全文HTML
--> --> -->从原子层面讲, 硅的化合价半径类似于磷的化合价半径, 且两原子相对原子质量接近, 使得两者之间具有很多联系. 黑磷烯作为磷单质的二维材料, 与硅这种元素相结合亦有许多应用. 首先是有关黑磷烯的制备方面. 从由最初的机械剥离方法[13,14]到近期报道的一种在硅基底上生长高结晶性黑磷薄膜的新方法, 黑磷烯在生产制备方面取得了极大地进展. Rajabali等[14,15]通过激光激发在硅衬底上形成高度结晶的薄片来形成磷烯. Xu等[16]通过气相生长策略在绝缘硅衬底上直接生长大规模晶体黑磷烯薄膜. 这种直接在硅衬底上生长的黑磷烯具有层数少、制造面积大、稳定性强、可调性强等优点.
其次是黑磷烯在等离子体方面的应用[17,18]. Prajapati等[19]利用硅-黑磷-TDMC涂层表面等离子体共振生物传感器提高灵敏度. Kumar等[20]探讨了硅混合黑磷纳米结构表面等离子共振生物传感器的灵敏度. Kumar等[21]采用五层黑磷-硅基隧道场效应晶体管克服热离子限制.
再次是黑磷烯在硅基电池方面的应用. 硅基材料用作阳极的硅基锂离子电池在使用和制造过程中存在一些弊端, 如材料粉碎、活性材料晶粒间导电损失[22,23]、活性物质损失等[22,24]. 这些缺陷制约了硅基锂离子电池的发展. 而黑磷烯由于其特殊性能[25-28], 在能量存储器件中具有应用前景. Zhang等[27]报道了锂离子在磷烯中的扩散速度很快, 在硅阳极中引入磷烯有望促进锂离子的吸收. Park和Sohn [29]的研究表明多层黑磷-碳复合材料用作电池表现出优异的循环性能. 一些研究者[30-34]将磷掺杂到作为硅基电池材料的硅晶格中进行研究, 结果表明磷是改善锂离子电池中硅阳极电化学性能的有效方法. Peng等[35]从石墨烯等纳米结构碳质材料表面封装中得到启发, 使用单层黑磷烯包覆硅颗粒来提升硅基负极材料的电化学性能.
由此可见, 磷烯与硅相结合的应用较为广泛, 在硅基底上生长黑磷烯时, 衬底硅对其电子性质有着不可忽视的影响. 当黑磷用作等离子体传感器及晶体管时, 硅层和黑磷层直接接触, 其层间影响对传感器灵敏度和场效应晶体管影响较大. 在黑磷烯用于包覆硅基锂离子电池阳极材料硅时, 两者相互接触. 所以研究硅原子与黑磷之间的相互作用十分必要. 目前, 使用第一性原理方法对磷烯与硅之间的研究仅局限于掺杂方面, Shojaei等[36]研究了硅掺杂单层和双层磷烯纳米带中的硅-硅键效应. 结果表明: 双层磷烯纳米带掺杂硅后, 结构都变成了间接间隙半导体. Olmedo等[37]研究了硅掺杂的磷烯纳米片物性, 结果表明, 掺杂使几何结构改变, 磷烯纳米片带隙减小. 但以上研究均基于实验方法, 侧重黑磷烯制备及应用方面, 并非直接探寻黑磷烯与硅原子之间的相互作用. 仅有的几个理论模拟也均为硅原子掺杂黑磷烯物性研究, 而层间接触、包覆材料等则需要通过吸附特性进行深入研究. 目前, 有关黑磷烯吸附Si原子的研究未见报道. 所以本文基于第一性原理研究黑磷烯吸附硅原子的电子特性, 并期望对其电子特性实现稳定调控. 希望该研究对于硅电池、半导体器件、等离子体生物传感器有指导作用.
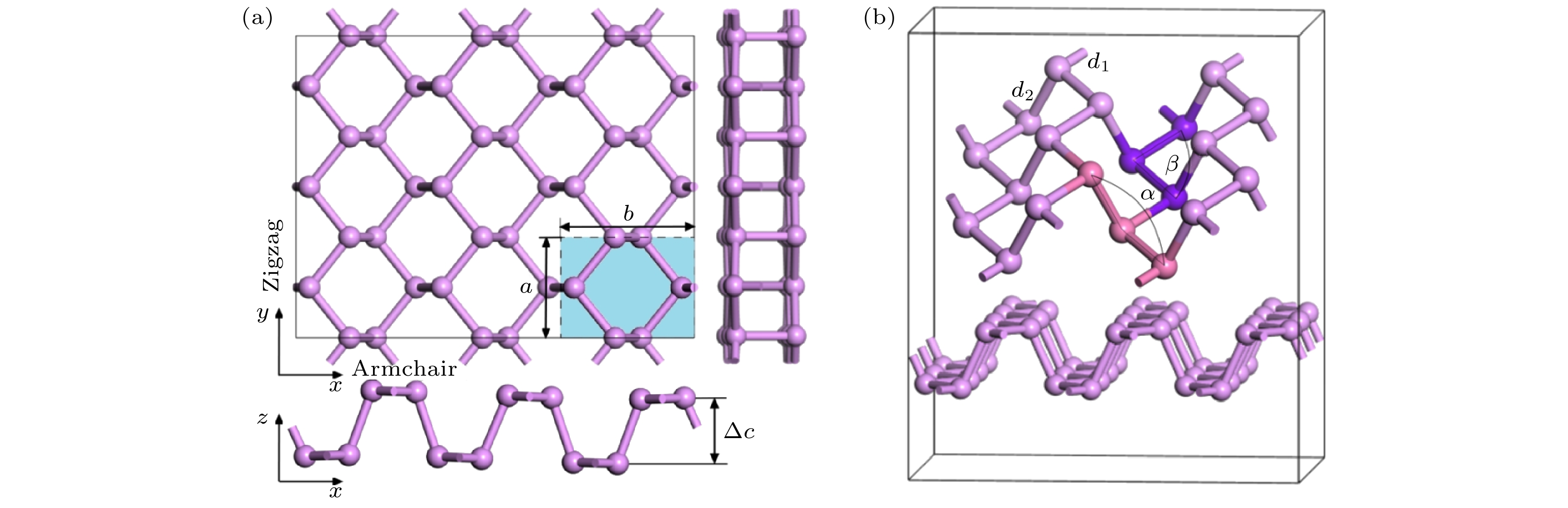 图 1 本征黑磷烯模型 (a)黑磷烯的主视图、俯视图、侧视图; (b)黑磷烯结构示意图
图 1 本征黑磷烯模型 (a)黑磷烯的主视图、俯视图、侧视图; (b)黑磷烯结构示意图Figure1. Intrinsic black phosphorene model: (a) Front view, top view, and side view of black phosphorene; (b) schematic diagram of the structure of black phosphorene.
