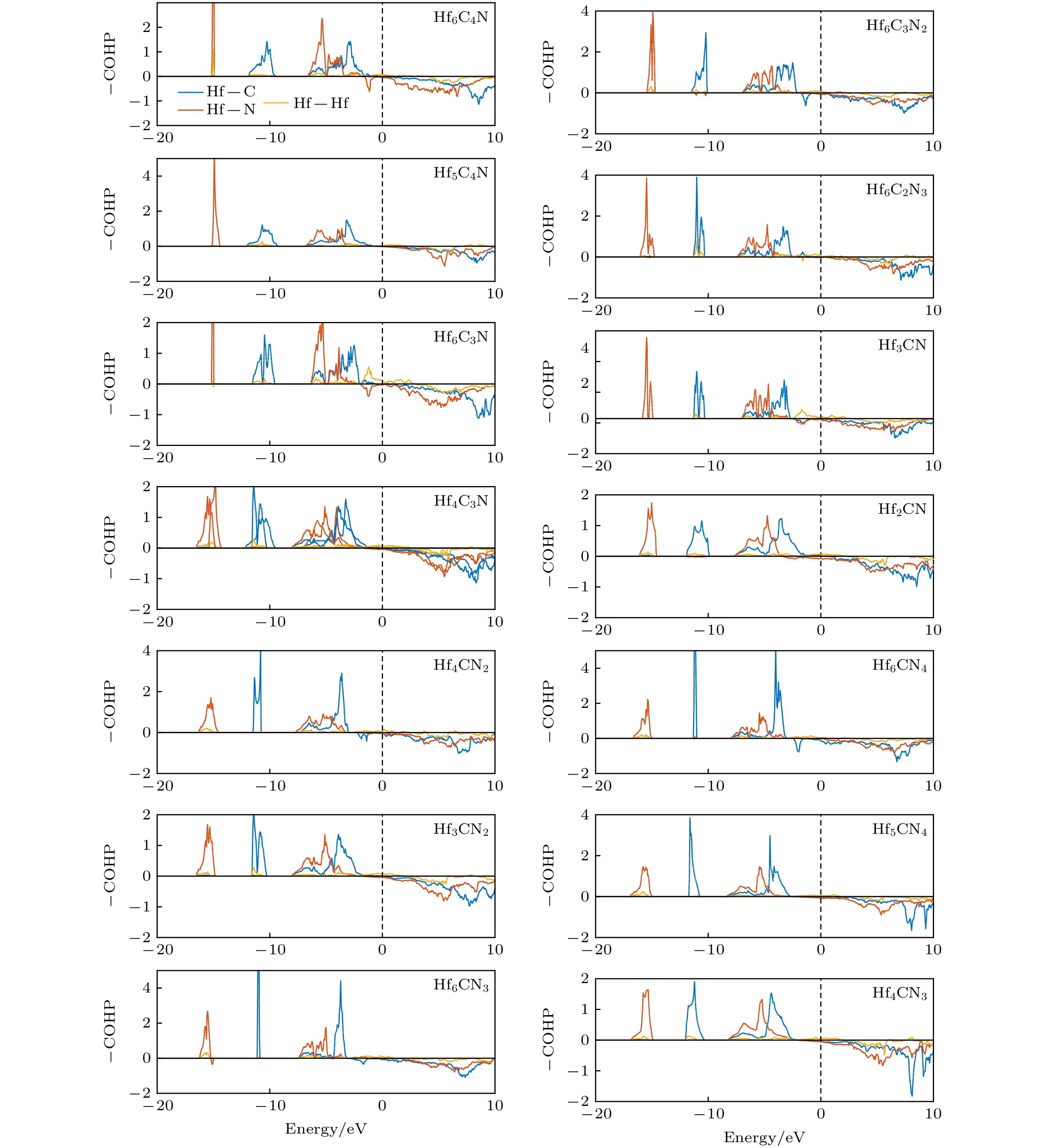 图 6 Hf-C-N化合物的晶体轨道哈密顿分布(–COHP), Fermi能级位于0 eV
图 6 Hf-C-N化合物的晶体轨道哈密顿分布(–COHP), Fermi能级位于0 eVFigure6. Crystal orbital Hamilton populations (–COHP) of Hf-C-N compounds. The Fermi level is at 0 eV.
