全文HTML
--> --> -->多孔碳材料, 尤其是石墨烯, 质量小且比表面积大, 是潜在的储氢媒介[2]. 纯石墨烯通过范德瓦耳斯力储氢, 可逆性好, 氢气充放快捷, 但存在吸附量偏少, 操作温度较低等缺点. 大量研究证明金属修饰可以有效提高石墨烯的室温储氢性能[9], 在众多金属中, 3d过渡金属对石墨烯储氢量的提高最为显著. 首先, 过渡金属具有多个空d轨道, 掺杂在石墨烯表面可通过Kubas作用[10]活化氢分子. Kubas作用[10]在2001年首次被提出, 是指氢分子的成键和反键轨道与过渡金属d轨道之间的重叠. 其次, 负载在石墨烯表面的过渡金属原子呈现正电荷, Niu等[11]表明带正电荷的金属离子可以将周围氢分子极化并通过静电相互作用吸附. 在Kubas和静电协同作用下氢分子的吸附能大约为–0.10 eV至–0.80 eV, 介于物理吸附和化学吸附之间[12]. 3d过渡金属修饰为突破现在的石墨烯储氢瓶颈指明了方向, 成为最活跃的前沿领域之一.
过渡金属原子修饰的石墨烯储氢材料在理论上被广泛研究[13,14]. 尽管其理论储氢量都超过了美国能源部的最终目标, 但实验实现却困难重重. 这是因为过渡金属有较低的迁移势垒和较强的金属间作用, 容易团聚成簇[15]. 另外, 第1个被吸附的氢分子通常会解离[16-19]. 在室温储氢过程中解离的H原子可能进一步迁移到石墨烯基底, 这时的储氢机理就会从物理吸附向化学吸附转变.明确H原子在体系中迁移的难易程度对人们深入认识储氢机理非常重要.但是很少有人关注氢分子在Sc, Ti, V单原子修饰石墨烯表面的解离和迁移.
为了增大金属与碳材料载体的结合能, 目前的研究主要集中在两方面: 一是引入缺陷, 二是在纯碳材料表面引入杂原子. 其中引入缺陷在实验上易于操作, 已经被合成并表征[20,21]. 很多实验和计算结果表明, 缺陷能有效地增强金属与石墨烯的作用, 从而有利于金属的分散[22-28]. 为此, 本文使用密度泛函理论详细考察单缺陷的引入对3d过渡金属Sc, Ti, V修饰石墨烯结构和储氢性能的影响. 首先对3d过渡金属原子与石墨烯的结构和结合能进行分析; 然后比较第1个H2的成键形态, 通过分析基底重构和化学键作用, 明确吸附机理; 最后根据最优氢分子吸附能范围估测体系的储氢量. 为明确缺陷对3d金属修饰石墨烯储氢的影响, 本文还对Sc, Ti, V修饰完整石墨烯进行了计算, 考虑了分子吸附、解离吸附以及解离能垒的大小. 本文的研究将有利于深入认识金属修饰碳材料的储氢机理, 并为设计结构稳定的高效储氢材料提供理论基础.
连续H2吸附能(Ec)和平均吸附能(Ea)定义分别为

3.1.Sc, Ti, V修饰石墨烯的结构
作为储氢材料, 首先应该具有稳定的结构.为综合比较引入单缺陷对单过渡金属原子在石墨烯的结合能的影响, 本文同时计算了Sc, Ti, V修饰完整石墨烯(pristine graphene, PG)和缺陷石墨烯(monovacancy graphene, MVG)两种结构结构. 图1所示为Sc, Ti, V修饰PG和MVG的优化结构.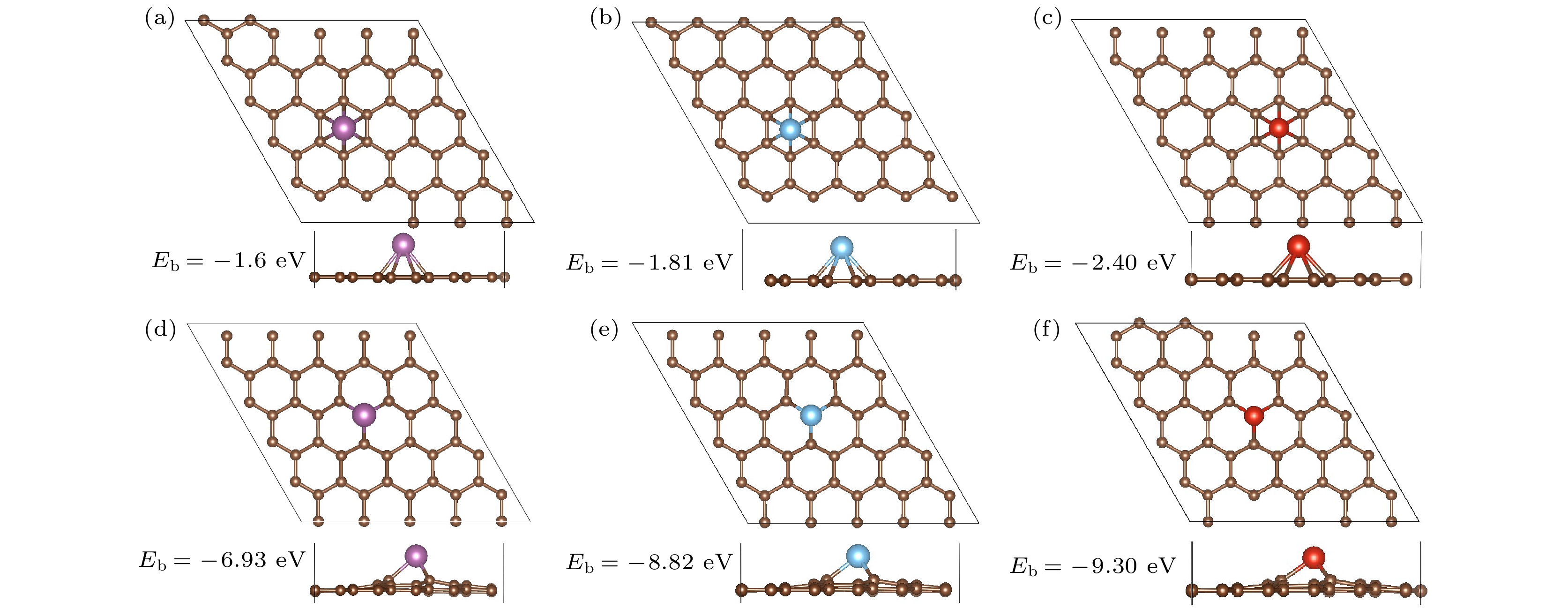 图 1 Sc, Ti, V修饰PG (a)—(c)和MVG (d)—(f)的优化结构(紫色小球表示Sc原子, 蓝色小球表示Ti原子, 红色小球表示V原子)
图 1 Sc, Ti, V修饰PG (a)—(c)和MVG (d)—(f)的优化结构(紫色小球表示Sc原子, 蓝色小球表示Ti原子, 红色小球表示V原子)Figure1. Optimized structures of Sc, Ti, V decorated PG (a)?(c) and MVG (d)?(f). The purple, blue and red balls represent Sc, Ti, and V atoms.
