全文HTML
--> --> -->通过对已有的铁磁材料进行合金化掺杂是提高材料居里温度的一个有效方法. Xue等[12]在单层CrCl3中利用同价W原子取代50%的Cr原子, 发现单层CrWCl6具有很强的面外磁各向异性以及强的自旋轨道耦合, 居里温度达到了76 K; Guan等[13]通过在二维MnS2铁磁半导体中引入Re原子, 构建等价合金化合物MnxRe1–xS2, 将居里温度提高到室温以上. 引入Mn-Re相互作用后, 也看到了自旋耦合的显著增强. 因此, 基于现有磁性二维半导体材料构建合金化合物很有可能实现提高居里温度的目的. 本文基于实验报道的单层CrI3构建了合金化合物CrTMI6, 其中的过渡金属TM遍历了元素周期表中第四、第五周期的过渡金属元素. 通过基于第一性原理的高通量计算, 选定了一个具有较大铁磁交换能的二维本征铁磁半导体CrMoI6作为进一步研究的对象. 通过声子谱计算和从头算分子动力学(AIMD)模拟证实了其动力学和热力学稳定性. 考虑了自旋轨道耦合后的理论计算表明, CrMoI6单层具有相当大的磁各向异性能, 易磁轴垂直于材料所在的二维平面. 蒙特卡罗模拟预测得到单层CrMoI6的居里温度约为92 K, 达到CrI3单层的2倍以上.
3.1.高通量筛选二维合金化合物CrTMI6
基于单层CrI3的晶体结构搭建二维合金化合物CrTMI6, 其中TM代表的是元素周期表中第四、第五周期的过渡金属元素, 如图1(a)所示. 具体来说, 从单层CrI3的2


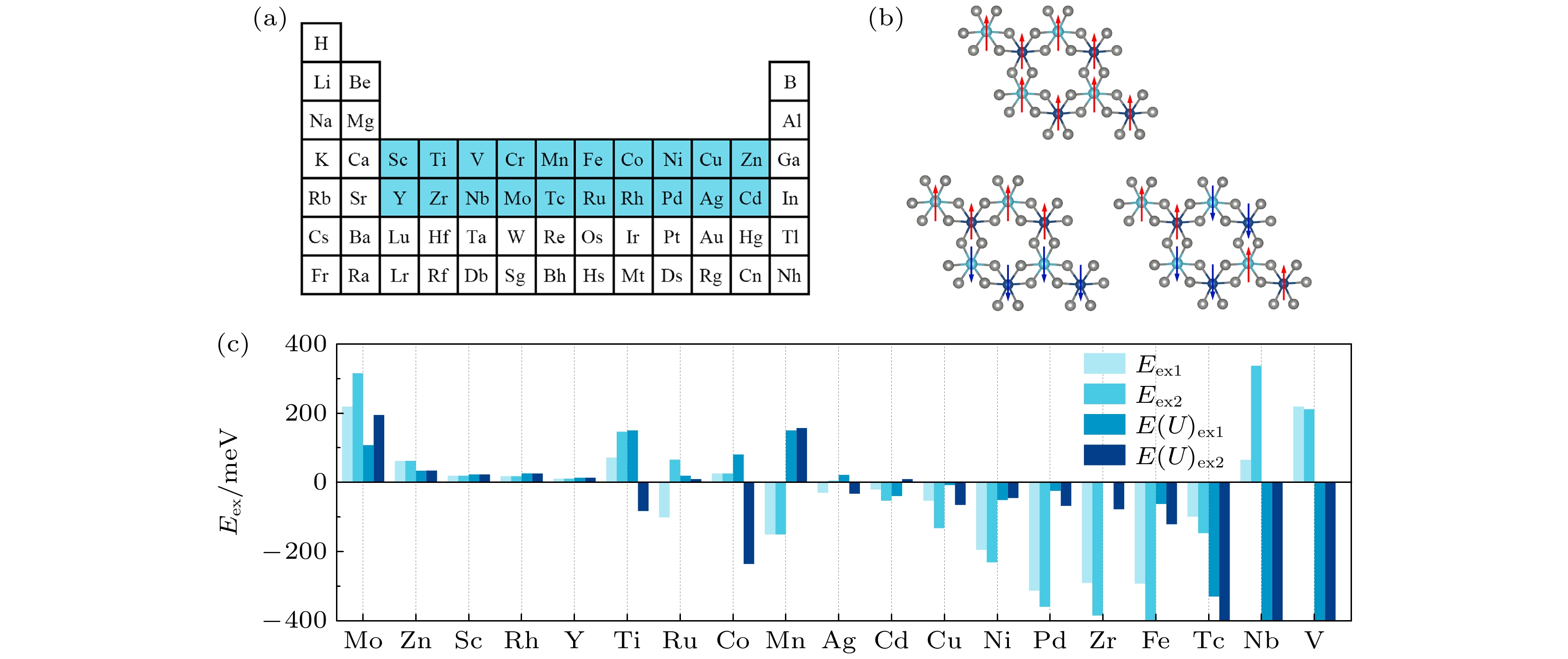 图 1 (a)二维等价合金化合物CrTMI6中TM元素的选取示意图; (b)高通量计算中考虑的单层CrTMI6的铁磁构型和两种反铁磁构型; (c)使用GGA+U方法前后单层CrTMI6材料的交换能. 其中Eex1 = EAFM1 – EFM, Eex2 = EAFM2 – EFM; E(U)ex1和E(U)ex2为使用GGA+U后的相应值
图 1 (a)二维等价合金化合物CrTMI6中TM元素的选取示意图; (b)高通量计算中考虑的单层CrTMI6的铁磁构型和两种反铁磁构型; (c)使用GGA+U方法前后单层CrTMI6材料的交换能. 其中Eex1 = EAFM1 – EFM, Eex2 = EAFM2 – EFM; E(U)ex1和E(U)ex2为使用GGA+U后的相应值Figure1. (a) Schematic diagram of the selection of TM element in the two-dimensional equivalent alloy compound CrTMI6; (b) ferromagnetic configuration and two antiferromagnetic configurations of monolayer CrTMI6 considered in high-throughput calculations; (c) exchange energy of CrTMI6 monolayer before and after GGA+U method applied, where Eex1 = EAFM1 – EFM, Eex2 = EAFM2 – EFM; E(U)ex1 and E(U)ex2 are the corresponding values using GGA+U.