首先对黑磷烯原胞进行几何结构优化, 结构优化后其晶格常数a = 3.313 ?, b = 4.374 ?, 与文献[44-48]相符. 然后对扩展后的黑磷烯超胞进行结构优化, 优化后的黑磷烯片层厚度Δc = 2.093 ?, 与文献[46]的模拟结果2.110 ?相比误差为0.081%; P—P原子的键长d1 = 2.210 ?, d 2 = 2.230 ?, 与文献[44, 48]的结果相符; 键角α = 103.016°, β = 98.539°与文献[48, 49]的模拟结果非常接近, ∠α与文献[50]所得的角度102.31°相比误差为0.69%. 说明本文参数设置合理.
形变和电场会影响材料的稳定性, 通常较大的形变会导致稳定性降低, 单原子结合能的定义式[51-55]为
此时黑磷吸附体系的吸附能Ead[56]可以表示为
3.1.本征黑磷烯电子特性
根据(1)式得到的单原子结合能为–5.774 eV. 计算黑磷电子态密度时, 布里渊区积分采用Monkhorst-Pack特殊K点对全布里渊区求和, 能带计算高对称K点路径为 Γ (0, 0, 0)→Y (0, 0.5, 0)→L (0.5, 0.5, 0)→X (0.5, 0, 0) →Γ (0, 0, 0). 从图2可以看出, 纯单层黑磷晶体价带顶(VBM)出现在Γ点, 导带底(CBM)非常靠近Γ点. 体现为近直接带隙半导体, 这点与文献[57, 58]得到的结论相同. 其带隙显示为1.578 eV, 与文献[48, 59-63]相同. 态密度(DOS)图表明黑磷烯在导带内及价带高能级部分能级主要由P原子的p轨道贡献, 而价带的低能级区主要由P原子的s轨道主导.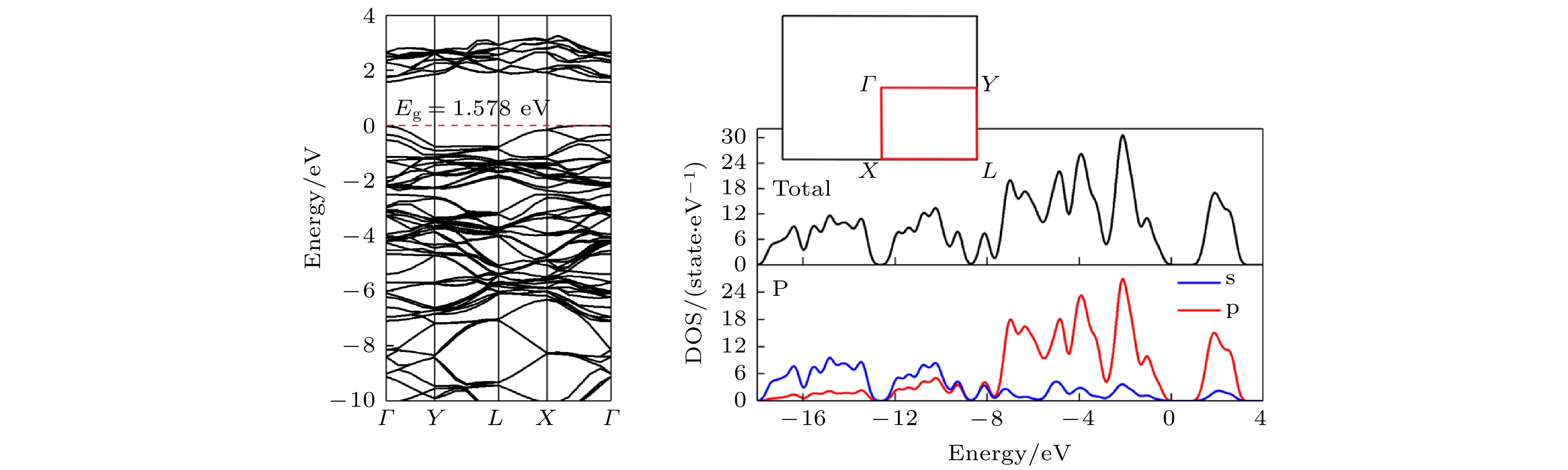 图 2 黑磷烯能带结构和DOS
图 2 黑磷烯能带结构和DOSFigure2. Band structure and DOS of black phosphorene.
2
3.2.吸附Si原子前后黑磷烯体系物性变化
33.2.1.黑磷烯吸附Si原子模型建立及吸附特性分析
在优化好的黑磷烯超胞结构P原子层上方吸附Si原子. 考虑图3所示的顶位(T)、桥位(B)和空位(H) 3个吸附位. 为了方便表述, 将黑磷烯吸附Si原子超晶胞体系中的磷原子依次编号, 如图3(c)所示. 对吸附体系来说, 吸附原子的百分比的影响非常重要[64]. 选取吸附位H, 建立了覆盖度为2.778%, 5.556%和8.333%的黑磷烯吸附Si原子体系结构模型, 在结构优化的基础上进行了电子特性计算. 结果表明吸附原子Si的覆盖度对于黑磷烯电子特性有着巨大影响, 覆盖度由2.778%增加到5.556%时, 带隙由0锐增为0.731 eV, 之后覆盖度增加为8.333%时, 黑磷吸附体系带隙又减小到0.216 eV. 由于覆盖度为2.778%的黑磷烯吸附Si原子模型使带隙改变较为明显, 引发了其由半导体至准金属的改变, 研究意义较大. 所以如未特殊说明, 本文接下来的计算均选取覆盖度为2.778%的黑磷烯吸附Si原子模型, 即超胞内含有1个Si原子和36个P原子.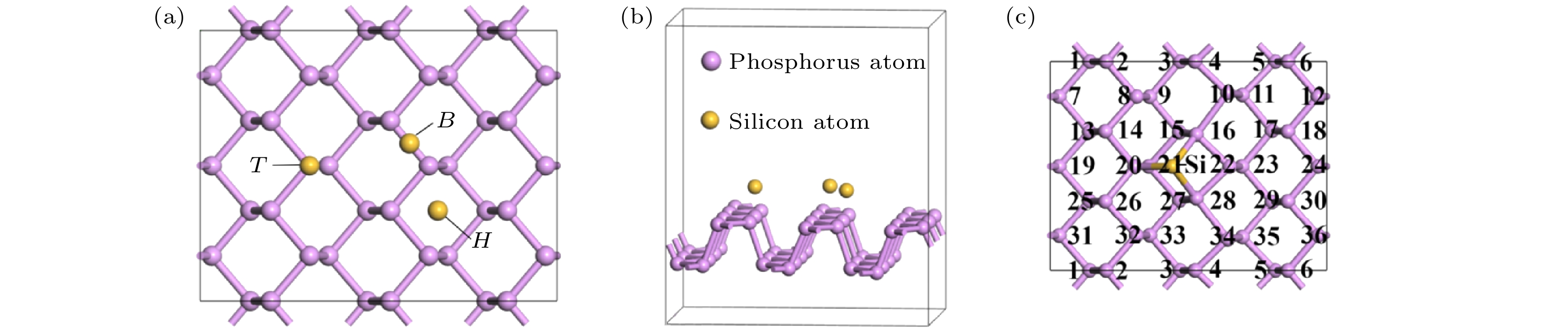 图 3 黑磷烯吸附Si原子模型 (a)主视图; (b)示意图; (c) P原子编号示意图
图 3 黑磷烯吸附Si原子模型 (a)主视图; (b)示意图; (c) P原子编号示意图Figure3. Si adsorbed on black phosphorene model: (a) Main view; (b) diagrammatic sketch; (c) numbering diagram of P atom.