随着原子序数的增加, 金属原子Sc, Ti, V与六元环的结合能增大, 分别为–1.60, –1.81, –2.40 eV, 与文献[26, 35]相近, 略大于文献[36]值0.20—1.50 eV. 金属原子Sc, Ti, V与石墨烯基面的距离d分别为1.78, 1.63和1.52 ?. 由于此时金属聚合能[37]均大于对应的结合能, 因此实验制备过程中金属倾向于团聚在一起, 而不是分散分布在PG表面. 实验研究也表明室温下钛簇在石墨烯上生长, 平均直径约为10 nm[38]. 当引入单缺陷后, 金属位于缺陷位, 这与文献[26]结果一致. 金属Sc, Ti, V与MVG的结合能分别为–6.93, –8.82和–9.30 eV, 是与PG结合能的4.33, 4.87和3.88倍, 与文献[26]结果(7.08, 9.03和8.29 eV)相近. 这说明单缺陷的引入可以有效增大3d过渡金属单原子与石墨烯的结合能, 从而稳定了金属原子, 为其催化及储氢应用提供了有效保证. 较大的结合能对应较短的TM—C键长, 如表1中的dTM—C数据所示.
| System | TM cohesive nenergy[37] (eV/atom) | Eb/eV | dTM—C/? | d/? | Δz/? | Charge/|e| |
| Sc/PG | –3.90 | –1.60 | 2.35—2.36 | 1.78 | +1.09 | |
| Ti/PG | –4.85 | –1.81 | 2.12—2.37 | 1.63 | +1.06 | |
| V/PG | –5.31 | –2.40 | 2.08—2.31 | 1.52 | +1.05 | |
| Sc/MVG | –3.90 | –6.93 | 2.07—2.08 | 1.96 | 0.58 | +1.37 |
| Ti/MVG | –4.85 | –8.82 | 1.84—2.14 | 1.67 | 0.52 | +1.37 |
| V/MVG | –5.31 | –9.30 | 1.77—2.07 | 1.47 | 0.41 | +1.24 |
表1Sc, Ti, V修饰PG和MVG的相关参数 (d为TM与石墨烯平面的距离; Δz为TM修饰前后, 基底在z方向的最大 形变量)
Table1.Relevant parameters of Sc, Ti and V decorated PG and MVG (d is distance between TM and graphene plane; Δz is the maximum deformation in the z direction of the substrate before and after TM modification).
从图1中结构的侧视图可以看出, 对于MVG, 金属原子周围的碳原子会发生0.41— 0.58 ?的微小形变. 由于Sc和Ti修饰后石墨烯形变产生的Δz较大, 导致金属Sc和Ti离平面的距离从1.78和1.63 ?增大到1.96和1.67 ?.
当过渡金属修饰石墨烯表面时, 金属中的电子会转移到石墨烯表面, 使金属呈现正电荷状态. 正电荷的金属位点可以极化周围的氢分子, 并且通过静电相互作用吸附多个氢分子. 从表1可以看出, 当过渡金属修饰PG时, 电荷大约在+1.05|e|— +1.09|e|范围, 当引入单个缺陷后, 电荷大约在+1.24|e|— +1.37|e|范围, 这说明过渡金属修饰单MVG上会转移更多的电子到碳材料上.
图2是Sc, Ti, V修饰石墨烯体系的态密度图(partial density of states, PDOS). 可以看出Sc, Ti, V的d轨道与C原子轨道的重合集中在费米能级附近, 且随着d电子的增多, 明显向左移动, 说明金属原子与石墨烯的作用增强, 这与前面金属结合能显示结果一致. 当存在缺陷时, Sc, Ti, V与C之间的轨道重叠范围在费米能级以下大大增加, 说明轨道之间有效重叠, d电子所在能级明显降低, 使得金属与基底之间作用增强.
