全文HTML
--> --> -->Lu等[15]用溶液法合成了一种环境友好、性能稳定的全无机卤化铋银AgBiI4太阳能吸收层材料, 表现出优异的空气稳定性. Kulkarni等[16]采用溶剂工程技术在较低温度下获得均匀无针孔的AgBi2I7薄膜, 组装成的太阳能电池具有光、热和湿度的长期稳定性. Ramachandrana等[17]通过溶解碘化银和碘化铋后真空退火制备碘化银铋薄膜, X射线衍射分析表明薄膜结构为Ag2BiI5, 具有1.71 eV的带隙, 使用其组装成的可见光探测器表现出良好的响应性、灵敏度和入射光强度依赖性. Yi等[19]报道了一种用于太阳能电池的新型Bi基类钙钛矿材料AgBi3I10, 其晶体结构由AgI6和BiI6八面体构成, 研究表明其光学带隙为1.80 eV, 同时具有良好的空气稳定性.
综合考虑丰度和成本因素, CuBiI三元化合物更具有研究价值. 目前仅报道过Cu2BiI5和CuBiI4两种组分的结构, 光伏领域的研究仅局限于CuBiI4化合物[20-23]. Hu等[21]利用低温溶液法合成了均匀致密的CuBiI4薄膜, 带隙为2.67 eV, 利用其组装成的太阳能电池在光照条件下保持良好的稳定性. Zhang等[22]在室温下采用直接金属表面元素反应的方法合成了均匀致密的CuBiI4薄膜, 禁带宽度为1.81 eV. CuBiI三元化合物是否还存在着其他稳定组分结构, 其电学和光学性质, 以及光伏性能等均值得进一步研究.
本文采用基于遗传算法的晶体结构预测软件USPEX (universal structure predictor: evolutionary xtallography)对CuBiI三元化合物(CuBi2I7, Cu2BiI5, Cu2BiI7, Cu3BiI6, Cu3Bi2I9, CuBi3I10, Cu4BiI7)的晶体结构进行了系统的研究. 共预测了12种热力学、晶格动力学和弹性力学稳定的CuBiI三元化合物晶体结构. 同时计算了它们的能带结构, 发现不同晶体结构的带隙值存在明显差异(1.13—3.09 eV). 在可见光区域中, Cu2BiI5, Cu2BiI7, CuBi2I7和Cu4BiI7表现出极强的光吸收能力, 光吸收系数均接近4 × 105 cm–1, 超过了CH3NH3PbI3. 此外, CuBiI三元化合物晶体结构(Cu2BiI7-P1)的SLME值高达31.63%, 表明了其具有光电吸收材料的潜质.
所有的计算均采用基于密度泛函理论(density functional theory, DFT)的第一性原理从头算程序包(vienna ab-initio simulation package, VASP)来完成[26]. 电子交换相关能和离子势分别采用 Perdew-Burke-Ernzerhof (PBE)参数化的广义梯度近似(generalized gradient approximation, GGA)[27]和投影缀加波(projected augmented wave, PAW)方法[28]来描述. 平面波截断能量设为600 eV. K点网格使用以G点为中心的Monkhorst-Pack网格方法[29], 网格间距为2π × 0.03 ?–1. 收敛标准设为总能量小于1 × 10–8 eV, 每个原子受力小于0.001 eV/?.
利用基于密度泛函微扰理论(density functional perturbation theory, DFPT)的PHONOPY程序计算CuBiI三元化合物的声子谱, 分析其动力学稳定性[30]. 根据应力-应变的方法计算弹性常数矩阵, 验证CuBiI三元化合物的力学稳定性[31,32]. 同时采用更加精确的杂化泛函(heyd-scuseria-ernzerhof, HSE)方法[33,34]计算电子结构、光吸收谱、光转换效率.
3.1.结构预测
采用机器学习方法, 以形成焓为标准, 基于稳定存在的BiI3, CuI, CuI2以及AgBiI三元化合物的组分比设计了7种CuBiI三元化合物, 分别为CuBi2I7, Cu2BiI5, Cu3BiI6, Cu4BiI7, Cu3Bi2I9, CuBi3I10, Cu2BiI7. 其中, Cu2BiI7的Cu离子为+2价, 其他CuBiI化合物的Cu离子为+1价. 然后以焓值为适应度函数, 基于“能量越低, 结构越稳定”的原则, 对这7种CuBiI三元化合物进行定成分晶体结构预测, 从预测结构中选择稳定和亚稳结构(亚稳结构与稳定结构之间焓值差在100 meV/atom以内).进一步计算了CuBi2I7, Cu2BiI5, Cu3BiI6, Cu4BiI7, Cu3Bi2I9, CuBi3I10, Cu2BiI7的稳定以及亚稳结构的声子色散谱, 确定了12个动力学稳定的晶体结构. 结合成分比以及对称性对12个动力学稳定结构进行命名, 分别为CuBi2I7-P1, CuBi2I7-P1-II, Cu2BiI5-P1, Cu2BiI5-Cm, Cu3BiI6-P3, Cu3BiI6-R3, Cu4BiI7-P3, Cu4BiI7-P1, Cu3Bi2I9-P1, CuBi3I10-P1, Cu2BiI7-P1, Cu2BiI7-P1-II. 图1给出了12个动力学稳定结构的声子谱, 在整个布里渊区范围内, 12个结构的声子谱均没有出现虚频, 声学和光学分支之间没有明显的间隙, 存在重叠部分, 可以有效增强声学支光学支的散射.
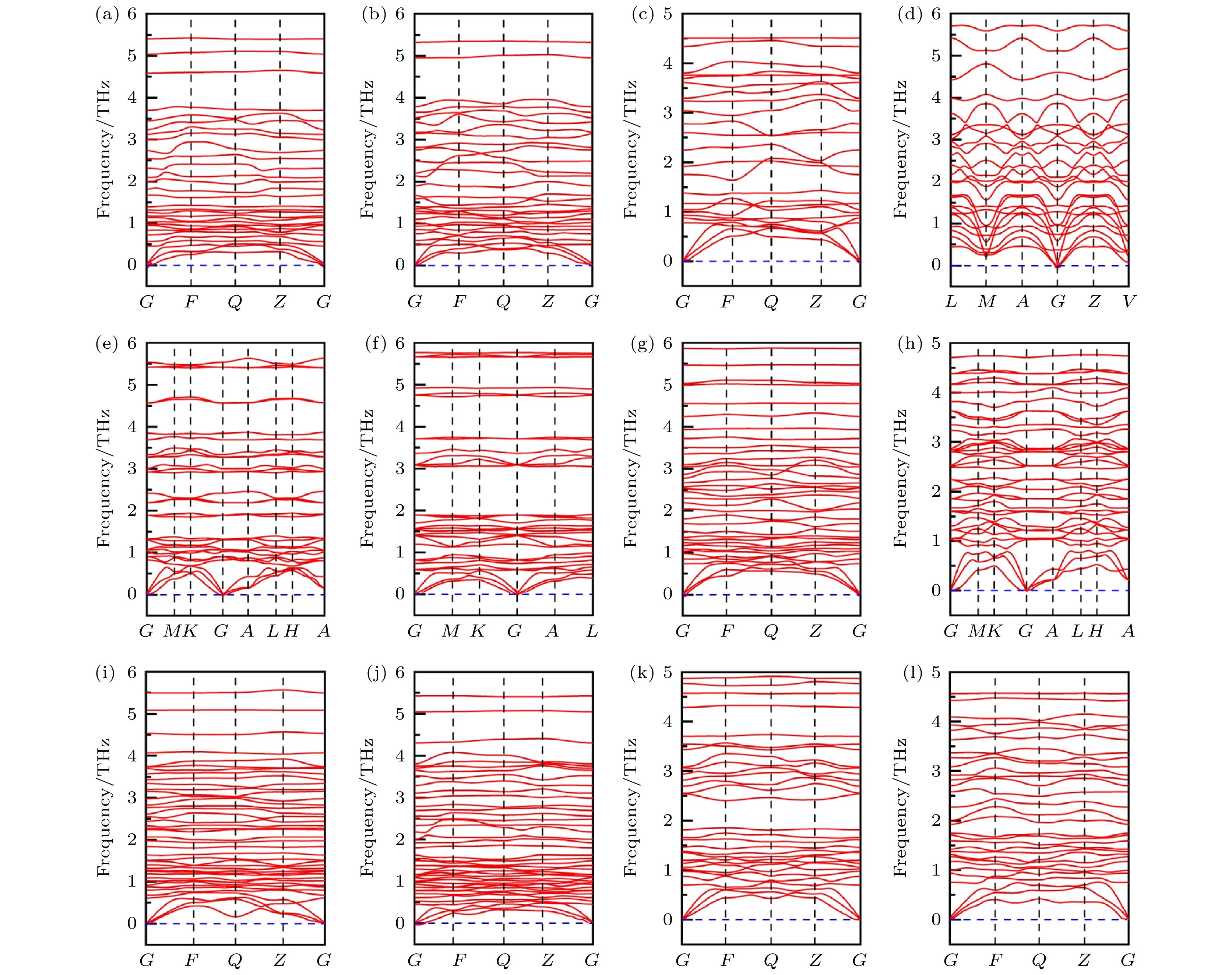 图 1 12个CuBiI三元化合物结构的声子色散谱图 (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II
图 1 12个CuBiI三元化合物结构的声子色散谱图 (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-IIFigure1. Phonon dispersion spectra for the 12 structures of CuBiI ternary compound: (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II.