基于GGA+PBE计算结果, 得到了9种铁磁材料(Eex1和Eex2均为正)和10种反铁磁材料(Eex1和Eex2均为负或任一为负), 如图1(c)和表1所示. 为了考虑d电子之间较强的库仑相互作用, 使用GGA + U[20,21]方法再次计算交换能. 在不同的过渡金属元素U值的选取上, 使用文献提供的每种过渡金属元素相应价态的参数来描述它们的d轨道电子, 具体数值见表2. 计算后发现部分材料的磁性耦合基态发生了变化: 有4种材料由铁磁性转变为反铁磁性, 分别为CrTiI6, CrCoI6, CrNbI6, CrVI6; 有两种材料由反铁磁性转变为铁磁性, 分别为CrRuI6, CrMnI6. 因此, 若按照GGA + U的结果来看, 得到了6种铁磁材料和13种反铁磁材料(图1(c)及表1). 值得注意的是, 在整个过程中, CrMoI6单层的磁性耦合情况没有发生变化. 而且, 在铁磁材料中, CrMoI6的总交换能最大, 即铁磁耦合最强. 因此, 在后面的内容中, 将围绕CrMoI6单层材料进行更为深入的研究. 为了结果的准确性, 上述所有不同结构加U后的能量都在对应磁性耦合状态下重新进行结构优化后得到.
| 材料 | 交换能/eV | |||
| Eex1 | Eex2 | E(U)ex1 | E(U)ex2 | |
| CrMoI6 | 219.40 | 315.84 | 108.58 | 196.34 |
| CrZnI6 | 62.14 | 62.06 | 33.52 | 34.48 |
| CrScI6 | 19.53 | 19.49 | 23.13 | 23.00 |
| CrRhI6 | 17.97 | 17.94 | 25.99 | 25.91 |
| CrYI6 | 10.65 | 10.66 | 13.90 | 13.88 |
| CrTiI6 | 72.12 | 146.71 | 150.49 | –82.80 |
| CrRuI6 | –101.15 | 66.06 | 14.58 | –8.83 |
| CrCoI6 | 25.84 | 25.88 | 80.72 | –235.76 |
| CrMnI6 | –150.41 | –150.73 | 150.10 | 157.28 |
| CrAgI6 | –29.97 | 4.74 | 21.69 | –32.62 |
| CrCdI6 | –20.45 | –52.78 | –39.27 | 9.51 |
| CrCuI6 | –52.43 | –132.37 | –7.68 | –65.38 |
| CrNiI6 | –194.93 | –231.25 | –51.18 | –45.46 |
| CrPdI6 | –313.25 | –359.92 | –24.18 | –67.84 |
| CrZrI6 | –290.64 | –385.45 | 2.73 | –77.70 |
| CrFeI6 | –292.42 | –399.20 | –62.40 | –121.16 |
| CrTcI6 | –98.88 | –146.83 | –301.14 | –1104.20 |
| CrNbI6 | 65.53 | 337.29 | –698.92 | –681.61 |
| CrVI6 | 219.46 | 211.92 | –776.31 | –718.52 |
表1单层CrTMI6的2×2超胞的交换能
Table1.Exchange energy of 2×2 supercells of CrTMI6 monolayer.