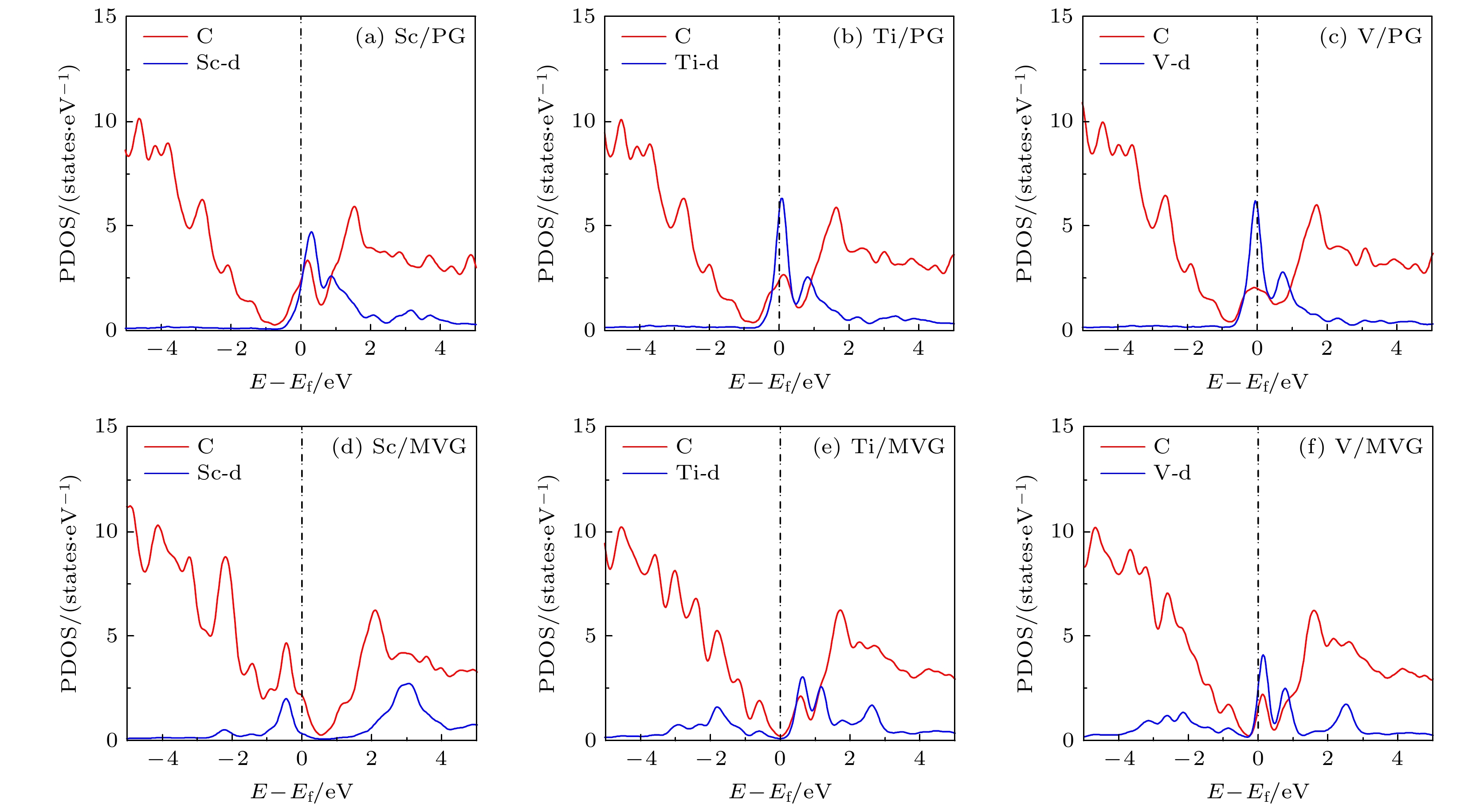 图 2 Sc, Ti, V修饰PG和MVG的PDOS图
图 2 Sc, Ti, V修饰PG和MVG的PDOS图Figure2. PDOS of Sc, Ti and V decorated PG and MVG.
2
3.2.1H2与Sc, Ti, V修饰石墨烯的相互作用
本节全面设计了Sc, Ti, V修饰石墨烯吸附1个H2的结构. 图3展示了PG对应的优化结构. 氢分子吸附在Sc/PG中Sc的侧上方, H—H键长为0.76 ?, 结合能为–0.57 eV. 解离吸附结构的能量比分子吸附结构的能量低0.20 eV. 两个H原子分别位于Sc原子的两侧. 对于Ti/PG, 优化后只能得到解离吸附结构, 氢分子的结合能为–1.34 eV. 说明H2在Ti/PG表面解离没有能垒. Ti修饰碳纳米管和富勒烯C60上吸附的第1个氢分子也会自发解离, 氢分子吸附能分别为–1.50 eV[39]和1.62 eV[40]. 图 3 Sc, Ti, V修饰PG吸附1个氢分子的优化结构(a)?(e)及H2解离、H原子迁移势能面图(f)?(h) (紫色小球代表Sc, 蓝色小球代表Ti, 红色小球代表V) (a), (b), (f) Sc/PG; (c), (g) Ti/PG; (d), (e), (h) Sc/PG
图 3 Sc, Ti, V修饰PG吸附1个氢分子的优化结构(a)?(e)及H2解离、H原子迁移势能面图(f)?(h) (紫色小球代表Sc, 蓝色小球代表Ti, 红色小球代表V) (a), (b), (f) Sc/PG; (c), (g) Ti/PG; (d), (e), (h) Sc/PGFigure3. (a)?(e) Optimized structures of 1H2-TM/PG (TM = Sc, Ti, V) and (f)?(h) the schematic diagrams of dissociation of H2 and the H migration process (Sc, Ti, and V atoms are represented by purple, blue and red balls): (a), (b), (f) Sc/PG; (c), (g) Ti/PG; (d), (e), (h) Sc/PG.
