全文HTML
--> --> -->目前, 广泛应用的含能材料主要是硝基类炸药. 硝基的π反键轨道是一个未被占据的低能轨道, 含能分子在受激发时电子主要被激发到该轨道上, 因此, 硝基会显著影响含能分子的光致解离过程. 硝基甲烷(NM)作为一种低感度的爆轰剂, 是研究常规炸药起爆和后续化学反应机理的理想体系[14]. 在纳秒紫外光的诱导下, NM分子硝基上的激发电子触发NM炸药的分解, 并引起一系列的链式反应[15-18]. 有证据表明, NM分子在光热等外界条件诱导下引发爆炸的链式反应第一步是C—N共价键断裂, 并形成含有不成对电子的硝基自由基中间态[5]. 环三亚甲基三硝胺(RDX)作为炸药和燃料有着广泛的应用[3,19], Gares等[20]利用深紫外共振拉曼光谱测量了RDX在229 nm激光下光解后的产物, 观察到N—NO2谱带强度的降低以及硝酸根离子(

综上, 利用激光诱导和光谱测量的方法对含能材料光诱导后的产物进行测定, 提出了多种可能的分解机制, 但起爆过程中涉及到的与激发态有关的瞬态过程, 如电子激发后的结构和能量变化等一系列超快过程没有给出清晰的物理图像, 这主要是源于含能材料的分解过程极其复杂, 且受时空分辨和光电探测能力的限制以及实验测量的高危险性, 许多现象和细节仍然在实验上无法直接获得.
从头算分子动力学和含时密度泛函理论相结合的方法是一种可靠的激发态计算方法[26-28], 该方法可有效研究激发载流子传输, 激发态原子结构弛豫和能级演化等[29]. 例如, 基于含时密度泛函的Surface hopping方法, Polyak等[30]对1,3-环己二烯分子的光解离过程进行了模拟, 发现该分子在激发后的首选反应路径为碳环上连接两个亚甲基的C—C键的断裂, 证明了分子解离的驱动力来源于C—C键伸缩振动所产生的动能, 整个激发态寿命为89 ± 9 fs; 基于含时密度泛函的Ehrenfest方法, Kolesov等[31]发现偶氮二异丁腈分子在光激发下可分裂成自旋相反的一对异丁腈自由基和一个氮分子, 与实验上通过紫外光激发观测到的解离产物相同, 证明了该方法可有效模拟有机分子的光解过程; 另外, 基于含时密度泛函理论下的透热激发态分子动力学方法, Nelson等[32]模拟了光学活性材料二乙烯基苯分子光诱导下的结构演化过程, 发现电子密度的重新分布导致了分子的键长发生周期变化, 且在室温(300 K)下分子的扭转角在250 fs内从24°减小到14°, 而在低温(10 K)下分子的扭转角在10 ps内从22°减小到4°. 因此, 我们将含时密度泛函理论用于含能材料体系的激发态研究, 以期获得含能分子受激后的几何结构和能量变化等一系列超快过程的微观物理图像. 本文中, 我们选取硝基甲烷(NM)、环三亚甲基三硝胺(RDX)和三氨基三硝基苯(TATB)三种典型含能分子为模型体系, 通过研究含能分子激发态结构弛豫过程中化学键变化和分子能级轨道的演化规律, 阐明激发态原子弛豫的动态过程和内在机理, 并揭示含能分子光解离的超快物理过程[33,34].
3.1.NM, RDX, TATB分子的基态几何结构与电子性质
首先, 我们通过结构优化给出了0 K下NM, RDX, TATB分子的基态构型, 如图1所示. 表1也列出了这几种含能分子的详细键长, 并同实验值进行了对比. 总体来说, 计算所得键长与实验测量值误差小于8%, 表明我们的计算方法能够很好地描述含能分子的基态构型. 但需要强调的是, 实验中主要是对含能材料分子晶体进行测量, 而本文则是针对含能分子进行模拟. 图 1 基态含能分子结构示意图 (a) 硝基甲烷(NM); (b) 环三亚甲基三硝胺(RDX); (c) 三氨基三硝基苯(TATB); 其中蓝色为N原子, 红色为O原子, 棕色为C原子, 白色为H原子
图 1 基态含能分子结构示意图 (a) 硝基甲烷(NM); (b) 环三亚甲基三硝胺(RDX); (c) 三氨基三硝基苯(TATB); 其中蓝色为N原子, 红色为O原子, 棕色为C原子, 白色为H原子Figure1. Structure diagrams of (a) nitromethane (NM); (b) cyclotrimethylenetrinitramine (RDX); (c) triaminotrinitrobenzene (TATB) at ground state. Blue ball is N atom, red ball is O atom, brown ball is C atom, and white ball is H atom.