表1给出了12个稳定结构的空间群、晶胞内原子数、体积以及形成能. 12种结构分别属于三斜晶系(CuBi2I7-P1, CuBi2I7-P1-II, Cu2BiI5-P1, Cu4BiI7-P1, Cu3Bi2I9-P1, CuBi3I10-P1, Cu2BiI7-P1, Cu2BiI7-P1-II)、单斜晶系(Cu2BiI5-Cm)和三方晶系(Cu3BiI6-P3, Cu3BiI6-R3, Cu4BiI7-P3). 为了判定这些结构的热力学稳定性, 按照以下公式计算了其形成能:
| Structure name | Space group | Number of/ (atoms·unit cell–1) | Volume/ (?3·unit cell–1) | ${{E} }_{\rm{form} }$/ (eV·atoms–1) | Structure name | Space group | Number of/ (atoms·unit cell–1) | Volume/ (?3·unit cell–1) | ${{E} }_{\rm{form} }$/ (eV·atoms–1) | |
| CuBi2I7-P1 | P1 | 10 | 474.24 | –0.362 | CuBi2I7-P1-II | P1 | 10 | 465.35 | –0.385 | |
| Cu2BiI5-P1 | P1 | 8 | 295.03 | –0.287 | Cu2BiI5-Cm | Cm | 16 | 742.54 | –0.290 | |
| Cu3BiI6-P3 | P3 | 10 | 404.63 | –0.265 | Cu3BiI6-R3 | R3 | 30 | 1318.62 | –0.244 | |
| Cu4BiI7-P1 | P1 | 12 | 428.29 | –0.237 | Cu4BiI7-P3 | P3 | 12 | 451.33 | –0.231 | |
| Cu3Bi2I9-P1 | P1 | 14 | 645.41 | –0.294 | CuBi3I10-P1 | P1 | 14 | 691.08 | –0.402 | |
| Cu2BiI7-P1 | P1 | 10 | 420.79 | –0.225 | Cu2BiI7-P1-II | P1 | 10 | 420.68 | –0.226 |
表112个CuBiI三元化合物结构的结构名称、空间群、晶胞内原子数、体积及形成能
Table1.Structure name, space group, number of atoms per unit cell, volume of the unit cell and formation energy for the 12 structures of CuBiI ternary compound.





为了进一步判定12个CuBiI三元化合物结构的力学稳定性, 计算了这些结构的弹性常数. 对于不同的晶系, 力学稳定性判断条件不同. 单斜结构包含13个独立的弹性常数, 三方结构存在8个独立的弹性常数, 可通过Born-Huang稳定性公式[31]来判定力学稳定性. 三斜结构具有21个独立的弹性常数, 由Born-Huang稳定性准则可知, 需要弹性矩阵的正定型来保证力学稳定性[32]. 12个结构的弹性系数Cij列于表2, 均满足于Born-Huang稳定性准则, 表明了12个结构的力学稳定性.
| Cij/GPa | CuBi2I7-P1 | CuBi2I7-P1-II | Cu2BiI5-P1 | Cu2BiI5-Cm | Cu3BiI6-P3 | Cu3BiI6-R3 | Cu4BiI7-P1 | Cu4BiI7-P3 | Cu3Bi2I9-P1 | CuBi3I10-P1 | Cu2BiI7-P1 | Cu2BiI7-P1-II | |
| C11 | 8.34 | 9.03 | 39.90 | 4.76 | 11.99 | 17.59 | 23.63 | 32.25 | 17.16 | 2.82 | 9.71 | 3.12 | |
| C22 | 12.61 | 14.16 | 29.93 | 35.09 | — | — | 18.34 | — | 20.42 | 9.34 | 14.12 | 10.34 | |
| C33 | 8.35 | 9.00 | 35.71 | 5.64 | 5.92 | 5.05 | 23.61 | 8.90 | 11.86 | 8.62 | 14.43 | 26.51 | |
| C44 | 3.52 | 3.62 | 10.18 | 1.73 | 1.01 | 3.53 | 6.87 | 1.41 | 3.91 | 3.27 | 6.46 | 7.16 | |
| C55 | 3.74 | 2.99 | 9.96 | — | — | — | 7.83 | — | 3.56 | 1.91 | 3.73 | 4.52 | |
| C66 | 2.41 | 4.43 | 6.77 | 1.24 | 4.42 | 6.13 | 8.72 | 11.26 | 6.01 | 1.93 | 3.13 | 3.18 | |
| C12 | 4.73 | 4.45 | 9.13 | 1.63 | 3.01 | 5.22 | 4.56 | 9.71 | 6.13 | 1.96 | 4.71 | 3.19 | |
| C13 | 2.61 | 2.57 | 14.13 | 2.77 | 1.33 | 2.99 | 7.76 | 3.30 | 3.14 | 2.11 | 5.61 | 6.45 | |
| C14 | –2.07 | –0.04 | 4.02 | — | 0.04 | 1.7 | –1.84 | 0.28 | 0.51 | –0.63 | –0.17 | –0.32 | |
| C15 | 0.18 | 0.21 | 0.18 | –0.67 | –0.15 | –0.38 | 2.85 | 0.06 | –0.79 | –0.21 | –2.11 | 0.54 | |
| C16 | 0.79 | 2.27 | 0.12 | — | — | — | –0.27 | — | –0.51 | 0.86 | –1.73 | –0.32 | |
| C23 | 2.69 | 2.76 | 14.94 | 2.42 | — | — | 5.49 | — | 6.79 | 2.98 | 7.51 | 7.18 | |
| C24 | –2.53 | 0.12 | 5.64 | — | — | — | –0.99 | — | 2.280 | –0.05 | –2.43 | 2.11 | |
| C25 | 0.28 | 0.16 | 0.17 | –0.12 | — | — | 2.68 | — | 0.49 | –0.09 | –3.12 | 1.54 | |
| C26 | 0.49 | 1.72 | –0.02 | — | — | — | 0.04 | — | 0.41 | 1.47 | 1.35 | 1.01 | |
| C34 | –1.69 | –0.27 | 5.74 | — | — | — | –0.56 | — | 2.53 | –0.89 | –1.51 | 0.36 | |
| C35 | –1.76 | 0.39 | 0.12 | –0.90 | — | — | 3.43 | — | –0.21 | –2.38 | –3.43 | 1.76 | |
| C36 | 1.47 | 1.17 | –0.04 | — | — | — | 1.72 | — | 0.80 | 0.81 | 0.13 | –0.48 | |
| C45 | –0.41 | 0.83 | 0.13 | — | — | — | –0.47 | — | 0.21 | 0.85 | 1.26 | 0.19 | |
| C46 | –0.10 | 0.42 | 0.01 | 0.12 | — | — | 1.75 | — | 0.34 | –0.62 | –2.13 | –0.69 | |
| C56 | –0.58 | –0.29 | 1.485 | — | — | — | –1.07 | — | 0.94 | –0.53 | 0.82 | 0.72 |
表212个CuBiI三元化合物结构的弹性系数(Cij)
Table2.Calculated elastic constants for the 12 structures of CuBiI ternary compound.