对于V/PG, H—H键长为0.79 ?, 氢分子结合能为–1.15 eV. 解离吸附结构的能量比分子吸附结构的能量低0.01 eV. 这些结果说明Sc, Ti, V修饰PG吸附1个氢分子时, 氢分子将解离. 解离吸附时形成稳定的TM—H键, 伴随着较大的氢分子吸附能, 因此H2的脱附很难实现. 图3(f)—(h)分别展示了H2解离和H原子迁移的能垒, 可以看到Sc/PG和V/PG表面H2解离能垒分别为0.12和0.08 eV. 这些数据说明H2非常容易发生解离. Sc/PG, Ti/PG和V/PG表面H原子迁移的能垒分别为2.84, 3.05和3.63 eV, 说明H原子的迁移在室温条件下是比较困难的.
图4展示了单MVG对应的优化结构. 单个氢分子吸附在Sc/MVG中Sc原子的顶部正上方, 结合能为–0.16 eV, 与文献[25]值–0.18 eV非常相近. H—H键长为0.76 ?. 分子吸附的能量比相应解离吸附的能量低2.07 eV. 对于Ti/MVG, 优化后只能得到分子吸附的结构, 氢分子结合能为–0.24 eV. 对于V/MVG, H—H键长为0.79 ?, 结合能为–0.31 eV. 分子吸附的能量比相应解离结构的能量低0.86 eV. 这些结果说明Sc, Ti, V修饰单MVG吸附1个氢分子时, 氢分子将以分子形式存在. 这对于物理储氢材料来说是非常重要的. 而且氢分子吸附能在–0.10 eV至–0.80 eV, 能够保证氢分子在室温下快速脱附. 文献[26]指出Sc, Ti, V修饰双MVG吸附1个氢分子时, 氢分子也以分子形式存在, 氢分子结合能分别为–0.26, –0.36和–0.63 eV. Chu等[41]也指出Ti修饰双MVG吸附1个氢分子的结合能为–0.31 eV. 通过与PG的对比可以明显得出, 单缺陷的引入有利氢分子以分子形式吸附. 这主要是由于引入缺陷后, TM原子外层电子减少(表1), 与氢分子作用时, 第二电离势较强, 无法将更多的电子反馈给氢分子.
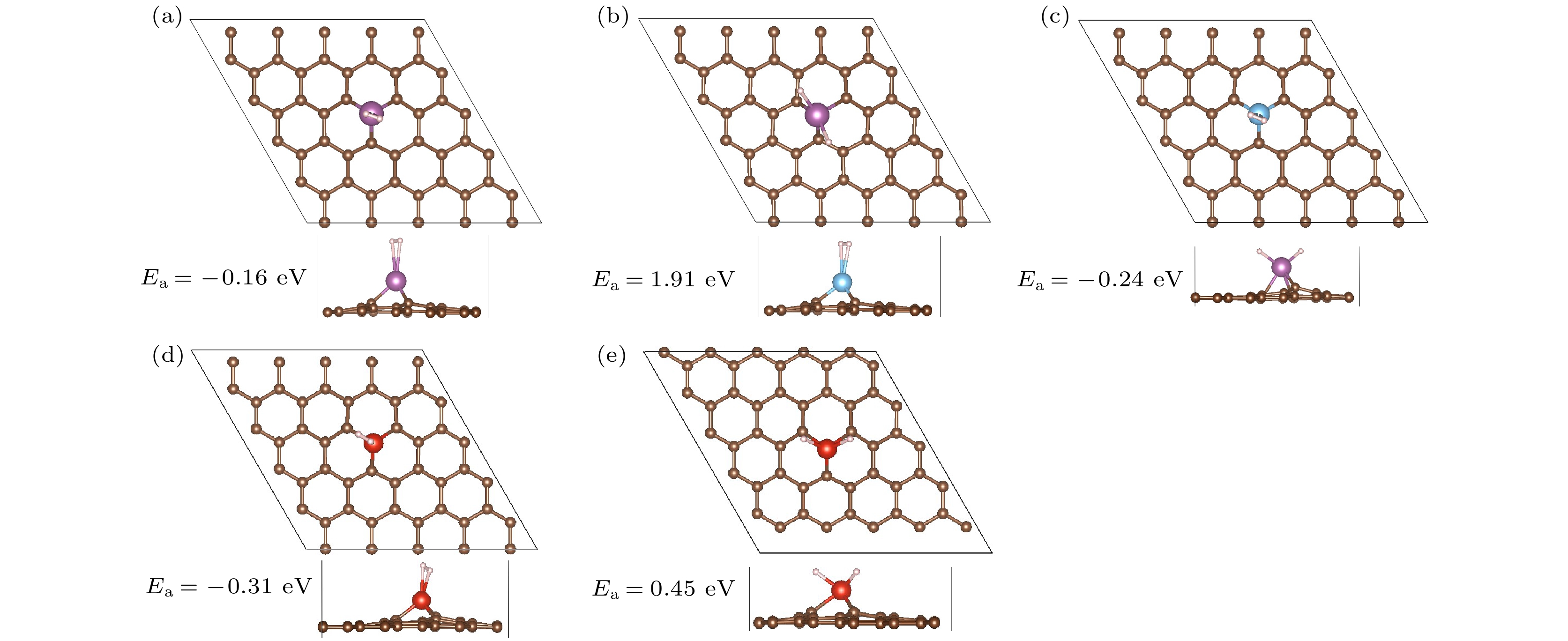 图 4 Sc, Ti, V修饰单MVG吸附1个氢分子的优化结构 (紫色小球表示Sc原子, 蓝色小球表示Ti原子, 红色小球表示V原子)
图 4 Sc, Ti, V修饰单MVG吸附1个氢分子的优化结构 (紫色小球表示Sc原子, 蓝色小球表示Ti原子, 红色小球表示V原子)Figure4. Optimized structures of 1H2-TM/MVG (TM = Sc, Ti, V). Sc, Ti, and V atoms are represented by purple, blue and red balls.