| NM | C—N | C—H1 | C—H2 | C—H3 | N—O1 | N—O2 | |
| Length/? | This work | 1.547 | 1.096 | 1.099 | 1.099 | 1.148 | 1.157 |
| Exp.[40] | 1.481 | 1.093 | 1.092 | 1.092 | 1.223 | 1.224 | |
| RDX | C—N | N1—N2 | N3—N4 | N5—N6 | C1—H1 | C1—H2 | |
| Length/? | This work | 1.443 | 1.443 | 1.471 | 1.472 | 1.182 | 1.183 |
| Exp.[41] | 1.464 | 1.351 | 1.352 | 1.398 | 1.085 | 1.087 | |
| TATB | C—C | C—Nnitro | C—Namino | N—O | N—H | O···H | |
| Length/? | This work | 1.425 | 1.436 | 1.318 | 1.216 | 1.021 | 1.646 |
| Exp.[42] | 1.441 | 1.442 | 1.316 | 1.243 | 0.925 | 1.780 | |
表1三种含能分子基态的键长信息
Table1.Bond lengths of energetic molecules at ground state
图2为含能分子基态的轨道能级排布, 以及最高占据态轨道(HOMO)与最低非占据态轨道(LUMO)的电荷密度分布图. 根据DFT计算, NM, RDX和TATB分子HOMO与LUMO之间的能隙分别为3.06 eV, 3.27 eV和2.62 eV, 与之前的理论计算值3.36 eV[43], 3.44 eV[44]和2.60 eV[45]相一致, 小于实验测量值3.80 eV[46], 3.60 eV[47]和6.60 eV[48]. 从电荷密度分布可知, NM分子的HOMO轨道主要由硝基上两个氧原子的孤对电子占据, 即HOMO轨道为氧原子的非键态, 而LUMO轨道的电荷密度主要分布在硝基, LUMO轨道为硝基的π反键态; RDX分子也具有类似的特征, 即HOMO主要分布在硝基的氧原子上, 为氧原子的非键态, LUMO分布在硝基上, 为硝基的π反键态; TATB分子的HOMO与LUMO轨道在六元环的碳原子和氨基上的氮原子上均有分布, 此外, HOMO和LUMO同样具有类似于NM和RDX分子的特点, 即HOMO被硝基上为氧原子的孤对电子占据, 而LUMO表现出硝基上π的反键态特点. 因此, 这三种含能分子的第一激发态都对应于电子从硝基氧原子的非键态到硝基π反键态的跃迁.
 图 2 三种含能分子的基态轨道能级排布及最高占据态轨道(HOMO)与最低非占据态轨道(LUMO)的电荷密度分布图 (a) 硝基甲烷(NM); (b) 环三亚甲基三硝胺(RDX); (c) 三氨基三硝基苯(TATB)
图 2 三种含能分子的基态轨道能级排布及最高占据态轨道(HOMO)与最低非占据态轨道(LUMO)的电荷密度分布图 (a) 硝基甲烷(NM); (b) 环三亚甲基三硝胺(RDX); (c) 三氨基三硝基苯(TATB)Figure2. The ground state molecular orbital (MO) energy levels and charge density of the highest occupied state orbitals and the lowest unoccupied state orbitals of (a) nitromethane (NM), (b) cyclotrimethylenetrinitramine (RDX), (c) triaminotrinitrobenzene (TATB).