2
3.3.Hf-C-N空位有序结构的电子性质
为了分析Hf-C-N空位有序结构的成键特性和空位对电子性质的影响, 对其态密度、分态密度和晶体轨道哈密顿分布进行了计算. 图5(a)—(h)为Hf-C-N空位有序结构的态密度和分态密度图, 在能级为–8至–2 eV, Hf-d轨道与C-p和N-p轨道之间存在大量重叠, 即存在强的杂化作用, 则Hf—C和Hf—N键存在强的共价性. 这是Hf-C-N空位有序结构具有非常高的模量和硬度的原因. 同时, 在Fermi面上存在自由电子, 证明其具有金属性. 这些成键特点和二元过渡金属碳、氮化合物[24-28]及HfC1–xNx[19]的相同. 对比Hf2CN (不含空位, 该结构及其电子性质已在文献[19]中报道)和Hf3CN (空位浓度为1/3)的总态密度, 分析空位对态密度的影响, 如图5(i)所示: 可以看到两者价电子的能级排布基本不变, 然而Hf3CN各能级处对应的价电子状态数小于Hf2CN的; 这是由于空位存在, Hf3CN的有效价电子浓度(VEC为7)小于Hf2CN的(VEC为8.5). 而空位对其他Hf-C-N空位有序结构的态密度也存在相似的影响.图6为Hf-C-N空位有序结构的晶体轨道哈密顿分布(–COHP), 同时对比了HfC1–xNx的–COHP. 图6中, 正值表示成键态, 负值表示反键态. 从图6可以看到, Hf-C-N空位有序结构与HfC1–xNx的–COHP相似, 杂化能级(–8 — –2 eV)范围内, 存在宽的成键区域, 即具有强的共价性, 与态密度的计算结果一致. –COHP的积分用ICOHP表示, ICOHP反映了化学键的强弱. 表4为Hf-C-N化合物的Hf—C, Hf—N和Hf—Hf键的–ICOHP的大小, 可以看到: 相同C/N比下, 含空位化合物(如Hf3CN)的Hf—C键和Hf—N键的键强要比不含空位的(如Hf2CN)更强, 且Hf—Hf金属键也更强. 然而, 随着空位浓度增大, Hf—C键和Hf—N键的总键数目减小(如表1所列Hf的配位数小于6), 则总体键强减弱, 因此抵抗外力的能力减弱, Hf-C-N化合物的体模量、剪切模量和弹性模量随之减小.
| Compound | –ICOHP | Compound | –ICOHP | ||||
| Hf—C | Hf—N | Hf—Hf | Hf—C | Hf—N | Hf—Hf | ||
| Hf6C4N | 3.373 | 3.567 | 0.529 | Hf6C3N2 | 3.181 | 2.990 | 0.571 |
| Hf5C4N | 3.373 | 3.033 | 0.459 | Hf4CN2 | 3.474 | 3.111 | 0.650 |
| Hf6C3N | 3.350 | 3.067 | 0.718 | Hf3CN2 | 3.551 | 3.091 | 0.541 |
| Hf4C3N | 3.319 | 3.029 | 0.454 | Hf6CN3 | 3.408 | 3.211 | 0.570 |
| Hf6C3N2 | 3.607 | 3.103 | 0.530 | Hf4CN3 | 3.321 | 3.159 | 0.520 |
| Hf3CN | 3.277 | 3.211 | 0.737 | Hf6CN4 | 3.675 | 3.179 | 0.591 |
| Hf2CN | 3.483 | 2.802 | 0.490 | Hf5CN4 | 3.319 | 3.017 | 0.500 |
表4Hf-C-N化合物的晶体轨道哈密顿分布的积分值(–ICOHP)
Table4.Integrated crystal orbital Hamilton populations (–ICOHP) of Hf-C-N compounds.
1) 搜索发现了8种新的Hf-C-N空位有序结构, 且都具有岩盐结构; 空位有序地分布在[Hf6]八面体间隙, 证明了Hf-C-N空位化合物可以以有序结构的形式稳定存在;
2) Hf-C-N空位有序结构具有非常高的模量和硬度; 相同C/N比时, 随着空位浓度的增大, 体模量、剪切模量、弹性模量减小;
3) 发现了一个空位硬化现象, Hf6CN4 (空位浓度为1/6)的硬度大于Hf5CN4(无空位)的硬度;
4) Hf-C-N空位有序结构的化学键具有强共价性和金属性. 随着空位浓度的增加, Hf—C和Hf—N共价键键强增大, 金属性增强, 但总体键强减弱, 使其模量减小.