一般来说, 氢分子解离吸附应该比分子吸附更有利. 但是对于本文中提到的TM修饰单缺陷的体系, 分子吸附构型能量更低. 接下来从结构重构与化学键的竞争效应来解释这个现象. 吸附氢分子前后能量的变化主要来自两方面: 一方面是H2吸附后TM/MVG的结构重构使体系能量升高, 另一方面是形变后的基底与H2的电子云相互作用使体系的能量降低. 为此, 本文定义了重构能(ER)和化学键能(E1).


如表2所列, 与分子吸附构相比, Sc/MVG和V/MVG的氢解离吸附结构形变的能量较大, 不可忽视. 例如, V/MVG在吸附H2后发生微小形变, 基底重构能为0.03 eV, 形变后的基底与H2的电子云相互作用能为–0.34 eV, 最终结果表现为V/MVG的氢分子结合能为–0.31 eV. 当H2解离吸附时, 基底形变能为1.28 eV, 形变后的基底与H2的电子云相互作用能为–0.83 eV. 最终结果表现为V/MVG的氢分子结合能为0.45 eV. 这说明Sc/MVG和V/MVG在吸/脱氢分子过程中结构基本保持不变, 这将极大地提高吸/脱附的动力学过程.
| Syestem | Ea/eV | ER/eV | E1/eV | Q/|e| | System | Ea/eV | ER/eV | E1/eV | Q/|e| | |
| 1H2-Sc/MVG | –0.16 | 0.00 | –0.16 | 1.38 | 2H-Sc/MVG | 1.91 | 2.55 | –0.64 | 1.58 | |
| 1H2-Ti/MVG | –0.24 | 0.51 | –0.75 | 1.37 | ||||||
| 1H2-V/MVG | –0.31 | 0.03 | –0.34 | 1.23 | 2H-V/MVG | 0.45 | 1.28 | –0.83 | 1.39 |
表21H2-TM/MVG (TM = Sc, Ti, V)的氢分子吸附能(Ea)、重构能(ER)、化学键能(E1)及TM的电荷
Table2.Hydrogen adsorption (Ea), structure reconstruction energy (ER), the chemical bond energy (E1) of 1H2-TM/MVG (TM = Sc, Ti, V), and chargesof TM(Q|).
分子氢吸附时, Sc, Ti, V原子上的电荷(+1.38|e|, +1.37|e|, +1.23|e|)与未吸附氢分子时(+1.37|e|, +1.37|e|, +1.24|e|)相比几乎没有变化. 说明分子氢吸附在Sc, Ti, V修饰的MVG的主要机理是静电相互作用. 本文还对Sc, Ti, V修饰MVG吸附1个氢分子的结合能与文献中Li+吸附氢分子的结合能进行比较. 众所周知, Li+和氢分子之间是纯的静电相互作用, 完全没有Kubas作用. 高斯软件CCSD (T)/6-311++G (3 df, 3 pd)水平下计算结果显示Li+与氢分子之间的作用能为0.20—0.24 eV[42]. Dmol3和CASTEP计算碱金属Li修饰的石墨烯的氢分子吸附能分别为0.23和0.28 eV[22,23].
氢分子解离吸附时, Sc和V原子的电荷为+1.58|e|和+1.39|e|, 说明Sc和V向H2反馈0.21|e|和0.15|e|, 即金属对H2有更多的反馈电子, 较多的反馈电子填充在H2的反键轨道, 使H2解离. 图5中的电荷差分密度图说明对于分子氢吸附构型, 电荷分布变化较小, 有明显的极化作用. 对于氢解离吸附的构型, 电荷变化非常明显.
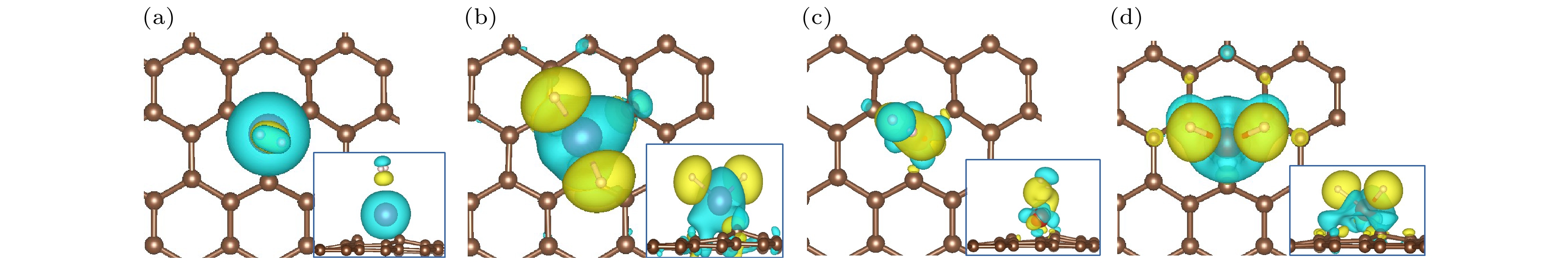 图 5 (a) H2-Sc/MVG, (b) 2H-Sc/MVG, (c) H2-V/MVG, (d) 2H-V/MVG的电荷差分密度图, 黄色部分表示电荷增加, 青色部分表示电荷减少, 界面值为0.003 e/?3
图 5 (a) H2-Sc/MVG, (b) 2H-Sc/MVG, (c) H2-V/MVG, (d) 2H-V/MVG的电荷差分密度图, 黄色部分表示电荷增加, 青色部分表示电荷减少, 界面值为0.003 e/?3Figure5. Charge density difference of (a) H2-Sc/MVG, (b) 2H-Sc/MVG, (c) H-V/MVG, and (d) 2H-V/MVG. The charge accumulation and depletion regions are indicated by yellow and blue, respectively. The isosurface value is 0.003 e/?3 .