2
3.2.光激发后NM, RDX, TATB分子结构演化
为研究NM, RDX, TATB分子在室温下的激发态行为, 我们首先将含能分子预热至300 K. 三种含能分子的几何结构在300 K时都有不同程度的变化, 其中表2重点列出了RDX分子与TATB分子结构的变化. 为方便讨论, 在RDX分子中我们用α1表示∠C1N2C2, α2表示∠N2C2N3, δ表示C1—N2—C2所成平面与N2—N5键构成的线面角. 与0 K时相比, 300 K下RDX分子的α1, α2, δ分别减小了1.6°, 5.0°, 5.5°, 表明RDX分子的六元环发生了一定程度的扭曲. 在TATB分子中, θ表示∠C2C3C4, γ1表示C2—C3—C4所成平面与C3—N3键构成的线面角, γ2表示C1—C2—C3所成平面与C2—N2键构成的线面角. 与0 K时相比, 300 K下TATB分子的θ只增大了0.6°, 硝基与氨基的扭转使得γ1与γ2分别减小6.6°, 4.7°, 呈现出部分扭曲的现象.| RDX | Angle/(°) | TATB | Angle/(°) | ||||
| 0 K | 300 K | Change | 0 K | 300 K | Change | ||
| α1 | 113.7 | 112.1 | –1.6 | θ | 118.6 | 119.2 | +0.6 |
| α2 | 114.8 | 109.8 | –5.0 | γ1 | 179.9 | 173.3 | –6.6 |
| δ | 140.6 | 135.1 | –5.5 | γ2 | 179.1 | 174.4 | –4.7 |
表2300 K与0 K下RDX和TATB的键角对比
Table2.Bond angles of RDX and TATB under 300 K and compared with 0 K
根据预热后的分子构型, 我们基于含时密度泛函理论模拟了NM, RDX, TATB分子光激发载流子的动力学过程, 此处的激发为第一激发态. 图3给出了激发后含能分子不同时刻的原子结构快照, 可以看出含能分子的结构弛豫在电子激发后会立刻发生, 并在200 fs内出现明显的解离现象. 具体地, 图3(a)中NM分子中甲基的C—H键随时间演化发生伸缩振动, 同时C—H键绕着C—N键转动, 40 fs时甲基上的氢原子会逐渐向硝基上的氧原子靠拢, 并在95 fs时解离形成新的中间态; 图3(b)显示的RDX分子亚甲基上的一个氢原子在60 fs时发生解离, 同时, 附近的一个硝基也在80 fs时发生解离, 该过程同张伟等[49]利用时间质谱分析激光诱导RDX的解离产物一致; TATB分子中芳香环上相邻的硝基和氨基之间的分子内氢键将会影响光诱导下分子的解离途径, 图3(c)给出了氨基上的氢原子在60 fs时向邻近的氨基靠近, 直至85 fs时发生解离, 并与相邻硝基结合形成—HONO基团, 实现分子内的氢转移并形成一个新的过渡态, 这表明光激发将有利于TATB分子上硝基和氨基部分的激活, 而不是苯环的裂解.