2
3.2.晶体结构
12个CuBiI三元化合物结构的主要特征在于, 存在Cu—I和Bi—I键以及I原子的配位多面体. 表3总结了12个结构的晶格常数以及Cu/Bi—I键长. 下面对12个晶体结构进行详细的描述.| Structure name | a/? | b/? | c/? | α/(°) | β/(°) | γ/(°) | Cu—I/? | Bi—I/? |
| CuBi2I7-P1 | 7.93 | 7.94 | 7.92 | 97.67 | 82.58 | 76.98 | 2.53—2.55 | 3.02—3.32 |
| CuBi2I7-P1-II | 8.05 | 7.85 | 7.75 | 97.64 | 100.86 | 100.78 | 2.54—2.55 | 3.03—3.22 |
| Cu2BiI5-P1 | 4.42 | 7.62 | 9.57 | 95.94 | 103.35 | 106.82 | 2.59—2.67 | 3.09—3.18 |
| Cu2BiI5-Cm | 16.64 | 4.33 | 12.22 | 90.00 | 122.51 | 90.00 | 2.57—2.72 | 2.84—3.50 |
| Cu3BiI6-P3 | 7.89 | 7.89 | 7.54 | 90.00 | 90.00 | 120.00 | 2.54—2.61 | 3.02—3.29 |
| Cu3BiI6-R3 | 11.40 | 11.40 | 11.72 | 90.00 | 90.00 | 120.00 | 2.52—2.56 | 3.05—3.35 |
| Cu4BiI7-P1 | 7.61 | 7.79 | 7.64 | 101.68 | 100.50 | 98.22 | 2.56—2.74 | 3.06—3.22 |
| Cu4BiI7-P3 | 8.32 | 8.32 | 7.52 | 90.00 | 90.00 | 120.00 | 2.64—2.68 | 3.09—3.22 |
| Cu3Bi2I9-P1 | 7.67 | 8.59 | 9.85 | 84.58 | 88.98 | 86.74 | 2.55—2.70 | 2.99—3.28 |
| CuBi3I10-P1 | 9.46 | 10.12 | 7.85 | 103.09 | 106.70 | 77.25 | 2.53—2.54 | 2.99—3.32 |
| Cu2BiI7-P1 | 7.33 | 7.90 | 7.92 | 104.09 | 108.49 | 81.74 | 2.59—2.64 | 3.05—3.34 |
| Cu2BiI7-P1-II | 9.00 | 7.78 | 7.20 | 109.91 | 89.24 | 64.61 | 2.58—2.70 | 2.98—3.34 |
表312个CuBiI三元化合物结构的晶格常数以及Cu/Bi—I键长
Table3.Lattice constants and Cu/Bi—I bond length for the 12 structures of CuBiI ternary compound.
图2给出了CuBi2I7 (CuBi2I7-P1, CuBi2I7-P1-II)晶体结构的主视图和俯视图, 两个结构均属于三斜晶系. CuBi2I7-P1结构沿着[010]方向, 配位八面体BiI6通过共享边的形式形成了八面体BiI6链; 在[100]方向, BiI6链彼此之间通过共享点连接成重复单元; 在[001]方向, 重复单元通过片状CuI3共享边或者共享点构成三维空间结构. CuBi2I7-P1-II结构在[010]方向与CuBi2I7-P1相似, 配位八面体BiI6通过共享边形成BiI6链; 在[100]方向, BiI6链之间通过共享点的方式连接成空间结构, 并且片状结构CuI3通过共享边和共享点的方式依附在BiI6链上.
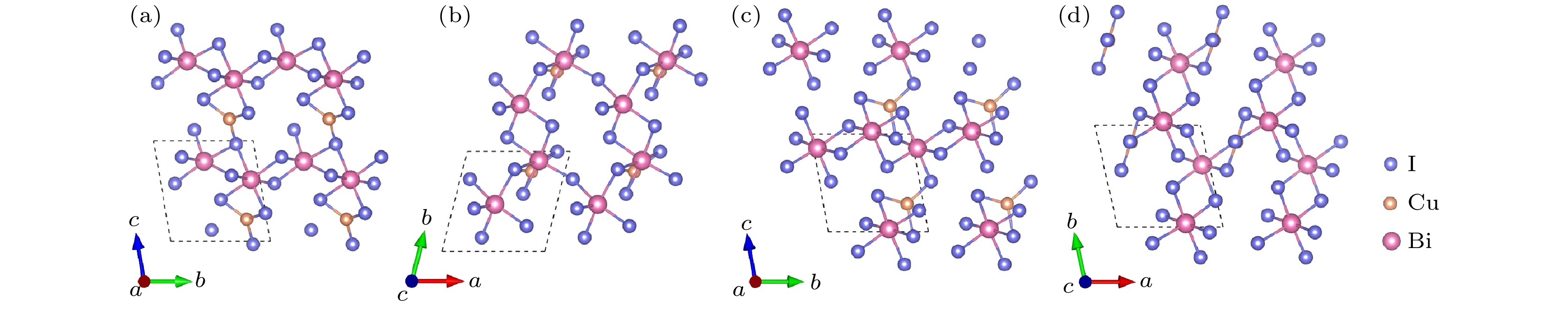 图 2 CuBi2I7-P1的晶体结构 (a) 主视图; (b) 俯视图. CuBi2I7-P1-II的晶体结构 (c) 主视图; (d) 俯视图
图 2 CuBi2I7-P1的晶体结构 (a) 主视图; (b) 俯视图. CuBi2I7-P1-II的晶体结构 (c) 主视图; (d) 俯视图Figure2. Crystal structure of CuBi2I7-P1: (a) Front view; (b) top view. Crystal structure of CuBi2I7-P1-II: (c) Front view; (d) top view
图3给出了Cu2BiI5 (Cu2BiI5-P1, Cu2BiI5-Cm)晶体结构的主视图和俯视图. Cu2BiI5-P1结构属于三斜晶系, 在[010]方向, 配位四面体CuI4通过共享点的方式形成CuI4链; 在[001]方向, 八面体BiI6以共享点的方式连接在CuI4链上; 在[100]方向, 配位四面体CuI4彼此之间通过共享点连接, 同样八面体BiI6以共享边的方式连接. Cu2BiI5-Cm结构属于单斜晶系, 该结构中Cu原子存在两种配位环境: 四面体CuI4与片状结构CuI3. 在[010]方向, 配位八面体BiI6以共享边的方式连接, 四面体CuI4和片状结构CuI3依次通过共享边的方式交替连接, [001]方向, 四面体CuI4与八面体BiI6以共享边的方式连接成重复单元.
 图 3 Cu2BiI5-P1的晶体结构 (a) 主视图; (b) 俯视图. Cu2BiI5-Cm的晶体结构 (c) 主视图; (d) 俯视图
图 3 Cu2BiI5-P1的晶体结构 (a) 主视图; (b) 俯视图. Cu2BiI5-Cm的晶体结构 (c) 主视图; (d) 俯视图Figure3. Crystal structure of Cu2BiI5-P1: (a) Front view; (b) top view. Crystal structure of Cu2BiI5-Cm: (c) Front view; (d) top view.
图4给出了Cu3BiI6 (Cu3BiI6-P3, Cu3BiI6-R3)晶体结构的主视图和俯视图, 两个结构均属于三方晶系. Cu3BiI6-P3为[001]方向的层状结构, 每层由配位八面体BiI6与片状结构CuI3构成: Bi原子位于顶点位置, 与周围的六个I原子形成配位八面体BiI6, Cu原子与周围三个I原子形成片状结构CuI3, 每个八面体BiI6与周围三个片状结构通过共享边构成重复单元, 并依次通过共享点的形式连接成层状结构. Cu3BiI6-R3结构是由八面体BiI6与片状结构CuI3构成的三维结构: 八面体BiI6与片状结构CuI3通过共享边连接成重复单元, 该重复单元在[010]或者[001]方向, 均通过八面体BiI6与三个片状结构CuI3的共享点构成三维空间结构.