| 材料 | U/eV |
| V | 3.1[22] |
| Cr | 3.5[22] |
| Mn | 3.9[22] |
| Fe | 4.0[22] |
| Co | 3.4[22] |
| Ni | 6.0[22] |
| Cu | 4.0[22] |
| Zn | 5.0[23] |
| Nb | 2.0[24] |
| Mo | 3.5[22] |
| Tc | 2.3[24] |
| Ru | 2.4[24] |
| Rh | 2.8[24] |
| Pd | 3.3[24] |
| Ag | 5.1[25] |
| Cd | 6.0[25] |
表2过渡金属元素TM所使用的U值
Table2.Value of U used for the transition metal element TM.
2
3.2.单层CrMoI6的结构特性及稳定性
针对单层CrMoI6, 在上述的基本构型之外, 进一步设计了6种可能的合金原子排列结构(图2(a)). 对于Cr原子和Mo原子, 使用表2中所列的对应值来适当地考虑强关联电子. 计算结果表明, C2构型的能量最低, 这一结构中存在Mo(Cr)原子的二聚体, 这可能是使该结构相对稳定的原因. 如图2(b)所示, C2构型中的Mo原子间的平均距离约为6.5 ?, Mo和Cr原子相互间隔或过于聚集都会造成能量的略微上升.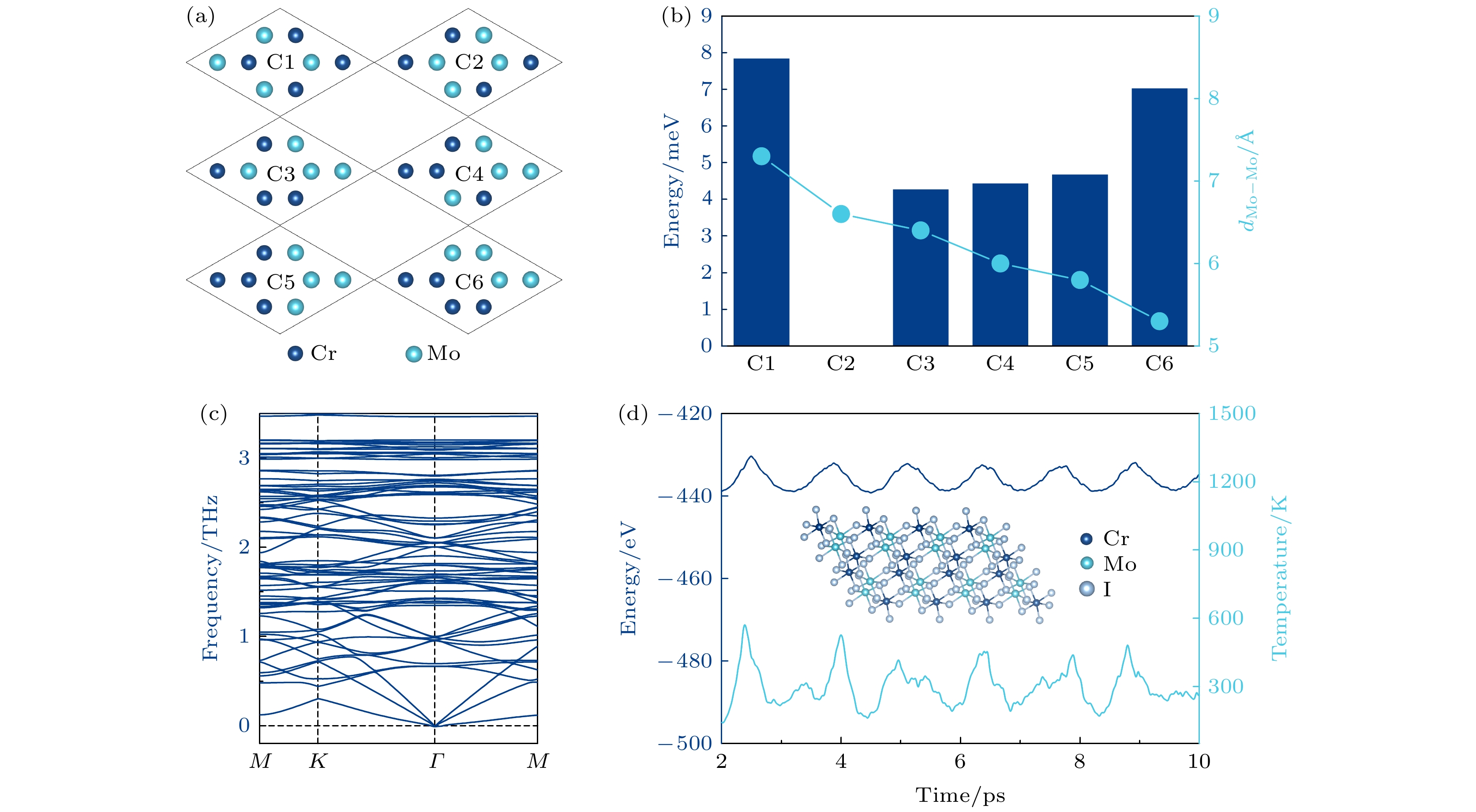 图 2 (a)单层CrMoI6可能存在的6种不同构型; (b) C1—C6各构型相对C2构型平均到每个化学式的能量差和Mo-Mo间的平均距离; (c)单层CrMoI6的声子谱; (d)时长10 ps, 温度300 K的分子动力学模拟下, 系统温度和总能量的变化以及模拟结束时的结构