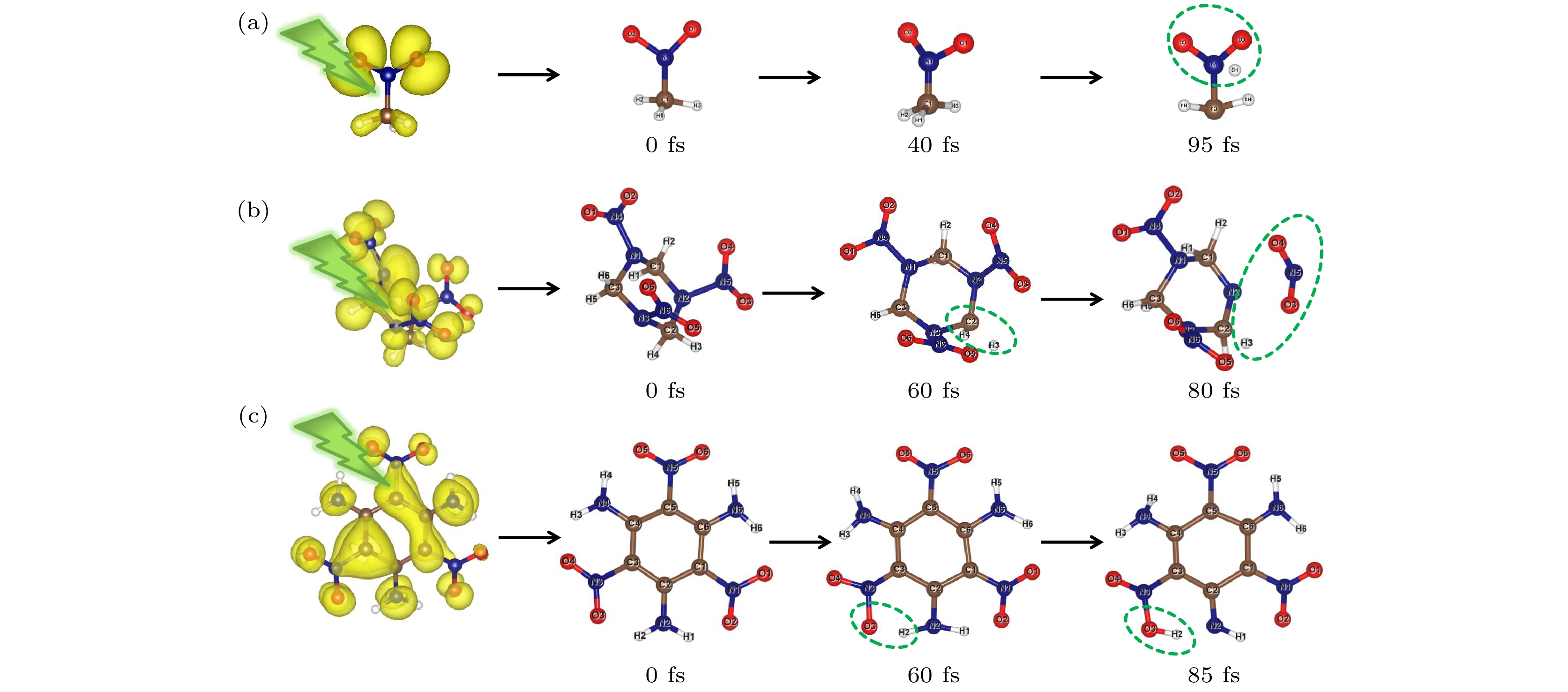 图 3 (a) 硝基甲烷(NM); (b) 环三亚甲基三硝胺(RDX); (c) 三氨基三硝基苯(TATB)在300 K时光致激发后分子结构随时间演化示意图
图 3 (a) 硝基甲烷(NM); (b) 环三亚甲基三硝胺(RDX); (c) 三氨基三硝基苯(TATB)在300 K时光致激发后分子结构随时间演化示意图Figure3. Time evolution of molecular structure at 300 K for (a) nitromethane (NM); (b) cyclotrimethylenetrinitramine (RDX); (c) triaminotrinitrobenzene (TATB).
为了分析光激发后含能分子上化学键的断裂过程, 我们给出了NM分子上连接硝基与甲基上C—N键和甲基上C—H键的键长随时间的演化, 如图4(a)所示. 在前65 fs C—N键发生伸缩振动, 键长先增加后减小, 频率为1240 cm–1, 之后C—N键进入新的振荡周期, 平均距离由1.50 ?减小到1.30 ?; 而对于甲基上的三个C–H键, 在前65 fs C—H键在平衡位置附近振荡, 随后激发电子的能量通过C—N键振动从硝基传递到甲基上, 导致甲基上的三个C—H键长明显增大, 在92 fs时C—H3键首先达到断裂的临界距离1.30 ?, 使得H3原子首先发生解离, 其后H1和H2原子也于104 fs, 105 fs相继解离. 这与Nelson等[50] 观察到硝基甲烷的光解离过程相一致, 即氢原子先解离并与硝基的一个氧原子行成新的中间态. 图4(b) 给出了RDX分子硝基与六元环上氮原子形成的三个N—N键的键长随时间的演变. 从图中可以看出, 在160 fs内, RDX分子的N1—N4, N3—N6键在平衡位置附近振荡; 而对于N2—N5键, 由于电子主要被激发到N5原子所在硝基的π反键轨道上, 激发电子的能量弛豫到邻近原子上, 使得N2—N5键键长显著增大, 并在78 fs时发生断裂, N2—N5键断裂的临界距离为1.73 ?. 对于钝感的TATB分子, 如图4(c)所示, 由于硝基与亚甲基上能量的重新分布, 六元环骨架发生弯曲振动, 环上的碳原子与硝基或氨基所形成的C—N键以0.20 ?的振幅在平衡位置振荡, 而氨基上的N2—H2键在80 fs前于平衡位置振荡, 之后逐渐增长并于85 fs时发生解离, N2—H2键断裂的临界距离为1.31 ?.