图6展示了Sc/V修饰MVG上氢以分子形式和解离形式吸附时的部分态密度图和相应重叠部分对应的主要分子轨道. 可以看出, 分子吸附构型的轨道中H的轨道和Sc/V的d轨道在–8 eV至–10 eV重叠, 态密度图中金属原子与H2电子云的重叠部分较少. 对于解离部分, 2H-Sc/MVG结构的轨道重叠部分在0至–4 eV附近, 2H-V/MVG结构的轨道重叠部分在0至–2 eV附近. 解离吸附的轨道重叠程度更大, 说明电子云相互作用较强. 根据体系重叠部分的能量, 本文找到了对应的主要轨道, 分别为2H的成键轨道与TM的d轨道的作用以及2H的反键轨道与TM的d轨道的作用. 由于解离吸附结构存在明显的H2和过渡金属d轨道的重叠, 因此第1个H2吸附时存在Kubas作用.
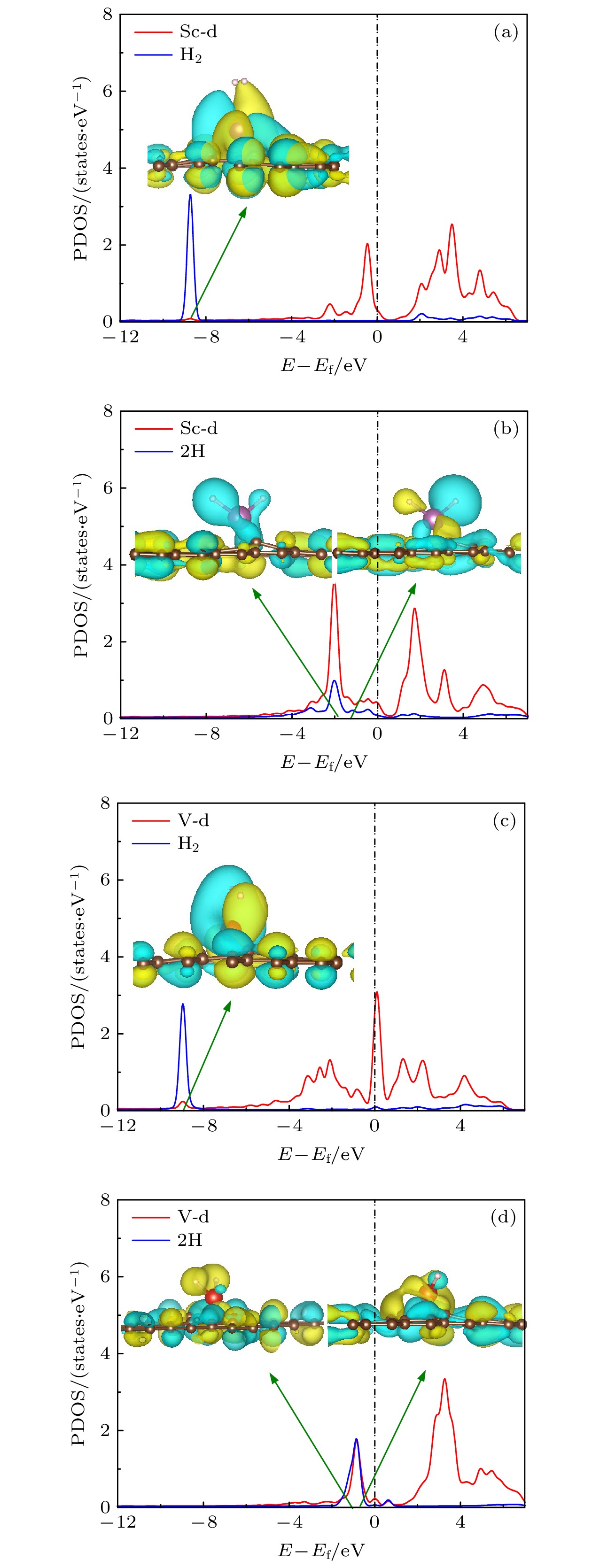 图 6 (a) H2-Sc/MVG, (b) 2H-Sc/MVG, (c) H2-V/MVG, (d) 2H-V/MVG的DOS图和主要的轨道图(黄色部分表示正相位, 青色部分表示负相位, 等值面为1.5 × 10–7)
图 6 (a) H2-Sc/MVG, (b) 2H-Sc/MVG, (c) H2-V/MVG, (d) 2H-V/MVG的DOS图和主要的轨道图(黄色部分表示正相位, 青色部分表示负相位, 等值面为1.5 × 10–7)Figure6. Main orbitals and PDOS of (a) H2-Sc/PG, (b) 2H-Sc/PG, (c) H2-V/PG, (d) 2H-V/PG. The positive and negative phases are indicated by yellow and blue regions, respectively. The isosurface is 1.5 × 10–7.
根据以上分析可以得出: Sc, Ti, V修饰的单MVG离子特性强, 当1个氢分子以分子吸附时, 结合能温和、电子转移较少、基底形变小, 主要机理是静电相互作用; 氢解离吸附的特点是吸附能大, 金属与H2之间有较大的电子转移, 基底形变较大, 存在Kubas作用.