黑磷烯吸附Si原子几何优化后的结构如图4所示. 可以看出Si原子吸附在黑磷烯上后, 吸附原子附近的黑磷烯平面略微向下凹陷, 对Si原子形成略微包裹的趋势. 根据(2)式计算了体系的吸附能, T位吸附能为3.507 eV, B位为2.751 eV, H位为3.657 eV, 均属于可逆化学吸附. 可知当Si原子吸附在P—P原子环中间的H位时体系能量最小, 吸附能最大, 表示Si原子吸附在黑磷烯表面越稳定. 将吸附高度d0定义为Si原子到BP表面的垂直距离. 为表征其吸附特性及成键稳定性, 将最短P—Si键键长, 吸附原子Si所在P原子环上的4个P—P键键长和吸附高度列于表1. 结合图4的结构优化图和表1可以看出, Si原子吸附在黑磷烯的B位上键长改变最大, 几何形变最强烈, 吸附高度最高. 吸附能也表明此结构稳定性最弱. 几何优化后, 在黑磷烯上T位吸附的Si原子被自动优化到H位上. 两个模型的键长相差不大, 几何结构相似, 吸附能相近. 因此, 后续的计算都将Si原子吸附在黑磷烯的H位.
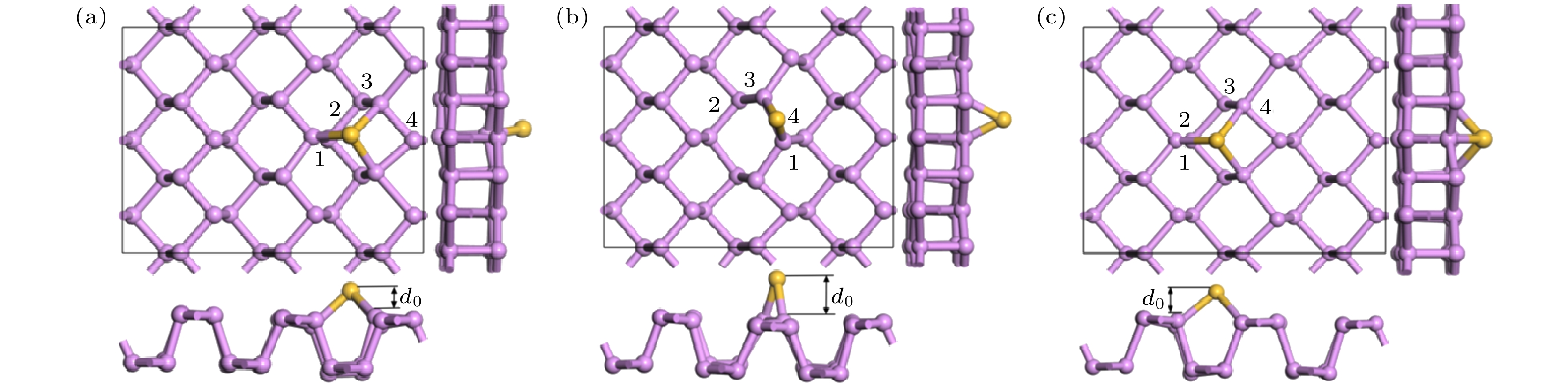 图 4 黑磷烯吸附Si原子模型几何优化结构 (a) T位; (b) B位; (c) H位
图 4 黑磷烯吸附Si原子模型几何优化结构 (a) T位; (b) B位; (c) H位Figure4. Geometry optimization of Si adsorbed on black phosphorene model: (a) T site; (b) B site; (c) H site.
| 吸附位 | P—Si/ | P—P(1)/? | P—P(2)/? | P—P(3)/? | P—P(4)/? | d0/? |
| T | 2.283 | 2.214 | 2.206 | 2.206 | 2.207 | 1.138 |
| B | 2.236 | 2.256 | 2.186 | 2.236 | 2.212 | 1.880 |
| H | 2.312 | 2.221 | 2.200 | 2.204 | 2.230 | 1.160 |
表1吸附原子所在原子环的P—P键键长与Si原子吸附高度
Table1.Relationship between P—P bond length and Si adsorption height.