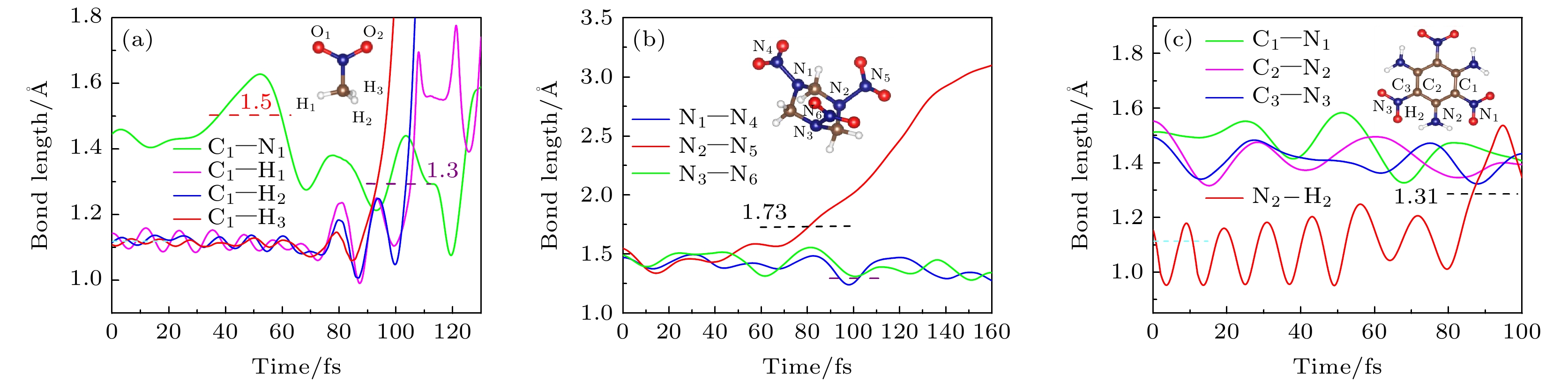 图 4 分子部分键长随时间的演化 (a) 硝基甲烷(NM); (b) 环三亚甲基三硝胺(RDX); (c) 三氨基三硝基苯(TATB)
图 4 分子部分键长随时间的演化 (a) 硝基甲烷(NM); (b) 环三亚甲基三硝胺(RDX); (c) 三氨基三硝基苯(TATB)Figure4. Time evolution of bond lengths: (a) Nitromethane (NM); (b) cyclotrimethylenetrinitramine (RDX); (c) triaminotrinitrobenzene (TATB).
2
3.3.激发态下NM, RDX, TATB分子轨道随时间的演化
为了深入理解光激发后含能分子的结构和键长随时间的演化规律, 我们进一步探究了含能分子光激发后的轨道能级随时间的演化, 如图5所示. 与基态相比, 图5(a)中NM分子在起始时刻的LUMO所在能级能量从–3.29 eV增加到–3.18 eV, HOMO所在能级能量从–6.35 eV减小到–7.25 eV, 说明初始温度从0 K升高到300 K, 会使得分子的能级展宽, 同时能级位置发生不同程度的变化. 随着时间的演化, 在0—60 fs内, HOMO能级缓慢上升而LUMO能级缓慢下降, 此时的振荡周期为22 fs, 表明激发电子和空穴不断向声子传输能量, 也说明该过程中载流子与声子发生了耦合作用, 通过这种耦合作用激发载流子不断向原子或者化学键传输能量, 这正如图4(a) 所示的在60 fs前C—N键键长不断增长; 在60 fs后HOMO能级进一步上升而LUMO能级进一步下降, 此时的振荡周期为22 fs, 激发电子的能量通过C—N键的伸缩振动从硝基传输到甲基上, C—N键伸缩振动振幅减小, C—H键伸缩振动振幅加剧, 在95 fs时, HOMO能级急剧上升而LUMO能级急剧下降, 此时氢原子已发生解离, 最终在104 fs时激发电子和空穴能级发生交叠. 图 5 分子轨道能级随时间的演化图 (a) 硝基甲烷(NM)分子HOMO, LUMO能级演化; (b), (c) 环三亚甲基三硝胺(RDX)分子HOMO, LUMO及其附近能级演化; (d) 三氨基三硝基苯(TATB)分子HOMO, LUMO能级演化. 绿色实线为最高占据态轨道, 对应激发空穴所在能级, 红色实线为最低非占据态轨道, 对应激发电子所在能级, 灰色实线为附近的其他能级
图 5 分子轨道能级随时间的演化图 (a) 硝基甲烷(NM)分子HOMO, LUMO能级演化; (b), (c) 环三亚甲基三硝胺(RDX)分子HOMO, LUMO及其附近能级演化; (d) 三氨基三硝基苯(TATB)分子HOMO, LUMO能级演化. 绿色实线为最高占据态轨道, 对应激发空穴所在能级, 红色实线为最低非占据态轨道, 对应激发电子所在能级, 灰色实线为附近的其他能级Figure5. Time evolution of excited energy level: (a) HOMO and LUMO of Nitromethane (NM); (b), (c) HOMO, LUMO and nearby orbitals of cyclotrimethylenetrinitramine (RDX); (d) HOMO and LUMO of Triaminotrinitrobenzene (TATB). Green solid line denotes the highest occupied molecular orbit corresponding to the excited hole, red solid line denotes the lowest unoccupied molecular orbit corresponding to the excited electron, gray solid lines denote other molecular orbit.
