Fund Project:Project supported by the National Key R&D Program of China (Grant Nos. 2016YFA0202601, 2016YFB0901502)
Received Date:02 March 2021
Accepted Date:03 June 2021
Available Online:15 August 2021
Published Online:20 October 2021
Abstract:Lithium-rich manganese-based ternary cathode material for lithium-ion batteries, Li1.208Ni0.333Co0.042Mn0.417O2, has excellent structural stability and electrochemical stability due to its high Ni content. In order to understand the physical properties of this lithium-rich material, its crystal structure, electronic structure and defect properties are calculated by employing the first-principles method based on the density functional theory. The obtained electronic structure shows that Li1.208Ni0.333Co0.042Mn0.417O2 is a magnetic semiconductor with a direct band gap of 0.47 eV. The analysis of the electronic state suggests that the electronic state at the valence band maximum (VBM) is the hybridization of px, py, pz orbitals of oxygen and the dxy, dyz, dxz orbitals of Ni-atom. The electronic state at the conduction band minimum has similar characteristics to those at the VBM, however, part of Ni-${3{\rm{d}}}_{{x}^{2}-{y}^{2}}$ and Mn-${3{\rm{d}}}_{{x}^{2}-{y}^{2}}$, and Mn-${3{\rm{d}}}_{yz}$ also contribute to the electronic hybridizations. The charge density difference calculations indicate that the bonding between O and transition metal atoms are through the mixture of covalent bond with ionic bond. The vacancy formation of a single metal atom is also calculated. The results show that the volumes of the defect systems containing metal vacancies are all reduced in comparison with the volume of perfect lattice. The volume change is the largest for the formation of Mn-vacancy, while the volume is almost unchanged with Co atoms extracted. The vacancy formation energies of the metals are Ef (Mn) > Ef (Co) > Ef (Ni), and the vacancy formation energy of Mn is significantly higher than those of Ni and Co, indicating that the presence of Mn provides a major structural stability for the material. The calculated charge density differences also show that the formation of metal vacancies influences only the charge distribution of the oxygen atoms around the vacancy, showing the local character of the vacancy effect. Since the formation of metal vacancy breaks the bonding between the metal and the surrounding oxygen atoms, the O-2p states near the Fermi surface are significantly increased as shown in the calculated electronic density of states. Such a picture suggests that the electrons on oxygen atoms in vicinity of the metal vacancies become freer. Keywords:Li-rich Mn-based ternary material/ electronic structures/ defect properties/ first principles calculations
表1空位形成前后Li1.208Ni0.333Co0.042Mn0.417O2的晶格常数, 原胞体积和空位形成能的计算值 Table1.Computational lattice constants, unit cell volume and vacancy formation energies of Li1.208Ni0.333Co0.042Mn0.417O2 before and after the defect formations.
表2各原子的电子轨道对价带顶和导带底上电子态的贡献 Table2.Contribution of electronic orbital of various types of atoms for the electronic states at VBM and CBM.
计算得到的Li1.208Ni0.333Co0.042Mn0.417O2材料的总电子态密度 (TDOS) 和各原子分态密度 (PDOS) 如图3(a)所示. 从图3(a)可以清楚地看到, 该材料是一个半导体, 而且是一个自旋极化的磁性半导体. 在图3(b)—图3(d)中, 分别给出了Li1.208Ni0.333Co0.042Mn0.417O2中空位周围的6个O原子的2p电子分态密度和在过渡金属空位 (即Ni, Co和Mn空位) 形成之前和形成之后的对比. 可以看到, 当金属空位形成后, 费米面附近的O-2p占据态 (DOS峰) 明显增加. 这是因为, 金属空位的形成打断了金属与周围O原子的成键, 导致周围这些氧原子上的电子更加自由, 能量上就更加接近于费米面. 带有Co, Ni 空位的体系的态密度特征类似, 即费米面附近有了很高的态密度值 (较自由的电子较多). 而Mn空位体系的DOS稍有不同, 其高态密度的峰离费米面稍远 (电子被束缚的较紧). 图 3 (a) Li1.208Ni0.333Co0.042Mn0.417O2材料的总态密度和各原子分态密度; (b)—(d) 金属空位形成前后材料中空位周围的6个氧原子的2p电子态密度和, 分别用黑色和红色实线表示 Figure3. (a) Total and atomic-decomposed partial density of states for Li1.208Ni0.333Co0.042Mn0.417O2; (b)–(d) sum of the density of states of 2p electronic states of the six oxygen atoms around the M-vacancy before and after the formation of M vacancy, respectively. Black and red solid lines represent DOSs before and after the vacancy formation, respectively.