3
3.2.2.黑磷烯吸附Si原子电子特性
图5和图6展示了本征黑磷烯和黑磷烯吸附Si原子的能带和态密度结构. 可以看出Si原子吸附在黑磷烯上的T, B和H位上时, 带隙分别为0.055, 0.072和0 eV. T位和B位吸附的黑磷烯体系带隙均为间接带隙且小于0.1 eV, 体现出准金属特性. H位吸附体系导带和价带之间发生重叠, 禁带消失, 电子可以无障碍地达到导带.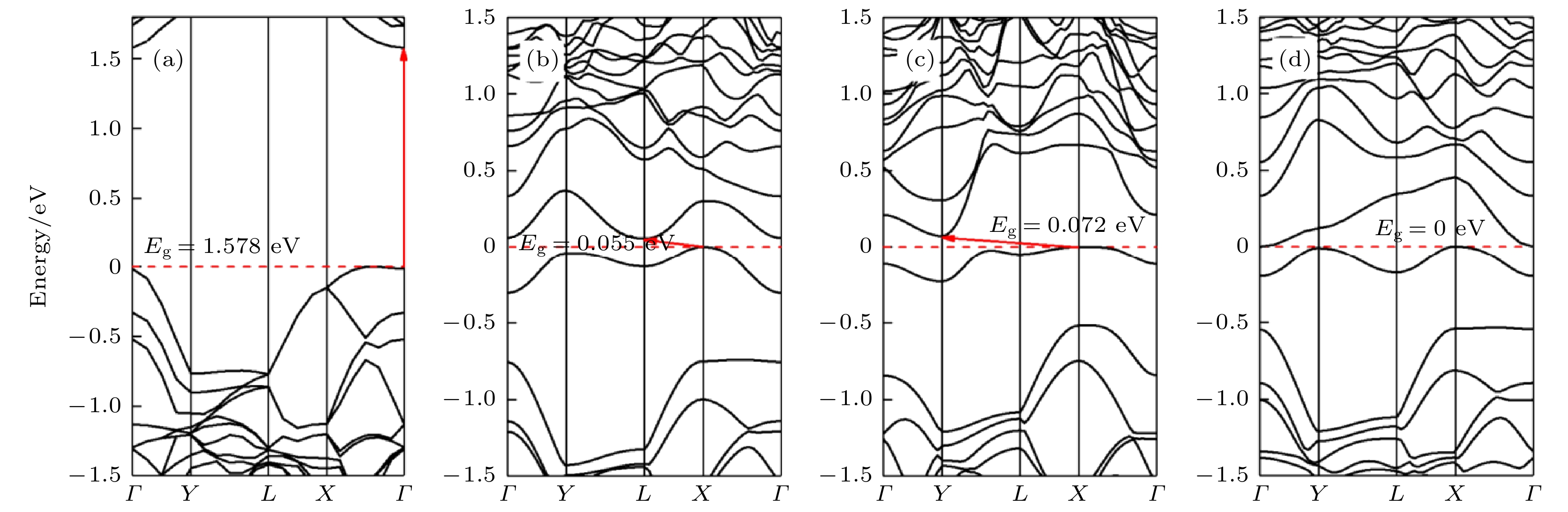 图 5 黑磷体系能带结构 (a)本征黑磷烯; (b) T位吸附; (c) B位吸附; (d) H位吸附
图 5 黑磷体系能带结构 (a)本征黑磷烯; (b) T位吸附; (c) B位吸附; (d) H位吸附Figure5. Band structure of black phosphorene system: (a) Intrinsic black phosphorene; (b) T site adsorption; (c) B site adsorption; (d) H site adsorption.
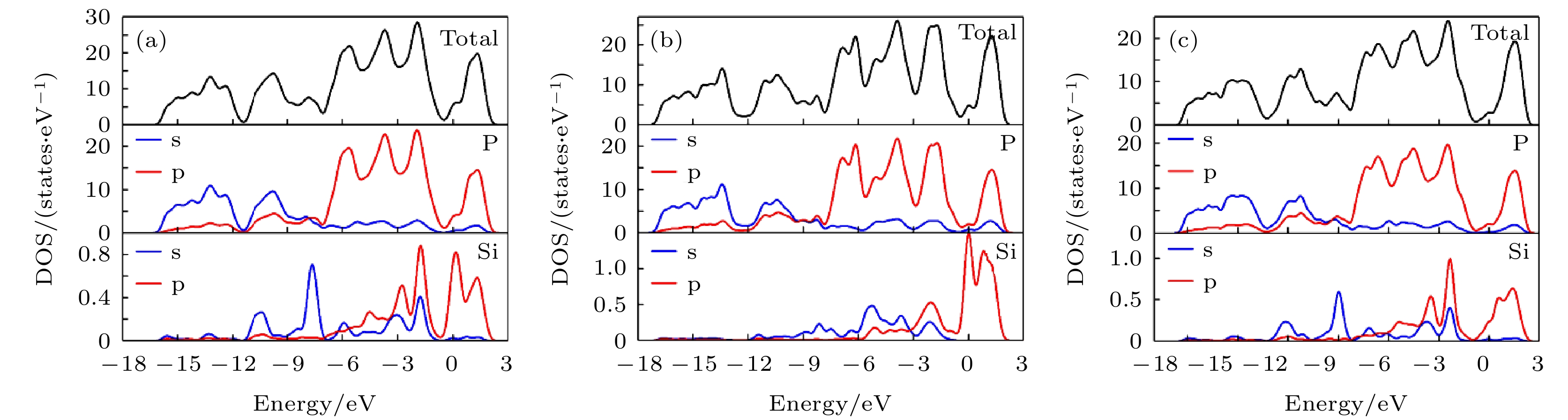 图 6 黑磷烯吸附体系DOS (a) T位; (b) B位; (c) H位
图 6 黑磷烯吸附体系DOS (a) T位; (b) B位; (c) H位Figure6. The DOS of Si adsorbed on black phosphorene system: (a) T site; (b) B site; (c) H site.
为了探究H位的Si吸附使黑磷烯结构禁带消失的机理, 研究了吸附体系每一个P原子在费米能级附近的DOS贡献, 并将贡献较多的原子按照图例颜色在模型中依次显示, 如图7所示. 从图7可以看出, 在费米能级处, 不完全是Si原子的贡献, 大部分是黑磷烯本身的贡献, 按照贡献由大到小排列依次为P10, 34 > P16, 28 > Si > P5 > P22 > P20 > P14, 26 > P4. 其余原子贡献低于0.05 eV, 固此处不予讨论. P原子的分波态密度(PDOS)曲线表明, 吸附Si原子后的黑磷烯在结构上仍然具有对称性, P10, 16, 14与P34, 28, 26的DOS曲线重合. 贡献较高的P原子集中在吸附原子附近沿zigzag方向的一排P原子(即图7中编号为4, 10, 16, 28和34的一排磷原子). 为了从几何构型、成键、轨道重叠等方面解释禁带消失的原因, 接下来给出了费米能级处DOS贡献较多的P原子间键长、Mulliken键级, 所有P原子的电荷布居数, 以及吸附体系的电荷差分密度及电荷密度.