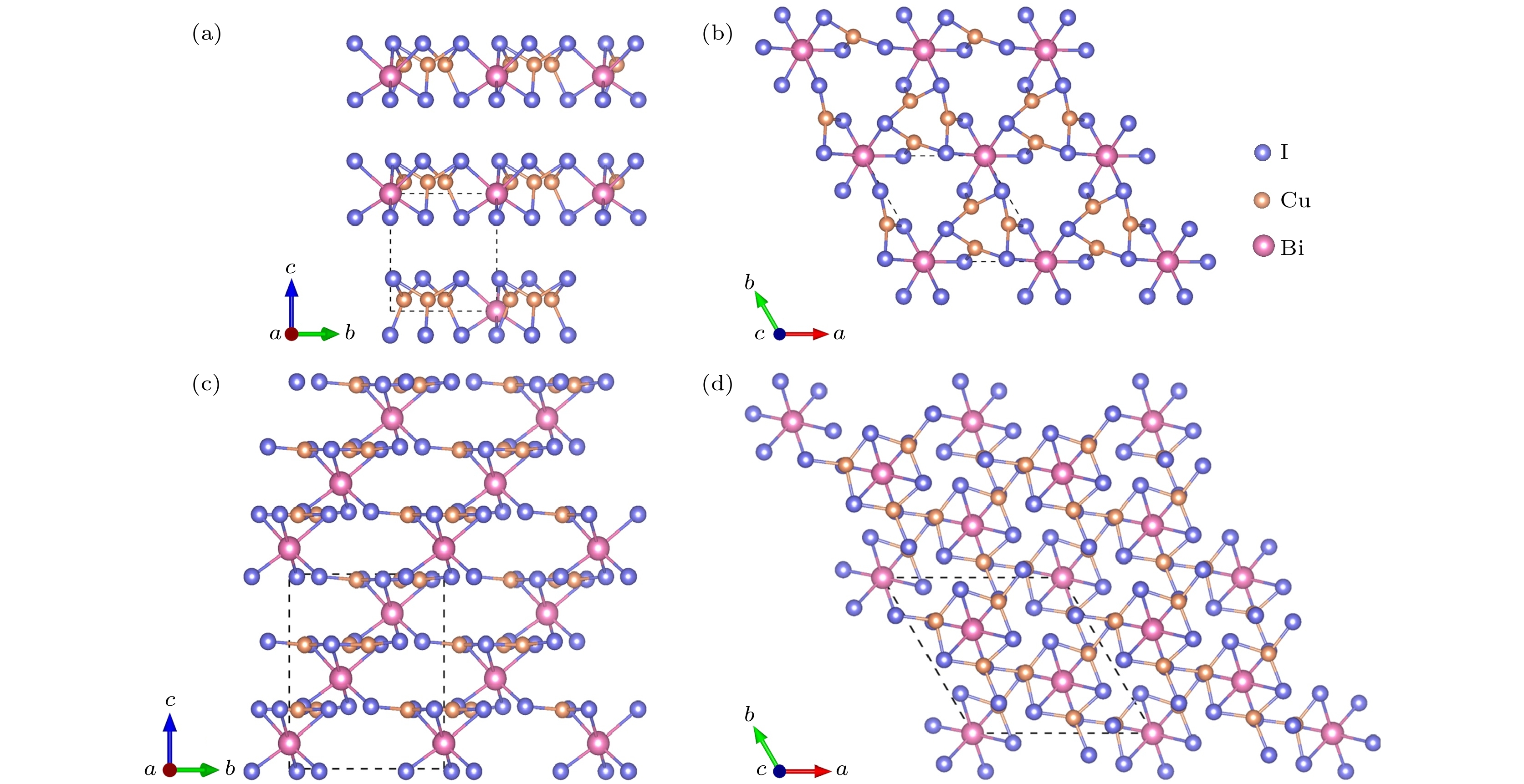 图 4 Cu3BiI6-P3的晶体结构 (a) 主视图; (b) 俯视图. Cu3BiI6-R3的晶体结构 (c) 主视图; (d) 俯视图
图 4 Cu3BiI6-P3的晶体结构 (a) 主视图; (b) 俯视图. Cu3BiI6-R3的晶体结构 (c) 主视图; (d) 俯视图Figure4. Crystal structure of Cu3BiI6-P3: (a) Front view; (b) top view. Crystal structure of Cu3BiI6-R3: (c) Front view; (d) top view.
图5给出了Cu4BiI7 (Cu4BiI7-P1, Cu4BiI7-P3)晶体结构的主视图和俯视图. Cu4BiI7-P1结构属于三斜晶系, 该结构中的Cu原子存在两种配位环境: 四面体CuI4与片状结构CuI3. 两个八面体BiI6之间通过两组共享边的片状结构CuI3与四面体CuI4相连成空间结构. Cu4BiI7-P3结构属于三方晶系, 该结构为[001]方向的层状结构, 每层由配位八面体BiI6与配位四面体CuI4构成: [100]方向或者[010]方向, 原子排列方式相同: 两个八面体BiI6之间均由两个共享边的四面体CuI4通过共享点或者共享边相连成层状材料.
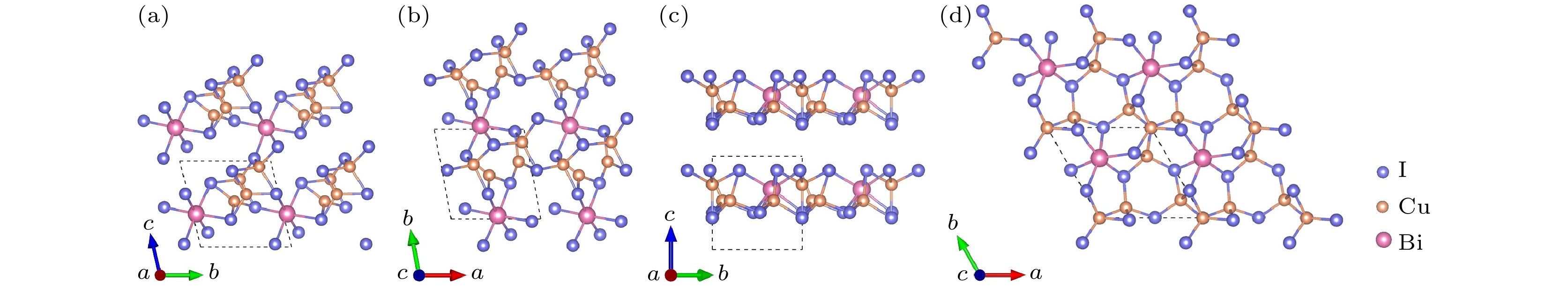 图 5 Cu4BiI7-P1的晶体结构 (a) 主视图; (b) 俯视图. Cu4BiI7-P3的晶体结构 (c) 主视图; (d) 俯视图
图 5 Cu4BiI7-P1的晶体结构 (a) 主视图; (b) 俯视图. Cu4BiI7-P3的晶体结构 (c) 主视图; (d) 俯视图Figure5. Crystal structure of Cu4BiI7-P1: (a) Front view; (b) top view. Crystal structure of Cu4BiI7-P3: (c) Front view; (d) top view.
图6给出了Cu3Bi2I9-P1和 CuBi3I10-P1晶体结构的主视图和俯视图. Cu3Bi2I9-P1与CuBi3I10-P1结构均属于三斜晶系. 对于Cu3Bi2I9-P1结构, 在[100]方向, 八面体BiI6彼此通过共享边形成BiI6链, 四面体CuI4通过共享点或边的方式连接在BiI6链上, 形成重复的单元; 在[001]方向, 该重复单元通过四面体CuI4之间的点共享构成的三维空间结构. CuBi3I10-P1结构为[100]方向的层状结构, 每层由配位八面体BiI6与片状CuI3结构构成: 在[010]或[001]方向, 八面体BiI6均通过共享边的方式连接, 而片状CuI3结构以共享边或共享点的方式嵌入八面体BiI6堆垛的间隙中, 以此构成了层状结构.
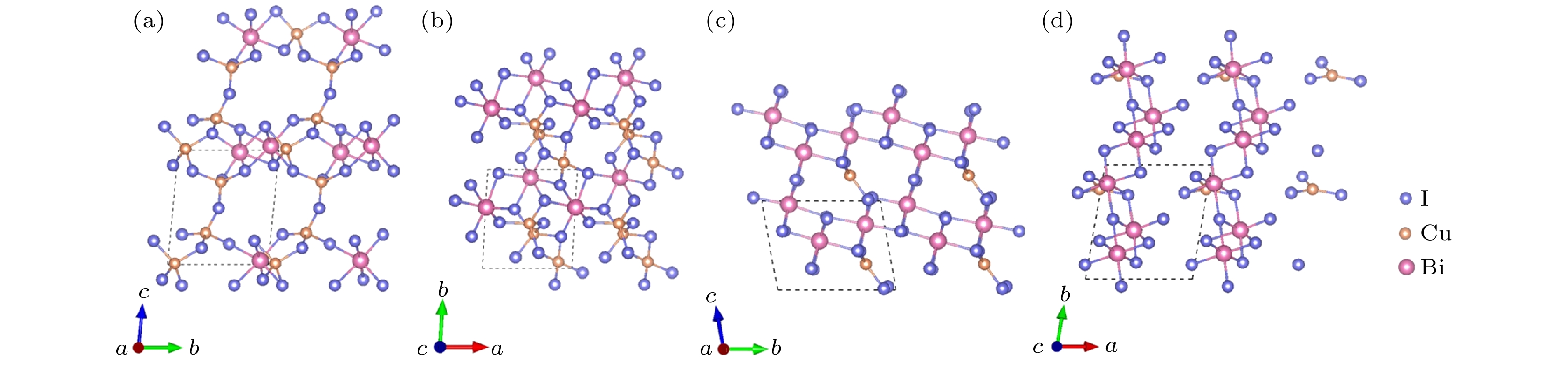 图 6 Cu3Bi2I9-P1的晶体结构 (a) 主视图; (b) 俯视图. CuBi3I10-P1的晶体结构 (c) 主视图; (d) 俯视图
图 6 Cu3Bi2I9-P1的晶体结构 (a) 主视图; (b) 俯视图. CuBi3I10-P1的晶体结构 (c) 主视图; (d) 俯视图Figure6. Crystal structure of Cu3Bi2I9-P1: (a) Front view; (b) top view. Crystal structure of CuBi3I10-P1: (c) Front view; (d) top view
图7给出Cu2BiI7 (Cu2BiI7-P1, Cu2BiI7-P1-II)晶体结构主视图和俯视图. Cu2BiI7-P1与Cu2BiI7-P1-II结构均属于三斜晶系, Cu2BiI7-P1结构为[100]方向的层状结构, 每层由配位八面体BiI6与配位四面体CuI4构成: 在[001]方向或[010]方向, 八面体BiI6与四面体CuI4通过共享边或共享点的方式交替相连成层状结构, 且四面体CuI4之间相互共享边. Cu2BiI7-P1-II与Cu2BiI7-P1相似, 也为[100]方向的层状结构, 不同之处在于[010]方向, 八面体BiI6与四面体CuI4之间仅通过共享边的方式连接, 且四面体CuI4之间不存在边共享.