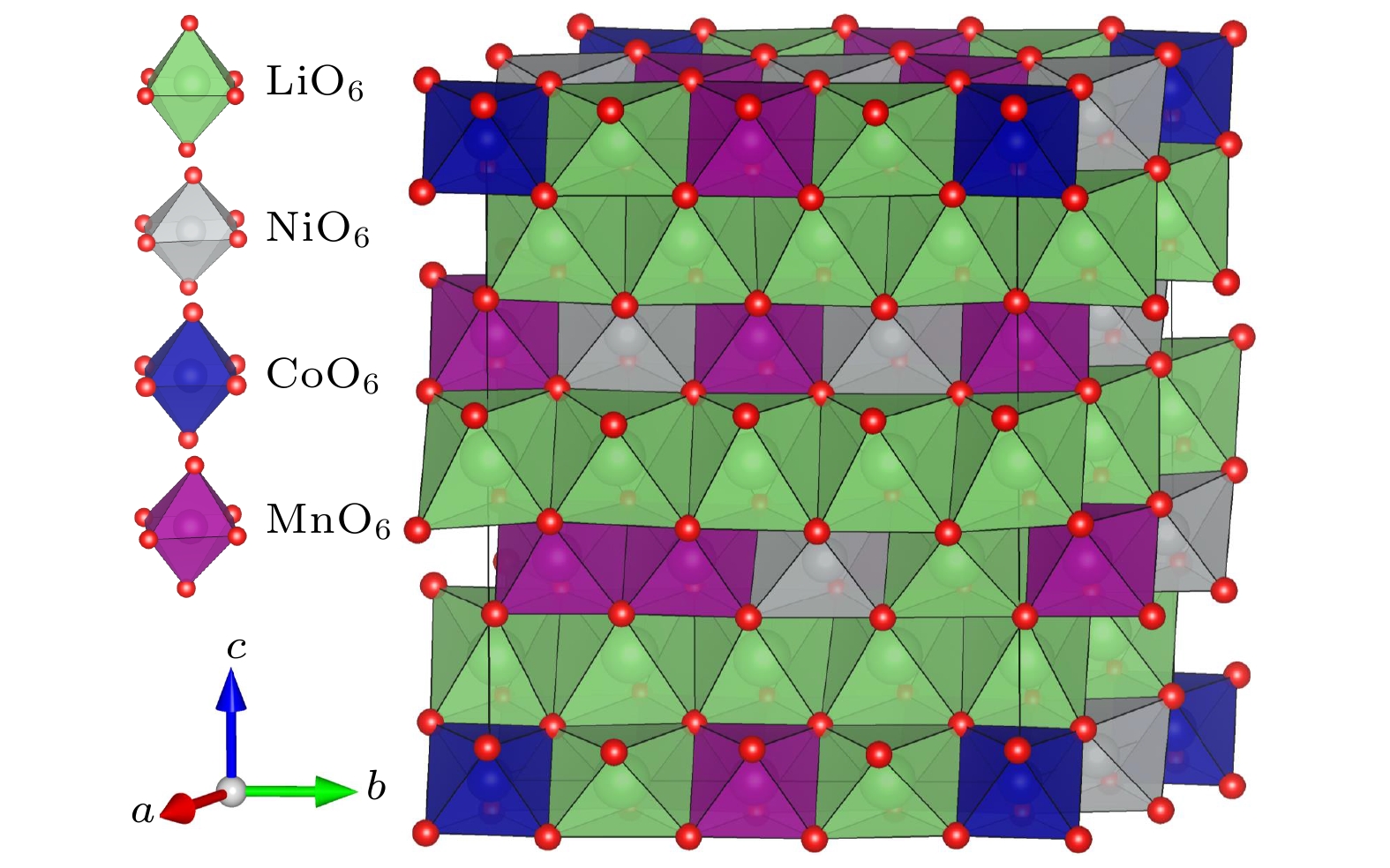 图 1 Li1.208Ni0.333Co0.042Mn0.417O2的晶体结构, 其中LiO6, NiO6, CoO6和MnO6八面体分别标识为绿色、灰色、蓝色和紫色
图 1 Li1.208Ni0.333Co0.042Mn0.417O2的晶体结构, 其中LiO6, NiO6, CoO6和MnO6八面体分别标识为绿色、灰色、蓝色和紫色


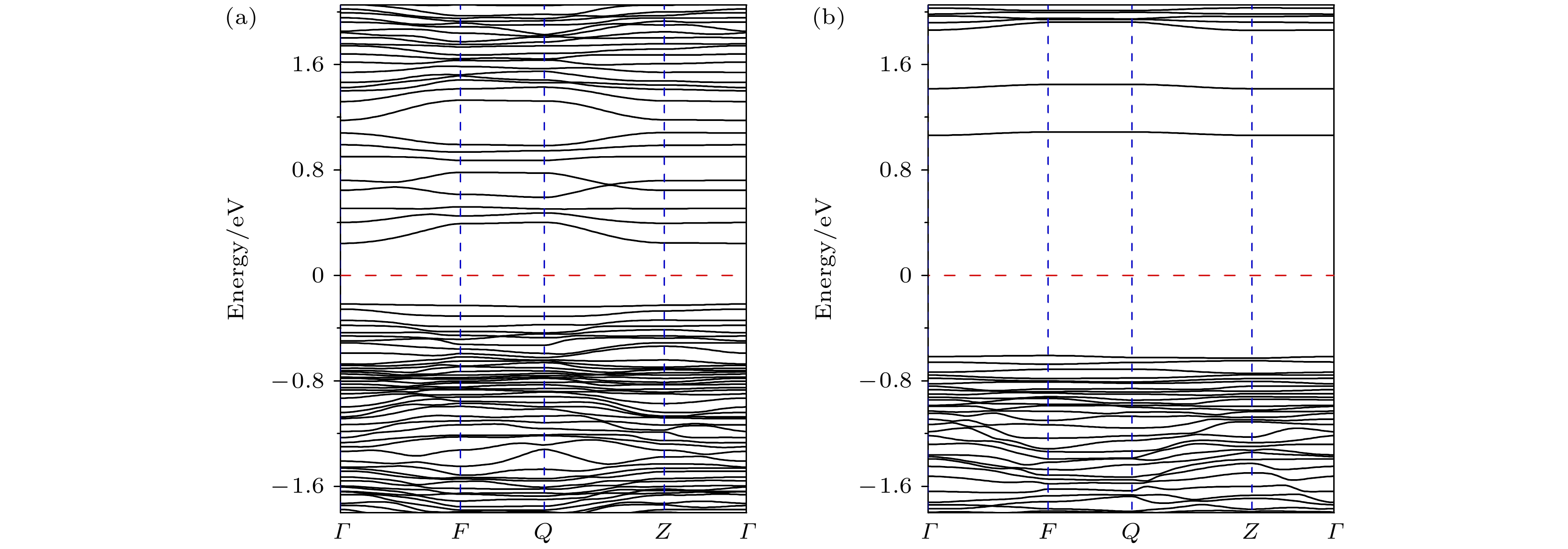 图 2 Li1.208Ni0.333Co0.042Mn0.417O2的能带结构 (a) 自旋向上的能带; (b) 自旋向下的能带. 