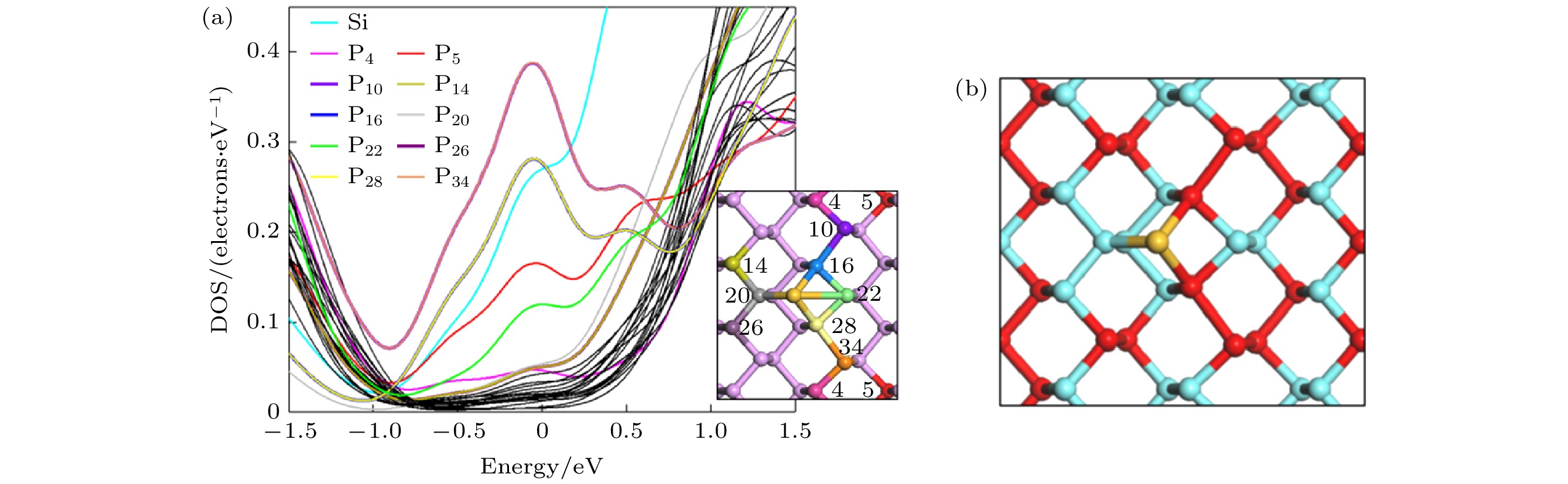 图 7 (a)单个P原子DOS图; (b) P原子得失电子示意图; 红色球体代表得到电子的P原子, 蓝色球体代表失去电子的P原子
图 7 (a)单个P原子DOS图; (b) P原子得失电子示意图; 红色球体代表得到电子的P原子, 蓝色球体代表失去电子的P原子Figure7. (a) DOS diagram of single P atom; (b) schematic diagram of gain and loss of electrons of P atom. The red sphere represents the P atom that gets electrons, and the blue sphere represents the P atom that loses electrons.
表2列出了P原子间键长及键级. 从几何构型来说, Si原子的吸附改变了黑磷烯的局域结构, 吸附原子附近的P原子位置及原子间键长改变剧烈. 吸附破坏了黑磷烯armchair方向的原始对称性, 但是仍旧保留了其zigzag方向的部分对阵性. 过吸附原子沿x方向做轴线, 黑磷烯的上半部分与下半部分几何形状完全一致. 这点也与DOS得到的结论一致. 正是由于armchair方向的原始对称性被破坏, 黑磷烯平面发生小范围形变, P—P键键长与键角变化, 导致磷原子间σ轨道重叠程度改变. 再杂化作用使得其电子特性变化, 禁带消失.
| P4—P10, P34—P4 | P10—P16, P28—P34 | P16—P22, P22—P28 | 本征P—P | P16, 28—Si | P20—Si | |
| 键长/? | 2.162 | 2.497 | 2.210 | 2.210 | 2.331 | 2.312 |
| 键级 | 0.48 | 1.00 | 0.45 | 0.47 | 0.42 | 0.33 |
表2P原子间键长及键级
Table2.Bond length and bond order between P atoms.
将吸附体系中磷原子Mulliken电荷布居数列于表3, 图8和图9显示了体系的电荷差分密度和电荷密度. 结合图表对电荷转移及成键类型进行分析. 从表3、图8和图9可以看出, Si原子吸附到黑磷烯表面, 失去了0.22e的电子, 全都转移到了黑磷烯上. Si附近的3个P原子P16, 28和P20的电荷布居数分别为–0.06e和0.06e, 表明它们得到和失去电子的数量为0.06e. 通过电荷差分密度图及电荷密度图也可证实, P原子与Si原子相互之间有强烈的电子迁移, 成键离子性很强. 磷原子中电子改变最为剧烈的原子当属P10和P34, 它们得到的电子数量为0.1e. 这点也体现在差分电荷密度和电荷密度图中, P10—P16和P28—P34之间几乎无电子转移, 离子性很弱. 这可能是由于吸附Si原子后, P10—P16和P28—P34键被拉长, 两原子轨道重叠程度降低, 分子轨道重新排布后更多电子聚集在P原子上. 它们的键级由本征P—P的0.47变为1, 说明这两对P原子成共价键. 正是由于吸附Si原子后, Si原子与黑磷烯间形成离子键. 强烈的电子转移更是改变了黑磷烯的电子体系, 加剧了黑磷烯本身P原子间的电荷转移, 改变部分原子成键类型. Si的吸附也在费米能级处引入了一条杂质能级. 这可能就是Si原子使黑磷烯体系能带结构发生变化, 使其由半导体完成向准金属转变的原因.
| 原子编号 | P10, 34 | P16, 28 | P11, 35 | P8, 12, 13, 25, 32, 36 | P1, 7, 9, 17, 24, 29, 31, 33 | P2, 15, 18, 19, 23, 27, 30 | P3, 4, 14, 26 | P5, 22 | P6 | P20 | P21 | Si |
| Total/e | 5.10 | 5.06 | 5.04 | 5.02 | 5.01 | 4.99 | 4.98 | 4.97 | 4.96 | 4.94 | 4.93 | 3.78 |
| Charge/e | –0.10 | –0.06 | –0.04 | –0.02 | –0.01 | 0.01 | 0.02 | 0.03 | 0.04 | 0.06 | 0.07 | 0.22 |
表3P原子的Mulliken电荷布居数
Table3.Mulliken charge population of P atom.