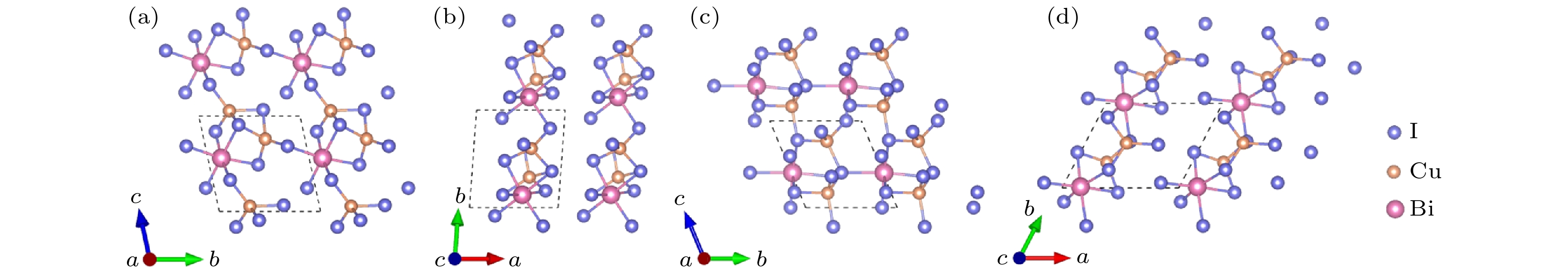 图 7 Cu2BiI7-P1的晶体结构 (a) 主视图; (b) 俯视图. Cu2BiI7-P1-II的晶体结构 (c) 主视图; (d) 俯视图
图 7 Cu2BiI7-P1的晶体结构 (a) 主视图; (b) 俯视图. Cu2BiI7-P1-II的晶体结构 (c) 主视图; (d) 俯视图Figure7. Crystal structure of Cu2BiI7-P1: (a) Front view; (b) top view. Crystal structure of Cu2BiI7-P1-II: (c) Front view; (d) top view
2
3.3.电子结构
分别采用PBE和HSE06计算了12个CuBiI三元化合物结构的能带结构, 如图8所示. 带隙值以及导带底和价带顶位置如表4所列. 与PBE计算值相比, HSE06计算值更大. CuBi2I7-P1, Cu2BiI5-Cm, Cu3Bi2I9-P1和CuBi3I10-P1结构为直接带隙半导体, 有利于光子的垂直跃迁. CuBi2I7-P1与Cu2BiI5-Cm价带顶(valence band maximum, VBM)和导带底(conduction band minimum, CBM)均在G (0 0 0)点, Cu3Bi2I9-P1的VBM和CBM在F (0 0.5 0)点, CuBi3I10-P1的VBM和CBM在Z (0 0 0.5)点. CuBiI三元化合物其他结构均为间接带隙半导体. 12个结构带隙值为1.13—3.09 eV. 其中, Cu2BiI5-P1 (1.56 eV), Cu2BiI7-P1 (1.13 eV)及Cu2BiI7-P1-II (1.40 eV)的带隙值相对较小, 接近于光吸收的最佳带隙值(1.40 eV), 适合作为太阳能电池中的光吸收材料. Cu3BiI6-R3 (2.81 eV)与Cu3BiI6-P3 (3.09 eV)的带隙值偏大, 接近于紫外光能量, 不利于可见光吸收. 其他结构的带隙均在可见光能量范围(1.64—3.19 eV)内, 且具有较宽的可见光吸收谱, 具备优良太阳能吸收材料潜质.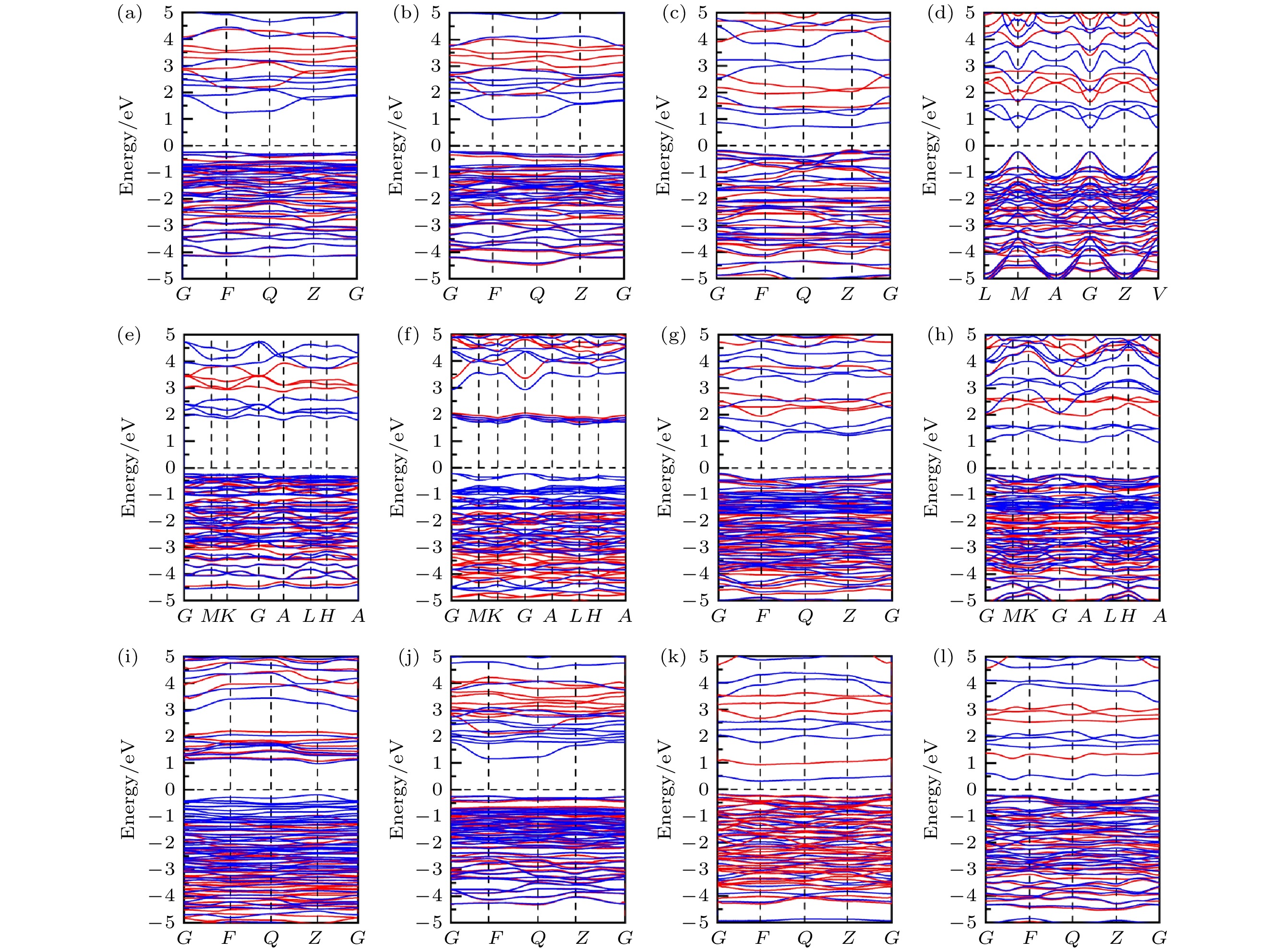 图 8 12个CuBiI三元化合物结构的能带结构图 (红色, HSE06方法计算结果; 蓝色, PBE方法计算结果) (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II
图 8 12个CuBiI三元化合物结构的能带结构图 (红色, HSE06方法计算结果; 蓝色, PBE方法计算结果) (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-IIFigure8. Band structure for the 12 structures of CuBiI ternary compound calculated by the PBE (blue lines) and HSE06 (red lines) methods: (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II.