图 2 (a)单层CrMoI6可能存在的6种不同构型; (b) C1—C6各构型相对C2构型平均到每个化学式的能量差和Mo-Mo间的平均距离; (c)单层CrMoI6的声子谱; (d)时长10 ps, 温度300 K的分子动力学模拟下, 系统温度和总能量的变化以及模拟结束时的结构Figure2. (a) Six possible configurations of CrMoI6 monolayer that may exist; (b) energy difference of C1?C6 configuration relative to C per chemical formula and the average bond distance between Mo-Mo; (c) phonon spectrum of CrMoI6 monolayer; (d) molecular dynamics simulation with duration of 10 ps, 300 K and the variation curves of system temperature and total energy, as well as the structure at the end of simulation.
结构稳定性是二维材料在实验制备和应用中的首要条件. 因此, 对单层CrMoI6的动力学稳定性和热稳定性进行了计算评估. 首先, 计算了单层CrMoI6的声子谱结构, 如图2(c)所示, 在整个布里渊区内, 声子谱没有虚频, 说明它具有良好的动力学稳定性. 图2(d)给出了300 K温度下单层CrMoI6在AIMD模拟过程中总能量的波动以及模拟10 ps后的晶体结构. 计算中使用了Nosé-Hoover方法[26]进行控温, 在正则系统中每个温度的步长为1 fs. 从图2(d)可以看出, 整个模拟过程中, 能量和系统温度波动都较小, 且所有原子都在平衡位置附近振动, 二维周期性结构始终能够得以保持. 以上计算结果表明, 单层CrMoI6具有良好的热稳定性, 预测可以在室温范围内稳定存在.