2
3.3.Sc, Ti, V修饰单MVG的储氢量
当继续吸附氢分子时, 由于位阻效应, 随着吸附氢分子数目的增加, 氢分子吸附能逐渐减小. 氢分子在TM/MVG体系的连续吸附能以及相关结构参数列于表3. 图7展示氢分子连续吸附能大于0.10 eV的优化结构. Sc修饰的MVG可以连续吸附6个氢分子. 前4个连续吸附能为–0.12 eV至–0.20 eV, Sc—H键长为2.39—2.96 ?. Ti/MVG可以连续吸附3个H2. 连续氢分子吸附能为–0.16 eV至–0.24 eV, Ti—H键为2.10 — 2.52 ?. V原子周围可以连续吸附3个氢分子. H—H键为0.77—0.82 ?, Ti—H键为1.93—3.36 ?. 吸附第4个氢分子时, 结合能较弱, 为–0.06 eV. Sc, Ti, V修饰单MVG可以分别吸附7, 3和4个H2. 文献[26]指出Sc, Ti, V修饰双MVG分别吸附4, 3, 2个H2. 当晶胞中存在4个缺陷位(缺陷浓度8%)时, 对应储氢量质量分数可达6.84%. 本文计算了4 × 4 × 1晶胞中存在4个缺陷位(缺陷浓度12.5%)形成的Sc4/MVG, 平均Sc原子结合能为6.33 eV, Sc—Sc键长为4.92 ?. 其储氢量质量分数可达9.86 %. 有文献报道Pd[25]和Sc[43]在石墨烯缺陷可以进行双面掺杂. 本文设计了双掺杂结构Sc8/MVG进一步增加储氢量. 优化结果显示平均Sc原子结合能为5.90 eV, 明显高于对应的金属聚合能3.90 eV, 是与PG结合能的3.69倍. 其储氢量质量分数估算高达13.96 %.| nH2-Sc/MVG | Ec/eV | Ea/eV | 键长/? | |
| H—H | Sc—H | |||
| n = 1 | –0.16 | –0.16 | 0.76 | 2.53—2.54 |
| n = 2 | –0.16 | –0.16 | 0.75—0.76 | 2.42—2.58 |
| n = 3 | –0.20 | –0.17 | 0.76—0.77 | 2.39—2.57 |
| n = 4 | –0.12 | –0.16 | 0.75—0.76 | 2.45—2.96 |
| n = 5 | –0.08 | –0.14 | 0.75—0.76 | 2.67—3.19 |
| n = 6 | –0.13 | –0.14 | 0.75—0.76 | 2.67—3.68 |
| n = 7 | –0.09 | –0.13 | 0.75—0.76 | 2.60—3.89 |
| nH2-Ti/MVG | Ec/eV | Ea/eV | 键长/? | |
| H—H | Ti—H | |||
| n = 1 | –0.24 | –0.24 | 0.78 | 2.30—2.31 |
| n = 2 | –0.16 | –0.20 | 0.75—0.78 | 2.14—2.32 |
| n = 3 | –0.20 | –0.20 | 0.75—0.81 | 2.10—2.52 |
| nH2-V/MVG | Ec/eV | Ea/eV | 键长/? | |
| H—H | V—H | |||
| n = 1 | –0.31 | –0.31 | 0.79 | 1.93—2.01 |
| n = 2 | –0.15 | –0.24 | 0.76—0.82 | 2.07—2.18 |
| n = 3 | –0.10 | –0.19 | 0.77—0.82 | 1.93—3.36 |
| n = 4 | –0.06 | –0.18 | 0.75—0.86 | 2.14—4.03 |
表3TM/MVG (TM = Sc, Ti, V)吸附多个氢分子的连续吸附能(Ec/eV)、平均吸附能(Ea/eV)以及H—H/Sc—H键长
Table3.Stepwise H2 adsorption energy (Ec in eV), the average H2 binding energies (Ea in eV), and bond lengths of H—H and Sc—H in TM/MVG (TM = Sc, Ti, V).