| Structure name | Eg/eV | VBM | CBM | Bader charge | SLME/% | |||
| HSE06 | PBE | Cu/(e·atom–1) | Bi/(e·atom–1) | I/(e·atom–1) | ||||
| CuBi2I7-P1 | 2.39 | 1.48 | 0 0 0 | 0 0 0 | 0.33 | 1.08 | –0.36 | 10.75 |
| CuBi2I7-P1-II | 2.13 | 1.21 | 0 0 0 | 0 0.5 0 | 0.33 | 1.09 | –0.36 | 9.50 |
| Cu2BiI5-P1 | 1.56 | 0.84 | 0 0 0.5 | 0 0.5 0 | 0.34 | 1.04 | –0.34 | 22.20 |
| Cu2BiI5-Cm | 1.87 | 0.89 | 0 0 0 | 0 0 0 | 0.29 | 1.07 | –0.33 | 7.50 |
| Cu3BiI6-P3 | 3.09 | 1.97 | 0.05 0 0 | 0 0 0.5 | 0.29 | 1.08 | –0.33 | 2.86 |
| Cu3BiI6-R3 | 2.81 | 1.85 | 0 0 0 | 0.5 0 0.5 | 0.31 | 1.01 | –0.32 | 5.49 |
| Cu4BiI7-P1 | 2.19 | 1.22 | 0 0 0 | 0 0.5 0 | 0.30 | 1.03 | –0.32 | 15.77 |
| Cu4BiI7-P3 | 2.21 | 1.21 | 0 0 0.06 | 0 0 0.5 | 0.32 | 1.06 | –0.33 | 13.61 |
| Cu3Bi2I9-P1 | 2.03 | 1.17 | 0 0 0.5 | 0 0 0.5 | 0.34 | 1.02 | –0.34 | 19.02 |
| CuBi3I10-P1 | 2.36 | 1.41 | 0 0.5 0 | 0 0.5 0 | 0.33 | 1.09 | –0.36 | 4.17 |
| Cu2BiI7-P1 | 1.13 | 0.50 | 0 0 0 | 0 0.5 0 | 0.37 | 1.09 | –0.26 | 31.63 |
| Cu2BiI7-P1-II | 1.40 | 0.60 | 0 0 0 | 0 0.5 0.5 | 0.35 | 1.06 | –0.25 | 28.30 |
表412个CuBiI三元化合物结构的带隙值(HSE06和PBE方法计算结果), 价带顶与导带底位置, Bader电荷转移以及SLME (spectroscopic limited maximum efficiency)值
Table4.Band gaps (Eg) calculated by the HSE06 and PBE method, positions of VBM and CBM, Bader charge and the spectroscopic limited maximum efficiency (SLME) values for the 12 structures of CuBiI ternary compound.
图9给出了CuBiI三元化合物12个结构的总态密度(total density of states, TDOS), 投影态密度(projected density of states, PDOS)和价带顶、导带底的电荷密度分布图. 12个结构的价带顶均由Cu-3d和I-5p轨道杂化而成. Cu3BiI6-R3的导带底主要由Bi-6p和I-5p轨道组成, Cu-3d有少许贡献; Cu2BiI7的两个结构(Cu2BiI7-P1, Cu2BiI7-P1-II)的导带底主要来自I-5p轨道, Cu-3d有少许贡献. 对于Cu2BiI7-P1结构, 在2.7—4.0 eV范围内Bi-6p和I-5轨道杂化. Cu2BiI7-P1-II结构在2.5—3.2 eV范围内, 也存在Bi-6p和I-5p轨道杂化; 其他结构的导带底均由Bi-6p和I-5p轨道杂化而成.
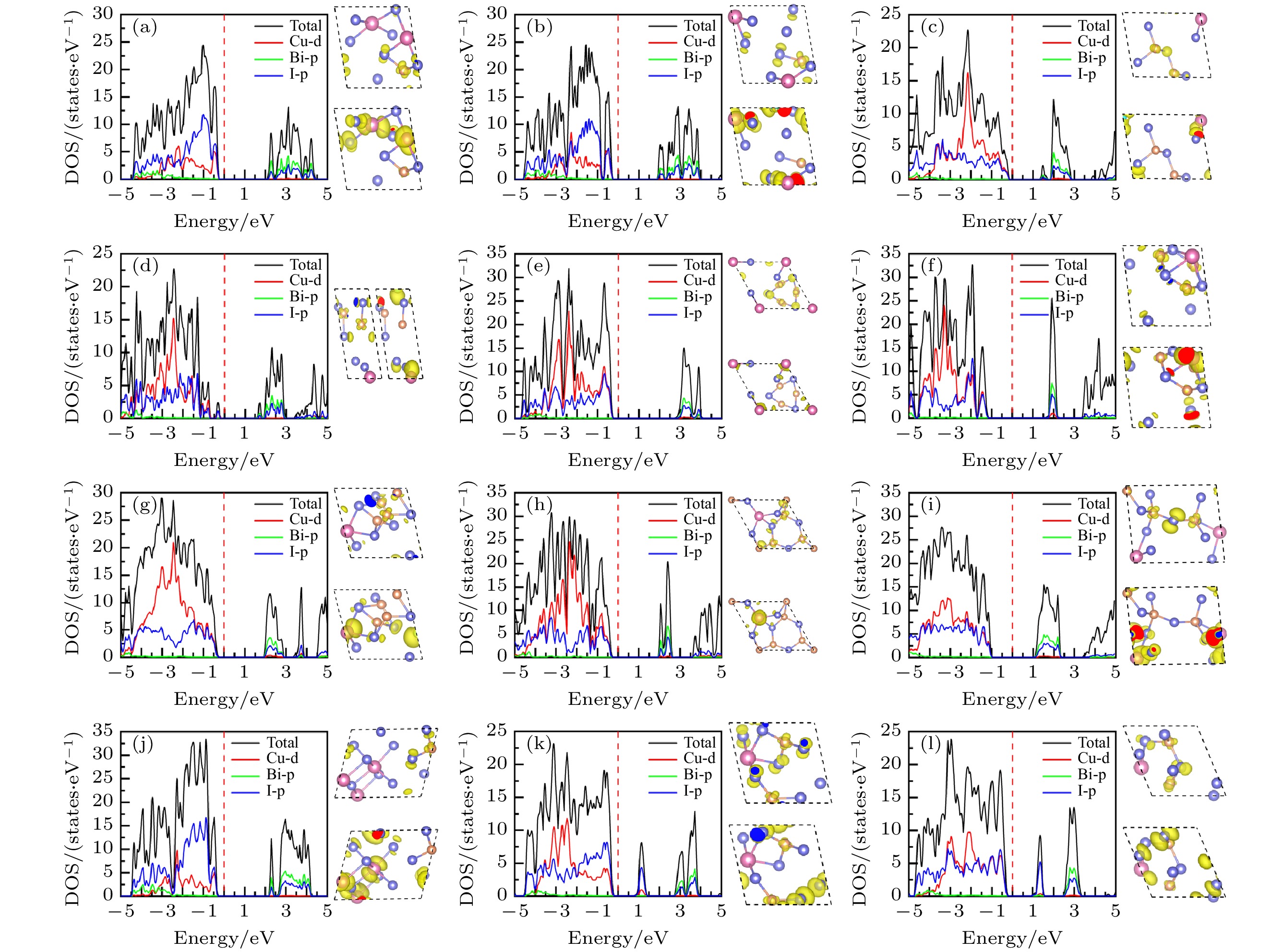 图 9 12个CuBiI三元化合物结构的总态密度、投影态密度图以及价带顶、导带底(从左到右或从上到下)的电荷密度分布图 (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II
图 9 12个CuBiI三元化合物结构的总态密度、投影态密度图以及价带顶、导带底(从左到右或从上到下)的电荷密度分布图 (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-IIFigure9. Total density of state (TDOS), projection density of state (PDOS) and charge density distribution (Left to right or top to bottom) at CBM and VBM for the 12 structures of CuBiI ternary compound: (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II.