2
3.3.单层CrMoI6的磁性耦合与能带
为了进一步验证单层CrMoI6更倾向于铁磁耦合, 考虑图3(a)所示的6种可能的反铁磁耦合结构. 计算结果表明, 铁磁态的能量最低, 相比反铁磁构型中能量最低的AFM1, 铁磁态平均到每个化学式的能量低约25 meV, 如图3(b)所示. 基于优化后的结构可以发现, Cr-I-Mo, Cr-I-Cr和Mo-I-Mo的键角均位于94°—95°区间内, 根据Goodenough-Kanamori-Anderson (GKA)规则[27-29], 这类键角在90°左右的磁性离子间的超相互作用(super-exchange interaction)一般会带来材料的铁磁序, 这可以解释上述计算得到的铁磁耦合下单层CrMoI6能量最低的结果.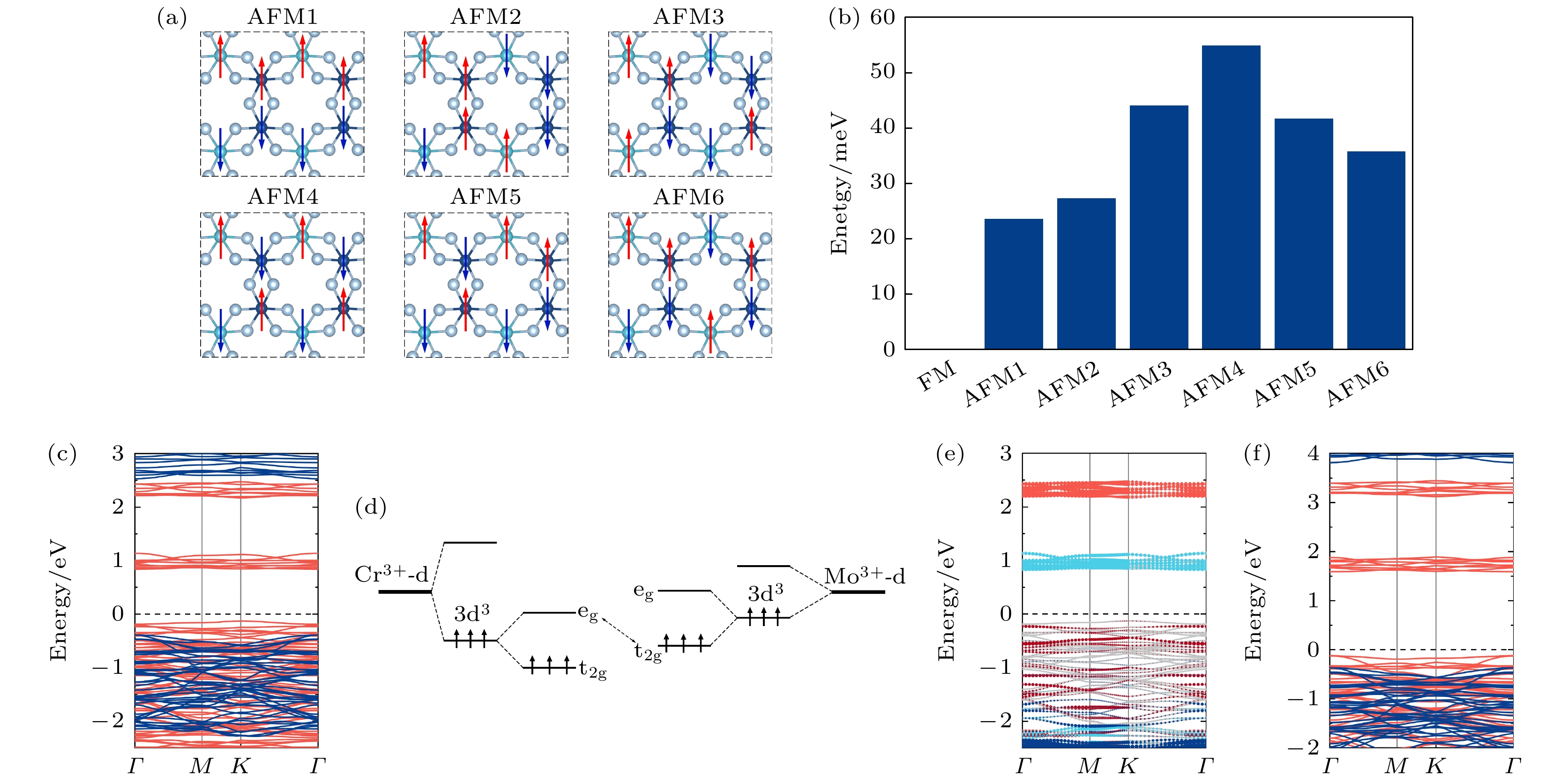 图 3 (a)单层CrMoI6的6种反铁磁构型示意图; (b)不同反铁磁构型相对于铁磁构型平均到每个化学式的能量差; (c)单层CrMoI6的能带结构示意图, 红色实线表示自旋向上通道, 蓝色实线表示自旋向下通道; (d) Cr, Mo合金体系的轨道演化及能级差示意图; (e)单层CrMoI6的自旋向上状态的分轨道能带结构, Cr的
图 3 (a)单层CrMoI6的6种反铁磁构型示意图; (b)不同反铁磁构型相对于铁磁构型平均到每个化学式的能量差; (c)单层CrMoI6的能带结构示意图, 红色实线表示自旋向上通道, 蓝色实线表示自旋向下通道; (d) Cr, Mo合金体系的轨道演化及能级差示意图; (e)单层CrMoI6的自旋向上状态的分轨道能带结构, Cr的



Figure3. (a) Schematic diagram of six antiferromagnetic configurations of CrMoI6 monolayerand (b) energy difference per formula of different antiferromagnetic configurations. (c) Band structure of CrMoI6 monolayer. The solid red and