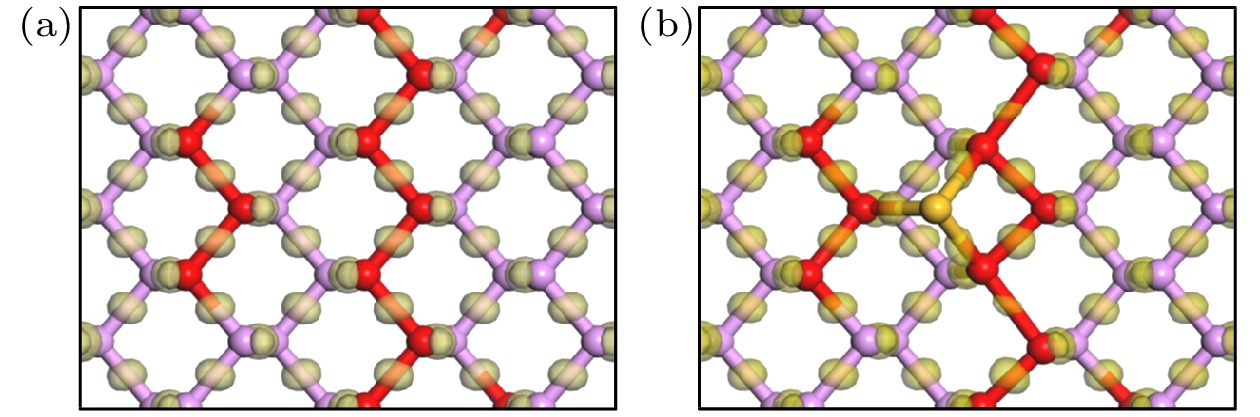 图 8 黑磷烯电荷差分密度图 (a)本征黑磷烯; (b)黑磷烯吸附Si原子
图 8 黑磷烯电荷差分密度图 (a)本征黑磷烯; (b)黑磷烯吸附Si原子Figure8. Differential charge density of the black phosphorene: (a) Intrinsic black phosphorene; (b) Si adsorbed on black phosphorene system.
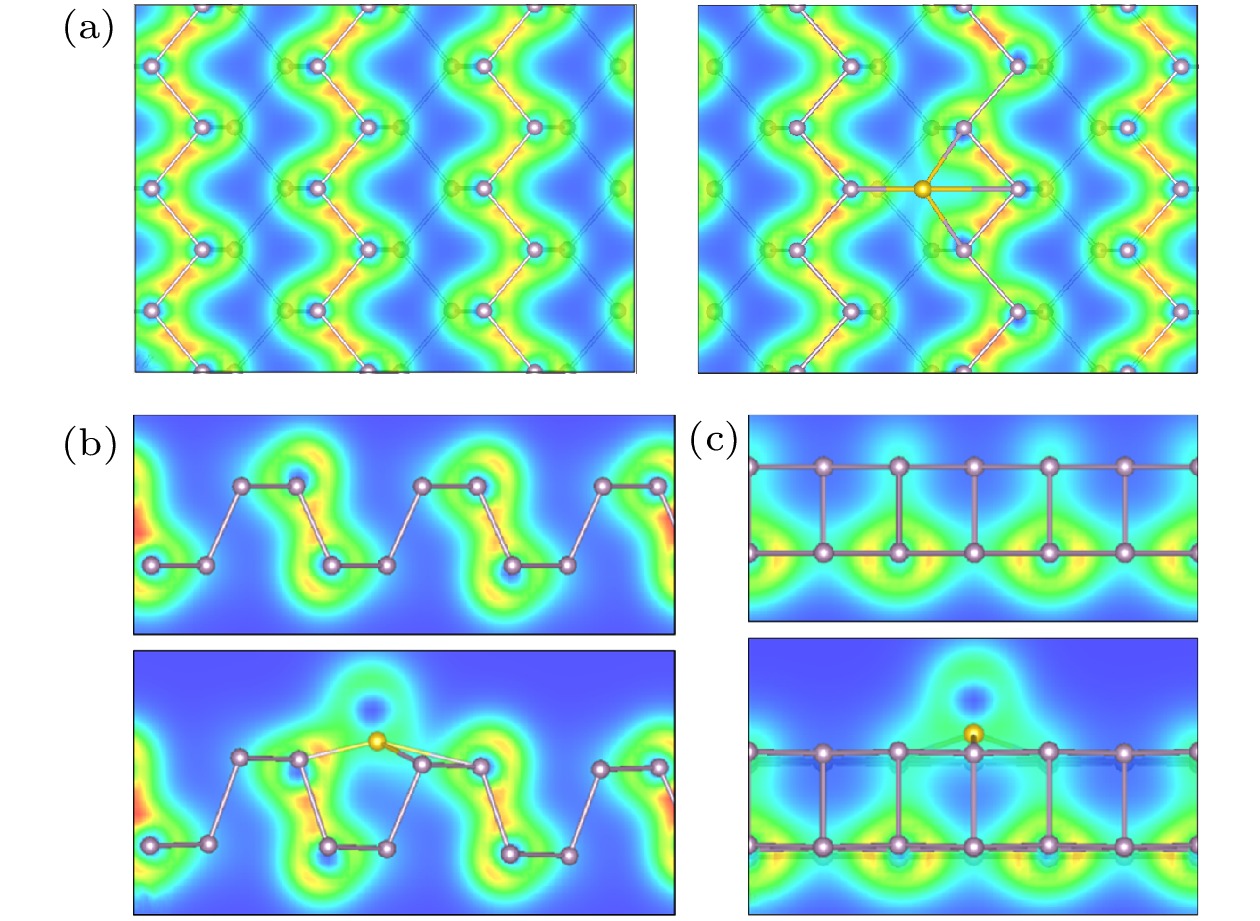 图 9 本征黑磷烯与黑磷烯吸附体系电荷密度图 (a)主视图; (b)俯视图; (c)侧视图
图 9 本征黑磷烯与黑磷烯吸附体系电荷密度图 (a)主视图; (b)俯视图; (c)侧视图Figure9. Charge density diagram of the adsorption system of intrinsic black phosphorene and black phosphorene: (a) Main view; (b) top view; (c) side view.