为了进一步分析化学键, 计算了CuBiI三元化合物12个结构的电子局域函数(electron localization function, ELF). 图10给出了CuBiI三元化合物12种结构的电子局域函数分布图. I原子核的ELF值接近于1, 电子在此处高度局域; Bi原子附近的ELF值约为0.50, 电子均匀分布; Cu原子核附近的ELF值约为0.25, 电子离域分布. 值得注意的是, Bi原子和I原子之间的ELF数值约为0.28, 而Cu原子和I原子之间的ELF约为0.22, 共价属性相对较低.
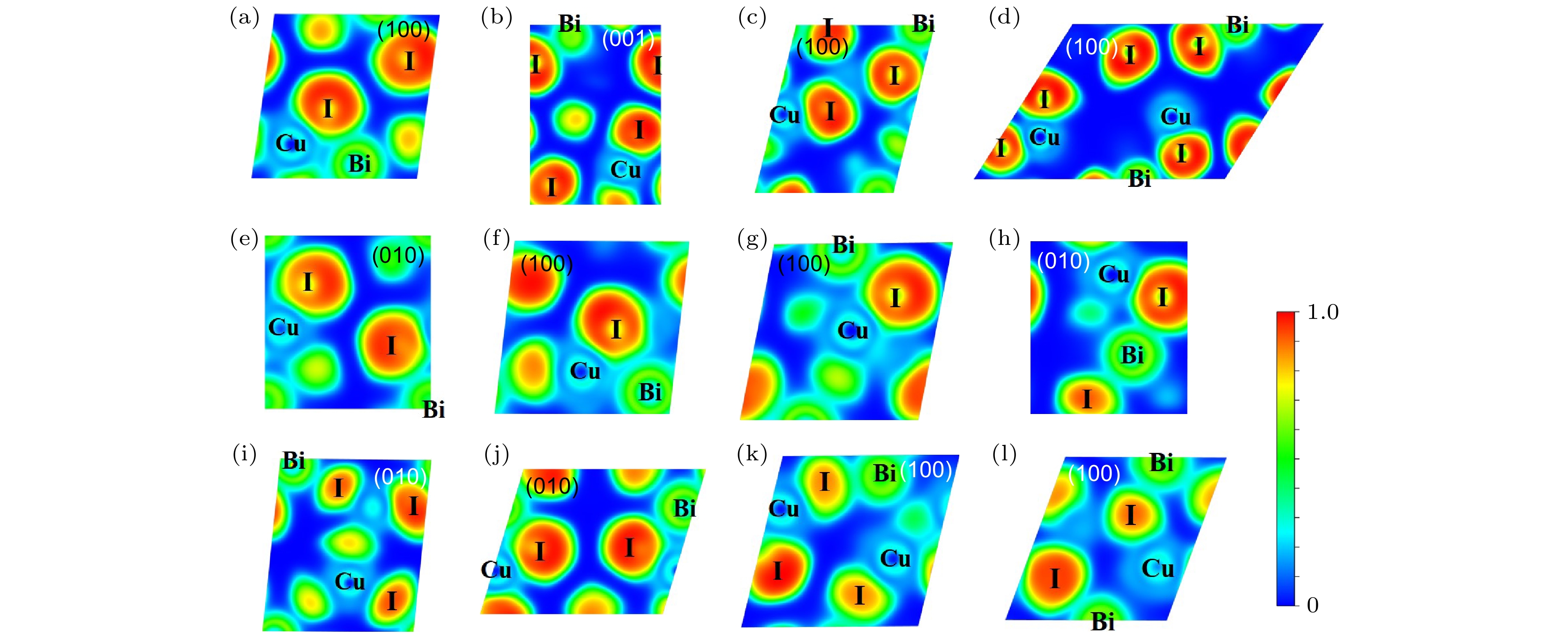 图 10 12个CuBiI三元化合物结构的电子局域函数分布图 (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II
图 10 12个CuBiI三元化合物结构的电子局域函数分布图 (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-IIFigure10. Electron localization function (ELF) for the 12 structures of CuBiI ternary compound: (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P3; (h) Cu4BiI7-P1; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II.
进一步计算了 Bader 电荷以分析Cu, Bi原子与I原子之间的电子转移情况, 计算结果见表4. Cu原子和Bi原子分别失去0.29e—0.37e和1.03e—1.09e电子, I原子获得0.25e—0.36e电子. Cu, Bi和I的Bader电荷与通常的纯离子键相互作用(例如Cu+/Cu2+, Bi3+, I–)偏离, 表明了Cu, Bi原子与I原子之间离子键和共价键相互作用共存. PDOS也证实了Cu/Bi—I的共价相互作用, Cu-3d/Bi-6p 轨道和 I-5p轨道存在着明显的耦合作用. 因此, CuBiI三元化合物12个结构中Cu—I, Bi—I键具有强的离子性, 弱的共价性.
2
3.4.光吸收性质
吸收系数α是反映光吸收强度的重要参数[35,36], 可通过以下公式得到:












为了更准确地描述CuBiI三元化合物12个结构的光学性质, 采用HSE06方法计算光吸收系数. 图11为CuBiI三元化合物中12个结构的光吸收谱. Cu2BiI7两个结构(Cu2BiI7-P1与Cu2BiI7-P1-II)的吸收起点位于红外光区, 吸收覆盖了整个可见光区. 相比之下, 其他结构的吸收光谱有显著的紫移, 吸收谱起点由其带隙值决定. 从吸收起点开始, CuBiI三元化合物中12个结构的光吸收强度均随着能量增加呈现上升的趋势. 在可见光能量范围内, 12种结构的吸收强度有着明显的差异. CuBi2I7-P1, Cu2BiI5-P1, Cu4BiI7-P1, Cu2BiI7-P1-II和Cu3Bi2I9-P1在可见光高能量区域的吸收系数接近于4 × 105 cm–1, 超过了CH3NH3PbI3[38]. 宽可见光范围内高吸收系数表明有能力捕获更多的光子, 产生电子和空穴对. 另外, Cu3BiI6-R3和Cu3BiI6-P3具有较宽的带隙值(2.81 eV和3.09 eV), 仅在可见光高能量处具有较高的吸光强度.
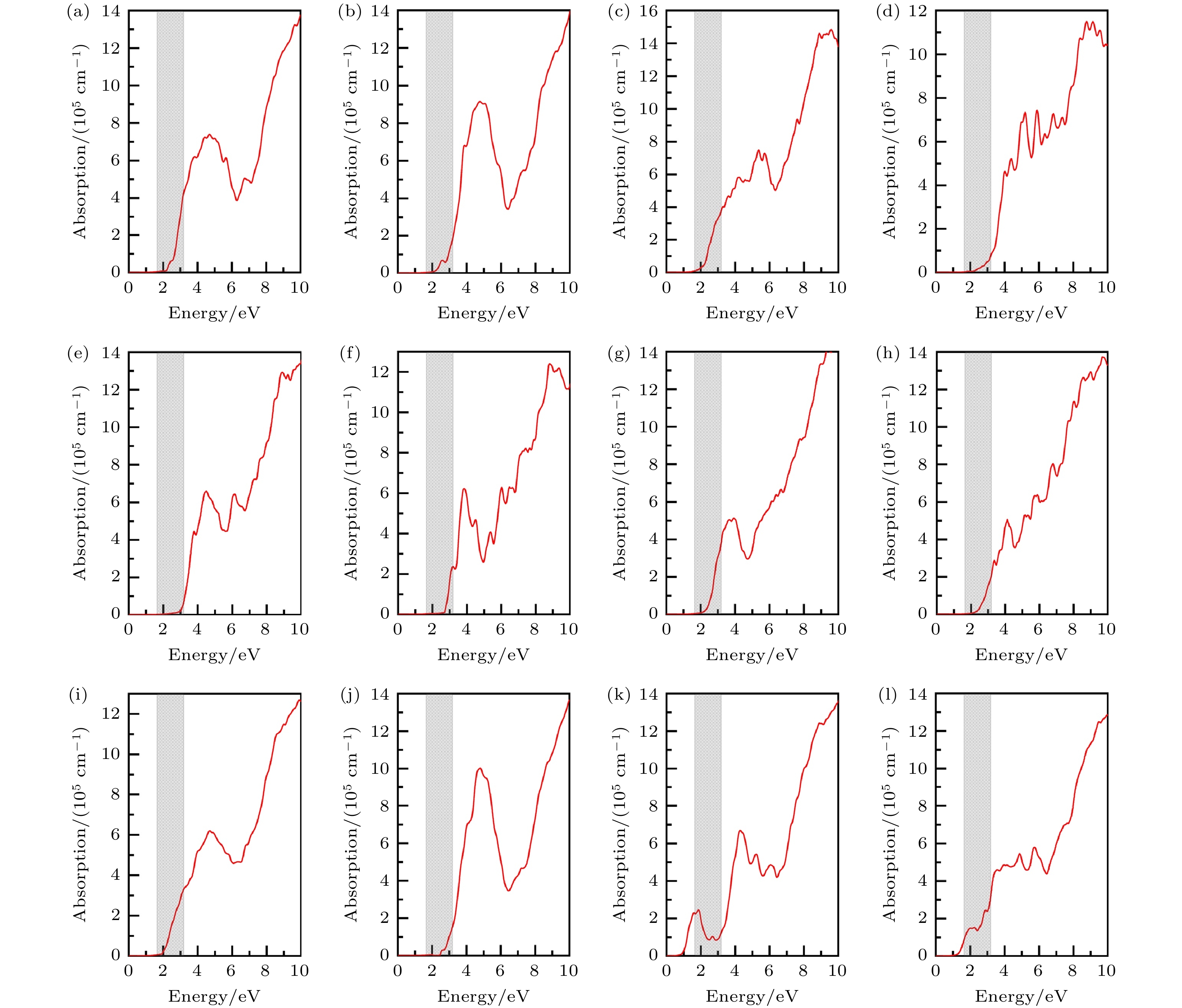 图 11 12个CuBiI三元化合物结构的光吸收谱, 灰色区域代表可见光能量范围(1.64—3.19 eV) (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II
图 11 12个CuBiI三元化合物结构的光吸收谱, 灰色区域代表可见光能量范围(1.64—3.19 eV) (a) CuBi2I7-P1; (b) CuBi2I7-P1-II; (c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-IIFigure11. Optical absorption spectrum for the 12 structures of CuBiI ternary compound. The gray area represents the Visible energy range (1.64–3.19 eV): (a) CuBi2I7-P1; (b) CuBi2I7-P1-II;(c) Cu2BiI5-P1; (d) Cu2BiI5-Cm; (e) Cu3BiI6-P3; (f) Cu3BiI6-R3; (g) Cu4BiI7-P1; (h) Cu4BiI7-P3; (i) Cu3Bi2I9-P1; (j) CuBi3I10-P1; (k) Cu2BiI7-P1; (l) Cu2BiI7-P1-II.