blue lines represent the spin-up and spin-down channels, respectively. (d) schematic diagram of orbital evolution and energy level difference of Cr and Mo alloy systems. (e) Band structure of CrMoI6 monolayer in the spin-up state. The


针对单层CrMoI6的铁磁耦合结构, 首先使用DFT+U的方法计算其电子能带结构, 如图3(c)所示. 单层CrMoI6的能带结构在自旋向上和自旋向下通道都表现出了半导体特性, 自旋向上通道的带隙为0.97 eV, 自旋向下通道的带隙为2.9 eV. 从整体来看, 价带顶和导带底均由自旋向上的电子提供, 和单层CrI3的能带结构类似[30]. 进一步分析发现, CrMoI6的结构可划分为多个CrI6和MoI6八面体, 八面体晶体场会使d轨道能级分裂为能量较低的















2
3.4.磁各向异性与居里温度
磁各向异性是二维长程磁有序存在的必要条件, 通过加入自旋轨道耦合修正计算了单层CrMoI6自旋取向沿不同方向的磁各向异性能(MAE). MAE沿x(100), y(010)和z(001)方向的角度依赖的计算公式如下: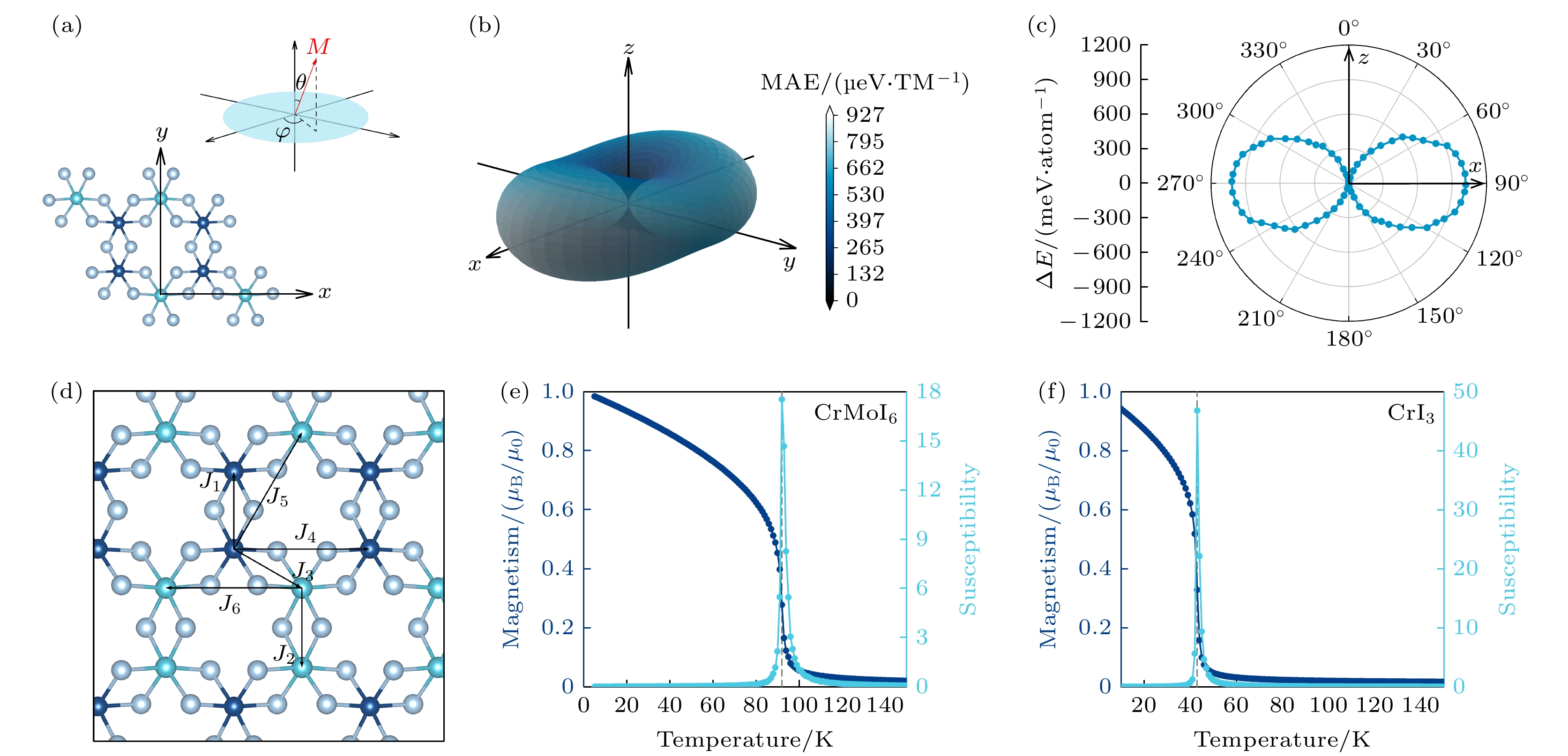 图 4 (a) θ和φ的示意角度及xy轴在结构平面上的方向; (b)全空间内的磁各向异性能分布; (c)垂直于y轴方向的xz平面内的磁各向异性能分布极坐标图; (d)交换相互作用J1—J6的示意图; (e)单层CrMoI6的磁矩及磁化率随温度的变化曲线; (f)单层CrI3的磁矩及磁化率随温度的变化曲线
图 4 (a) θ和φ的示意角度及xy轴在结构平面上的方向; (b)全空间内的磁各向异性能分布; (c)垂直于y轴方向的xz平面内的磁各向异性能分布极坐标图; (d)交换相互作用J1—J6的示意图; (e)单层CrMoI6的磁矩及磁化率随温度的变化曲线; (f)单层CrI3的磁矩及磁化率随温度的变化曲线Figure4. (a) Schematic