2
3.3.拉伸形变对黑磷烯吸附Si原子体系影响
33.3.1.黑磷烯拉伸模型建立及结构稳定性分析
众所周知, 黑磷烯具有强各向异性和负泊松比等特性. 许多****的计算结果表明, 其电学性能, 如带隙等对外加形变非常敏感[2,8,54,58,60-63,65-68], 会引起黑磷直接带隙与间接带隙的转变[60,62,63,68], 甚至会引起其由半导体到金属的改变[58]. 也有许多研究者对黑磷烯进行拉伸和压缩实验[69-72]. Peng等[68]发现, 磷烯可以分别承受高达10 N/m和30%的拉伸应力和应变. 已有研究表明, 黑磷烯所受应变在15%以内时, 其晶体结构没有任何明显损伤[73,74]. 基于此, 对黑磷烯吸附Si原子体系施加形变, 希望实现其带隙调控. 通过改变晶格参数的方式沿armchair方向对黑磷烯体系施加2%—10%的拉伸形变, 所有结构均被充分驰豫. 黑磷烯的拉伸形变的程度用形变量ε来表征, 将其定义为ε = (c – c0)/c0, 式中c0和c分别表示黑磷烯原胞的晶格长度和拉伸后的黑磷烯晶格长度[75]. 由于黑磷烯具有强各向异性和奇特的负泊松比的现象, 充分考虑泊松比的影响, 选取拉伸形变量为2的黑磷烯进行试算. 黑磷烯在armchair和zigzag两个方向上的泊松比分别为0.01和0.16[76], 垂直于黑磷烯片层方向上的泊松比为–0.027[77]. 在对黑磷烯的armchair方向施加的形变量ε = 2%时, 在zigzag方向同时施加–0.16Δc = –0.020 ?的形变, 垂直于平面的z方向施加0.027Δc = 0.007 ?. 试算得到的能带结构如图10所示, 图10(a)—(d)分别表示考虑泊松比前后, 拉伸形变为2%的纯黑磷烯、黑磷烯吸附Si原子, 电场作用的纯黑磷烯、电场作用的黑磷烯吸附Si原子的能带结构. 从图10可知, 泊松比对于黑磷烯体系的能带结构影响较小, 其曲线形状几乎没有改变, 带隙也仅具有极其微小的改变. 这表明泊松比对黑磷的电子特性影响较小. 如果未特殊说明, 本文计算结果均未考虑泊松比.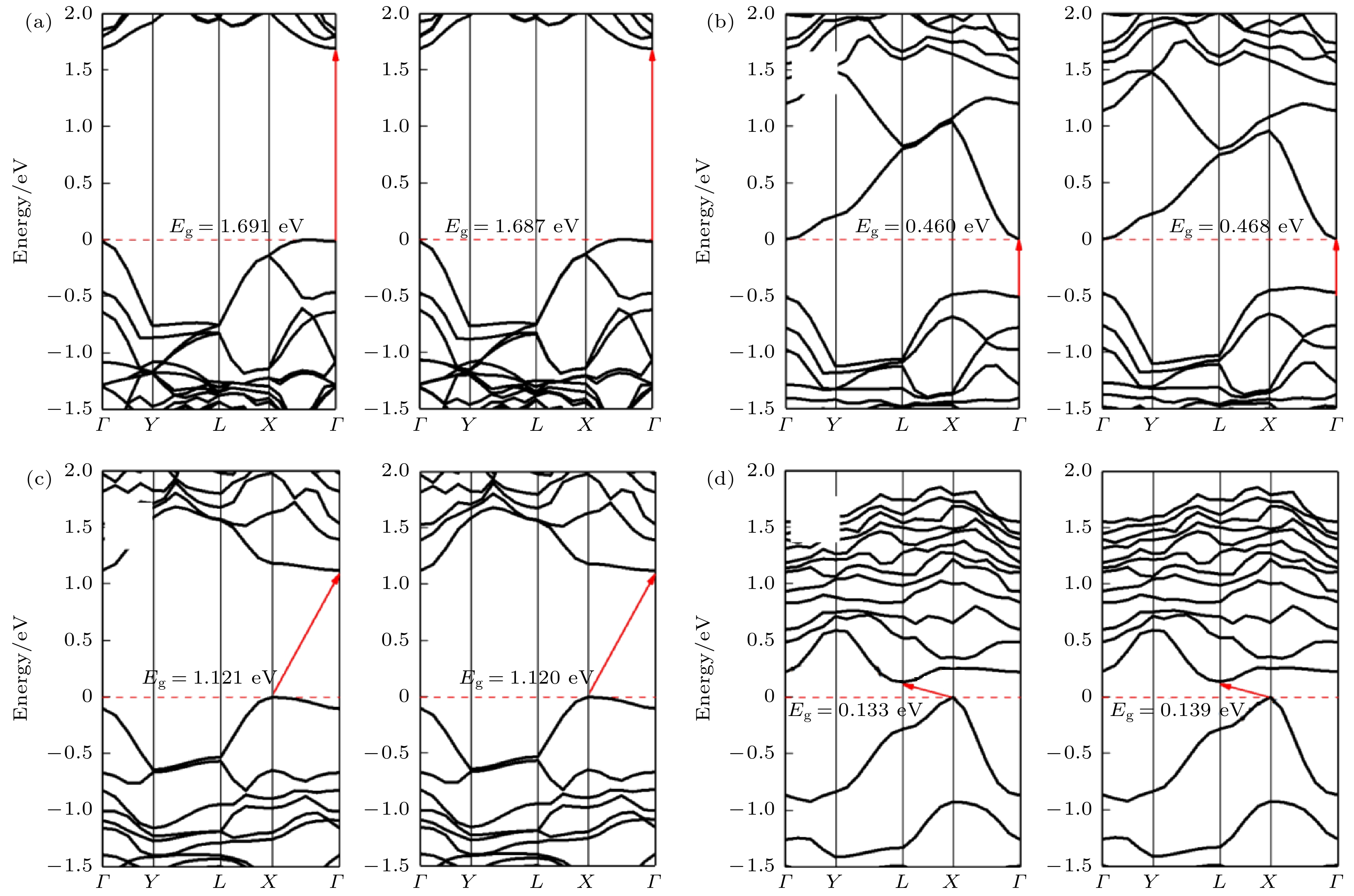 图 10 考虑泊松比前后、拉伸形变量为2%的黑磷烯能带结构 (a)纯黑磷烯; (b)黑磷烯吸附Si原子; (c)电场与形变共作用的纯黑磷烯; (d) 电场与形变共作用的黑磷烯吸附Si原子
图 10 考虑泊松比前后、拉伸形变量为2%的黑磷烯能带结构 (a)纯黑磷烯; (b)黑磷烯吸附Si原子; (c)电场与形变共作用的纯黑磷烯; (d) 电场与形变共作用的黑磷烯吸附Si原子Figure10. Band structure of black phosphorene with 2% tensile deformation before and after considering Poisson’s ratio (a) Pure BP, (b) Si absorbed on BP, (c) pure BP with co-action of electric field and deformation, (d) Si absorbed on BP with co-action of electric field and deformation.