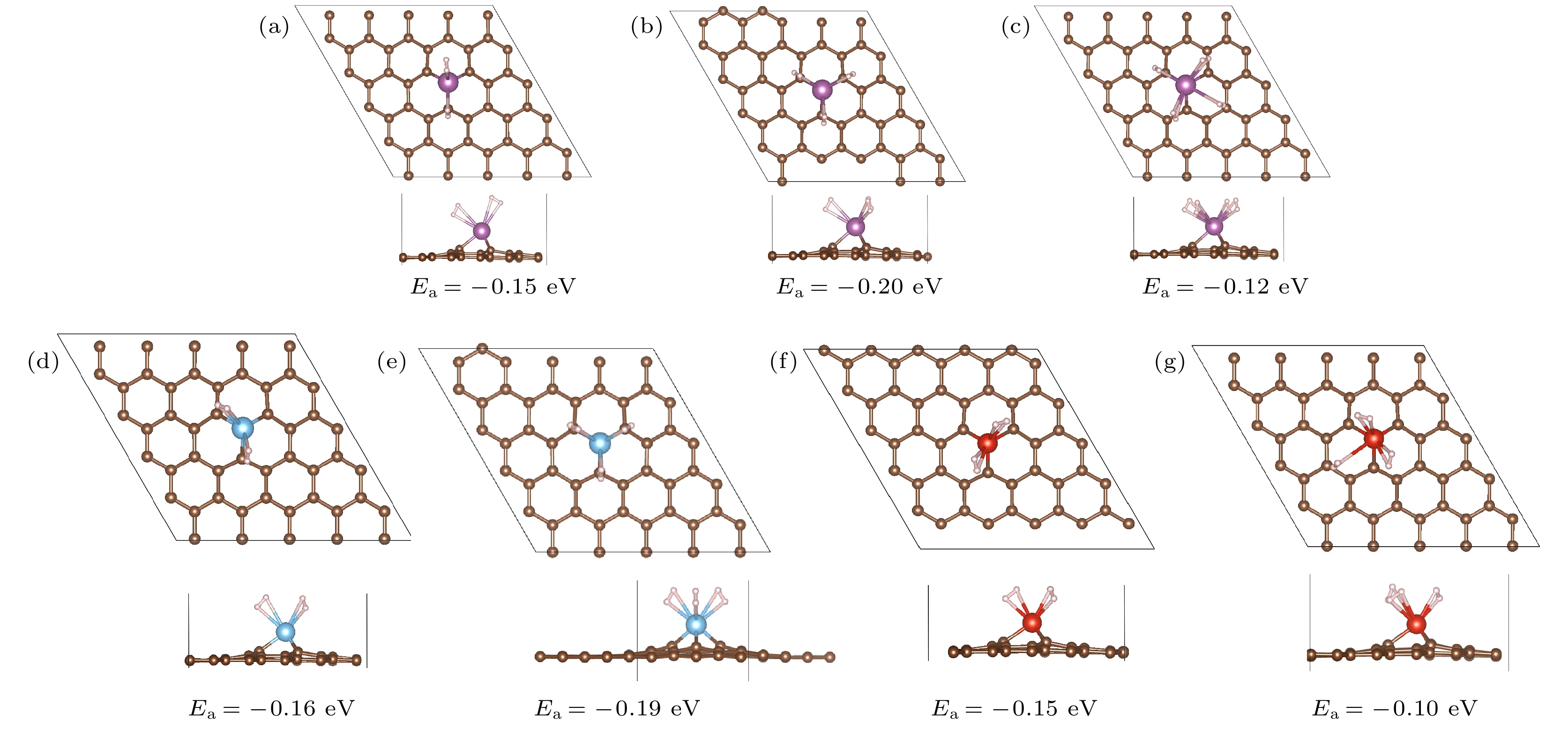 图 7 Sc, Ti, V修饰MVG连续吸附多个氢分子的优化构型(紫色小球表示Sc原子, 蓝色小球表示Ti原子, 红色小球表示V原子)
图 7 Sc, Ti, V修饰MVG连续吸附多个氢分子的优化构型(紫色小球表示Sc原子, 蓝色小球表示Ti原子, 红色小球表示V原子)Figure7. Optimized structures of nH2-TM/MVG (TM = Sc, Ti, V). Sc, Ti, and V atoms are represented by purple, blue and red balls.
平均氢分子吸附能常作为吸附剂与氢分子作用的描述符直接与理想吸附能范围比较[25,39,40]. 如表3所列, 本文中Sc, Ti, V修饰MVG分别吸附7, 3和4个H2时平均氢分子吸附能分别为–0.13, –0.20和–0.18 eV, 处于室温和中等压力下循环吸/脱吸过程的最佳能量范围. 从图8可以看出, PG上的金属位点对氢分子的平均吸附能为–0.50 eV至–1.34 eV, 明显比相应MVG上金属位点的氢分子吸附能大. 由于氢分子解离并形成了TM—H键, 因此部分氢分子在常温常压环境下较难脱附.
 图 8 Sc, Ti, V修饰完整和MVG的平均氢分子吸附能随H2分子数量的变化曲线, 其中–0.10 eV至–0.80 eV之间的绿色区域为最佳H2存储的能量窗口
图 8 Sc, Ti, V修饰完整和MVG的平均氢分子吸附能随H2分子数量的变化曲线, 其中–0.10 eV至–0.80 eV之间的绿色区域为最佳H2存储的能量窗口Figure8. Variation in the average binding energies per H2 molecule for TM/MVG (TM = Sc, Ti, V) as a function of the number of H2 molecules. The green region from –0.10 eV to –0.80 eV represents the energetic window for an optimal H2 storage.
本文从计算角度提出引入单缺陷可以解决过渡金属修饰石墨烯遇到的金属团聚和氢分子解离的难题.在实验上, Zhu等[44]通过原位刻蚀富勒烯分子(C60)构建了一种富含五边形缺陷的碳纳米材料. 近年来, 大量研究开发了单位点催化剂[45]. 期望Sc, Ti, V修饰单MVG也可以借助原位刻蚀和单原子催化剂制备的方法成功合成并应用于储氢实验.
1)当引入单缺陷后, 金属Sc, Ti, V的结合能分别为–6.93, –8.82和–9.30 eV, 是对应PG的4.33, 4.87和3.88倍, 而且明显高于对应的金属聚合能. 说明单缺陷的引入可以增加3d过渡金属单原子与石墨烯的结合能, 从而有效稳定金属原子, 为这类材料的催化及储氢应用提供了有效的保证.
2)当过渡金属修饰PG时, 电荷在+1.05|e|—+1.09|e|范围之间, 当引入单个缺陷后, 电荷大约在+1.24|e|—+1.37|e|范围之间, 这说明过渡金属修饰在单MVG上会转移更多的电子到碳材料上.
3) Sc, Ti, , V修饰PG吸附1个氢分子时, 氢分子将以解离形式存在. Ti/PG表面的H2解离没有能垒. Sc/PG和V/PG表面H2解离能垒分别为0.12和0.08 eV. 氢分子解离后迁移能垒为2.84—3.63 eV. 当引入单缺陷后, 氢分子将以分子形式存在.
4)分子氢吸附在Sc, Ti, V修饰的MVG的主要机理是静电相互作用. Sc/MVG和V/MVG不仅可以通过物理吸附氢分子作为潜在的储氢材料, 而且吸附和脱附过程中结构基本保持不变, 这将极大地提高吸脱附的动力学过程.
5)在最佳H2存储的能量窗口–0.10 eV至–0.80 eV之间, Sc, Ti, V修饰单MVG分别吸附7, 3和4个H2时平均氢分子吸附能分别为–0.13, –0.20和–0.18 eV. PG上的金属位点对氢分子的平均吸附能为–0.50 eV至–1.34 eV, 部分氢分子在室温和中等压力下将难以脱附.