angles of θ and φ and the direction of the xy axis on the structure plane; (b) distribution of magnetic anisotropy energy in the whole space; (c) polar coordinate diagram of magnetic anisotropic energy distribution in the xz plane perpendicular to the y axis; (d) schematic diagram of exchange interaction J1–J6; The magnetic moment and susceptibility of (e) CrMoI6 monolayer and (f) CrI3 monolayer as a function of temperature.
居里温度是铁磁材料实际应用的一个关键参数, 使用基于海森伯模型的蒙特卡罗方法计算单层CrMoI6的居里温度. 考虑到Cr原子和Mo原子的磁矩及交换相互作用的不同, 将二者区分开来考虑, 取最近邻Cr






2
3.5.应力对磁性耦合的影响
在实验和应用过程中, 二维材料容易因衬底等外部因素的影响而受到应力作用, 并导致应变的产生, 这种应变往往会影响二维材料的物理性能. 因此, 本文进一步研究了应变对单层CrMoI6的磁性影响. 首先施加等比例的双轴应变, 体系总能量和交换能Eex的变化如图5(a)所示. 拉伸时反铁磁和铁磁的交换能会进一步增加, 而压缩时则会降低. 我们认为这是磁性离子间的直接相互作用和超相互作用竞争的结果. 磁性离子间的直接相互作用倾向于反铁磁耦合, 而前面提到的键角接近90°的超相互作用则带来铁磁耦合. 当拉伸时, 如图5(b)和图5(c)所示, 磁性离子间距增加, 直接相互作用减弱, 所以铁磁耦合增强. 而压缩时, 磁性离子间距减小, 直接相互作用增强, 所以铁磁耦合强度减弱. 