同样地, 由于初始温度和能级展宽的影响, 与基态相比, 图5(b)中RDX分子起始时刻的LUMO所在能级能量从–2.69 eV增加到–2.42 eV, HOMO所在能级能量从–5.95 eV减小到–6.85 eV, RDX分子被激发后激发电子能级能量整体呈下降趋势, 而其附近非占据态轨道能量呈现上升趋势; 与此同时, 图5(c)中激发空穴能级能量整体呈上升趋势, 而其附近占据态轨道能量呈现下降趋势, 表明激发载流子在进行能量弛豫的过程中, 还存在与附近电子态或分子能级的耦合作用, 但最终激发载流子的能量都以热能的形式传输给声子. 在前60 fs, LUMO能级缓慢下降而HOMO能级变化较小, 能带隙小幅度减小, 这也与图4(b)给出的氢原子在60 fs时解离相一致; 在60 fs后LUMO能级下降加速而HOMO能级发生明显上升, 此后激发电子能级能量不断降低, 激发空穴能级能量不断升高, 能带隙急剧减小, 并在122 fs时激发电子和空穴能级发生交叠.
图5(d)为TATB分子激发后分子轨道随时间的演化图, 300 K时, LUMO所在能级能量从–2.71 eV增加到–1.98 eV, HOMO所在能级从–5.33 eV减小到–6.51 eV, 光激发后, HOMO所在能级能量总体呈上升趋势, LUMO所在能级能量呈下降趋势. 其中在起始20 fs内, LUMO能级能量快速下降, 从–1.98 eV下降到–3.81 eV, 此时γ2减小了4.0°. 在20—90 fs范围内, 激发空穴能级能量明显上升, 激发电子能级下降缓慢, 此过程中苯环发生扭曲形变, 氨基上的一个氢原子发生解离并完成分子内的氢转移, 由于分子构型的明显改变, 在90 fs后, 激发电子能级表现出了上升的趋势. 相比NM和RDX, TATB分子的激发能级演化有明显的较低频率(声子频率)振荡, 表明钝感炸药TATB分子中电子态与声子耦合较弱, 更不易被激发解离.
进一步, 通过含能分子第一激发态下HOMO, LUMO及其附近轨道能级的演化给出了分子的解离过程的物理解释. 发现初始温度升高到300 K, 使得分子能级展宽, 能级位置发生不同程度的变化. 随着时间的演化, 激发电子能级不断下降, 激发空穴能级不断上升, 并伴随有不同频率的振荡, 表明电子-声子耦合作用在激发载流子的能量弛豫过程中发挥了重要作用, 激发载流子的能量通过电声耦合以热能形式传输给原子, 从而导致N—H, C—H或N—N化学键的断裂. 分子发生明显解离或者扭转之后, 激发电子或激发空穴的能级发生不同寻常的变化, 比如HOMO与LUMO交叉或者LUMO轨道的上升, 这与分子构型的明显改变相关联. 在此过程中, 我们还发现RDX分子中激发电子附近的能级有上升趋势, 而激发空穴附近能级有下降趋势, 表明电子-电子耦合也在载流子的能量弛豫过程中发挥了作用. 因此, 本文从分子水平上描述的硝基类炸药被光激发后飞秒尺度内的超快激发态动力学过程, 验证了Ehrenfest定理可以有效地模拟光诱导反应的起始步骤, 加深了对含能材料激发态结构演化和光激发解离反应的理解, 为揭示含能材料超快复杂的爆轰过程提供了一种有效的理论手段.