2
3.5.功率转换效率
传统的肖克利-奎塞尔(Shockley-Queisser, SQ)法预测光电转换效率依赖于材料的带隙, 并未考虑光吸收谱, 因此SQ方法预测的光电转换效率在很多情况下并不准确的. 相对于SQ方法, 光谱有限最大效率(spectroscopic limited maximum efficiency, SLME)法运用了热动力学方法, 考虑了特定的吸收光谱以及电子空穴复合过程对光电转换效率的影响, 得到的光电转换效率会更加符合实际情况[39,40]. 本文采用了SLME方法计算太阳能电池的最大理论功率转换效率:




图12为SLME方法预测的CuBiI三元化合物12个结构的光电转换效率. 当吸收层的厚度很小(t < 0.10 μm)时, 由于吸收层对光的吸收很弱, 导致光电转换效率也较低; 当吸收层厚度较大(t > 1 μm)时, 光电转换效率与吸收层厚度不再具有依赖关系, 基本保持最大光电转换效率. 在实验中, 太阳能电池一般会做成薄膜结构, 厚度大概在0.50 μm左右. 表4给出了t = 0.50 μm时CuBiI三元化合物12个结构的光电转换效率. 光电转换效率与能带值以及可见光区域光吸收强度有着密切的联系. Cu2BiI7-P1与Cu2BiI7-P1-II的带隙值小, 位于红外光能量范围内, 在整个可见光吸收范围内(1.64—3.14 eV)均有较强的吸收强度, 且Cu2BiI7-P1为直接带隙, 在受到光电子激发时, 价带顶的电子直接跃迁到导带, 因此Cu2BiI7-P1与Cu2BiI7-P1-II的光电转换效率最大, 分别为31.63%和28.30%. 相反, Cu3BiI6-P3, Cu3BiI6-R3和CuBi3I10-P1结构的带隙值大很多, 与理想的太阳能电池吸收材料的带隙(1.40 eV)相差很大, 且在可见光范围内的光吸收系数小, 即对太阳光的利用率很低, 导致其光电转换效率极小(2.86%, 5.49%和4.17%). 此外, Cu2BiI5-P1, Cu4BiI7-P1, Cu3Bi2I9-P1结构在可见光范围内高的光吸收系数(4 × 105 cm–1)使得其具有较大的光电转换效率.
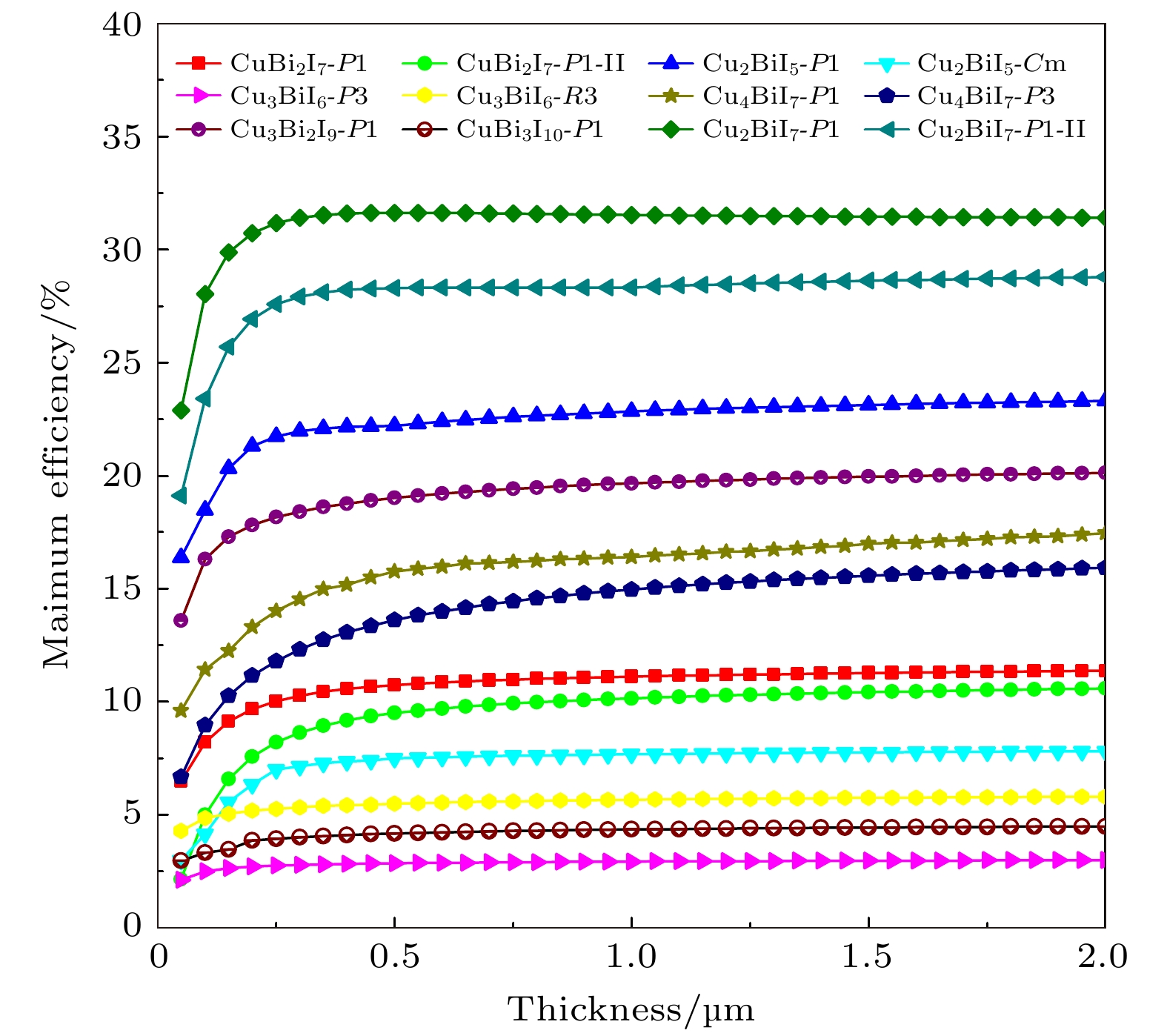 图 12 SLME方法预测的12个CuBiI三元化合物结构的光电转换效率与吸收层厚度的关系
图 12 SLME方法预测的12个CuBiI三元化合物结构的光电转换效率与吸收层厚度的关系Figure12. Photoelectric conversion efficiency of 12 structures of CuBiI ternary compound with respect to the absorption layer thickness predicted by SLME method.