另外, 我们发现键角偏离平衡位置也会减弱超相互作用带来铁磁耦合效果, 从图5(b)和图5(c)可以看到, 无论拉伸还是压缩, TM—I—TM的键角都会偏离原来的值, 造成图5(a)中铁磁耦合的减弱, 只是这种影响弱于离子间距改变对直接相互作用造成的影响. 图5(d)为在x, y单一方向施加应变的结果, 交换能的变化趋势和双轴应变的情况类似, 但变化幅度不同. 能量沿x方向变化较为平缓, 而y方向变化较为陡峭, 很显然这是由沿y方向的应变对磁性离子的间距影响更大造成的.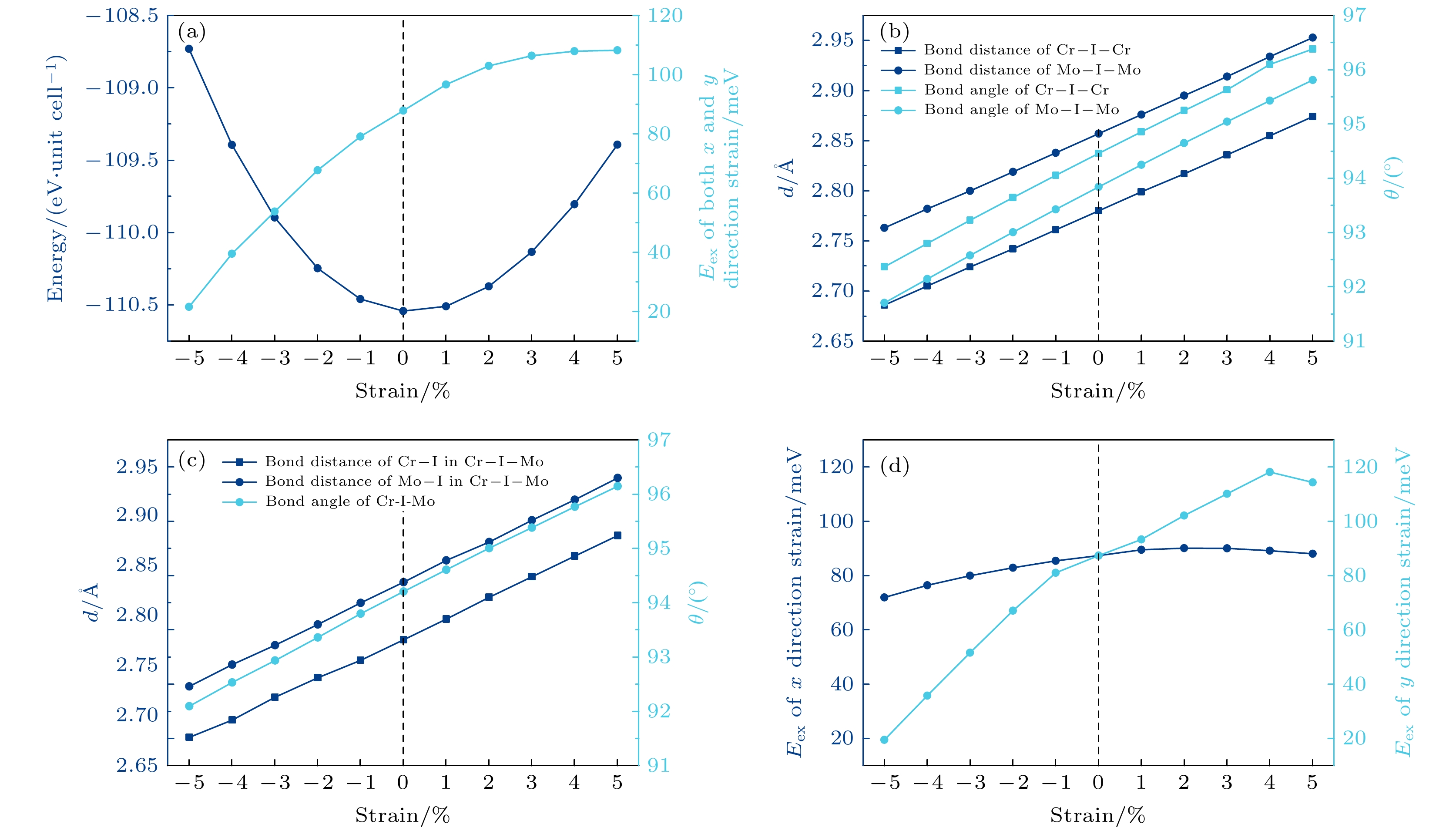 图 5 (a)双轴应变下CrMoI6单层的交换能以及总能量的变化; (b)施加拉伸及压缩应变时Cr—I—Cr和Mo—I—Mo的键长与键角随应变大小的变化; (c)施加拉伸及压缩应变时Cr—I—Mo的键角以及其中Cr—I和Mo—I的键长随应变大小的变化; (d)单轴应变下的交换能随应变大小的变化
图 5 (a)双轴应变下CrMoI6单层的交换能以及总能量的变化; (b)施加拉伸及压缩应变时Cr—I—Cr和Mo—I—Mo的键长与键角随应变大小的变化; (c)施加拉伸及压缩应变时Cr—I—Mo的键角以及其中Cr—I和Mo—I的键长随应变大小的变化; (d)单轴应变下的交换能随应变大小的变化Figure5. (a) Variation of exchange energy and total energy of CrMoI6 monolayer under biaxial strain; (b) variation of bond lengths and bond angles of Cr—I—Cr and Mo—I—Mo with strain under tensile and compressive strain; (c) variation of the bond angles of Cr—I—Mo and the bond lengths of Cr—I and Mo—I as a function of strain under tensile and compressive strain; (d) variation of exchange energy with strain under uniaxial strain.