红色虚线表示费米能级
图 2 Li1.208Ni0.333Co0.042Mn0.417O2的能带结构 (a) 自旋向上的能带; (b) 自旋向下的能带. 红色虚线表示费米能级




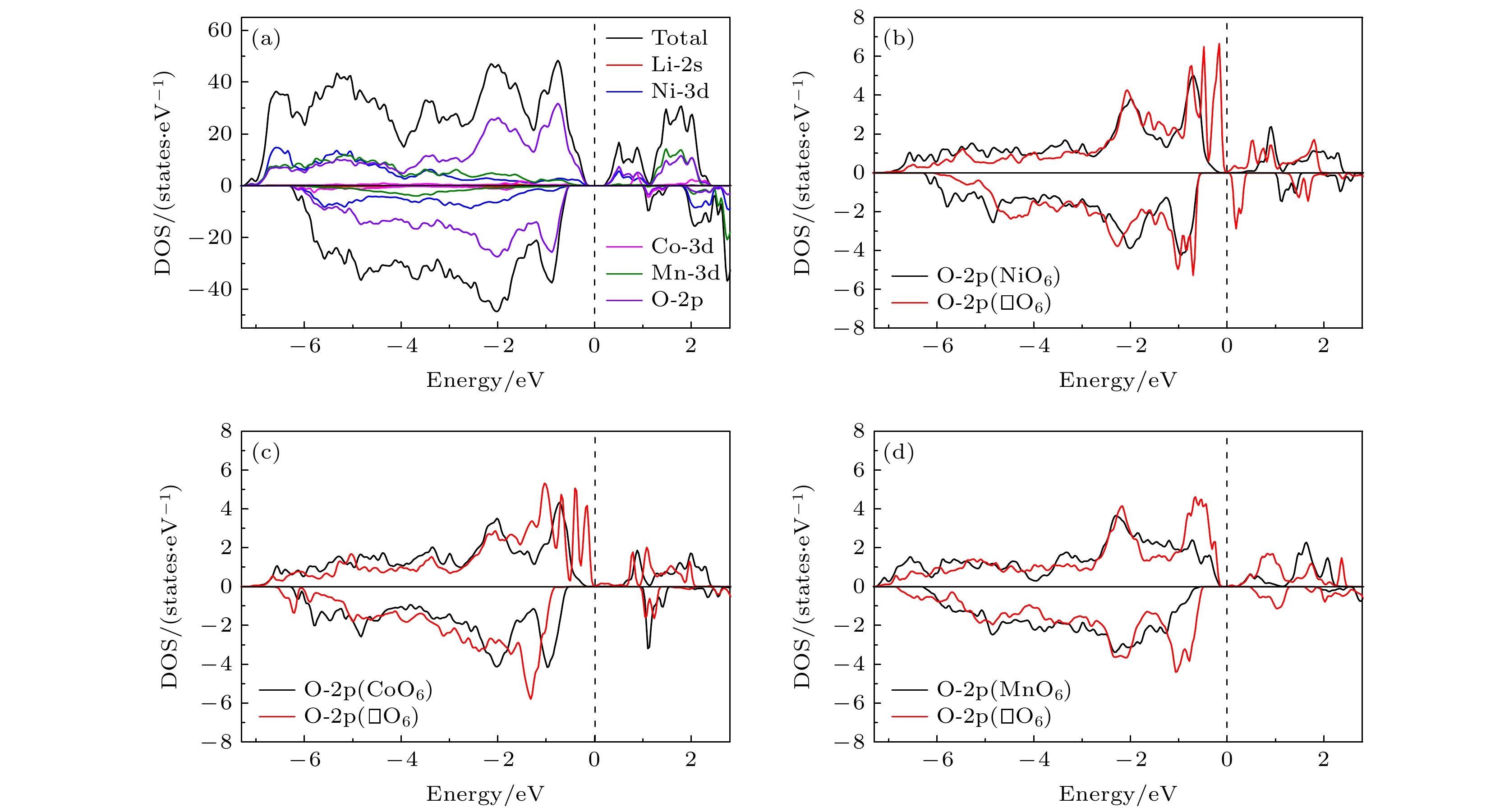 图 3 (a) Li1.208Ni0.333Co0.042Mn0.417O2材料的总态密度和各原子分态密度; (b)—(d) 金属空位形成前后材料中空位周围的6个氧原子的2p电子态密度和, 分别用黑色和红色实线表示
图 3 (a) Li1.208Ni0.333Co0.042Mn0.417O2材料的总态密度和各原子分态密度; (b)—(d) 金属空位形成前后材料中空位周围的6个氧原子的2p电子态密度和, 分别用黑色和红色实线表示




 图 4 材料中三种过渡金属差分电荷密度对比 (a)—(c) 空位形成前; (d)—(f) 空位形成后. 所画的平面是金属附近4个O所在的平面. 图中实线和虚线分别表示电荷聚集
图 4 材料中三种过渡金属差分电荷密度对比 (a)—(c) 空位形成前; (d)—(f) 空位形成后. 所画的平面是金属附近4个O所在的平面. 图中实线和虚线分别表示电荷聚集