根据(1)式, 首先计算了未吸附Si的纯黑磷烯体系在不同拉伸程度下的单原子结合能; 根据(2)式, 计算得到了拉伸黑磷烯吸附Si原子吸附能; 具体结果见表4.
| 形变量/% | 0 | 2 | 4 | 6 | 8 | 10 |
| 结合能/eV | –5.774 | –5.755 | –5.719 | –5.703 | –5.701 | –5.688 |
| 吸附能/eV | 3.970 | 3.914 | 3.864 | 3.683 | 3.652 | 3.657 |
表4拉伸形变作用下纯黑磷烯单原子结合能和黑磷烯吸附Si原子吸附能
Table4.Monoatomic binding energy of black phosphorene and adsorption energy of Si adsorbed on black phosphorene under tensile deformation.
从表4可以看出, 在当前研究范围内, 对于纯黑磷烯来说, 其他体系的结合能的绝对值呈现出随形变量增大而略微减小的趋势, 但变化较为微小, 说明形变对于黑磷烯体系能量影响较小. 类似地, 对于黑磷烯吸附Si原子体系来说, 形变略微降低了吸附能力, 但所有模型均属于化学吸附, 吸附过程存在电子转移, 伴随化学键的生成, 体系内原子重新排列.
3
3.3.2.黑磷烯吸附Si原子拉伸体系电子特性
不同拉伸程度的纯黑磷烯能带如图11(a)—(e)所示. 拉伸改变了黑磷烯的准直接带隙. 当形变量增加为6%时, 黑磷烯开始显现出间接带隙. 形变量为10%的体系的CBM能带较为疏散, 说明光产生电子的有效质量低, 迁移率高, 光生电子跃迁到CBM的概率更大. 除了8%的体系较为特殊, 其余体系的带隙大体呈现出随形变量的增加先增加后略微降低的趋势. 而形变量为8%时, 带隙骤降. 这是由于在拉伸程度为8%时, 黑磷烯平面发生较大的几何变形, 其结构俯视图如图12所示. 这种波纹状的几何形变加剧了黑磷烯P原子间电荷转移, 使其电子结构发生改变. 观察能带结构还可发现, 拉伸系统的价带能量几乎保持不变, 导带距电子势垒的远离或者接近导致了带隙的减小或增大. 这说明此时带隙的改变主要依赖于应变引起的导带底部能量变化.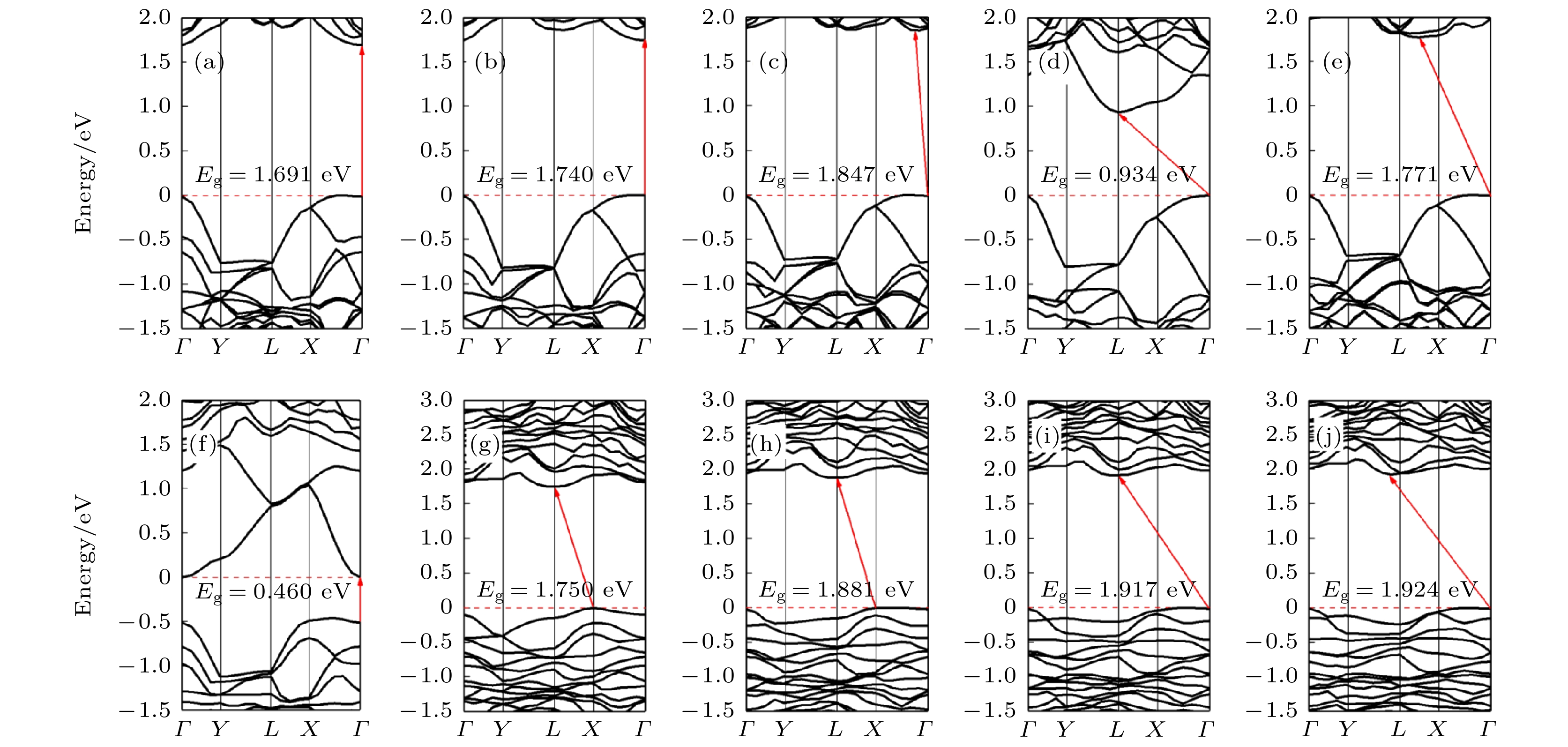 图 11 (a)—(e)拉伸形变量为2%—10%的黑磷烯能带结构; (f)—(j)拉伸形变量为2%—10%的黑磷烯吸附Si原子体系能带结构
图 11 (a)—(e)拉伸形变量为2%—10%的黑磷烯能带结构; (f)—(j)拉伸形变量为2%—10%的黑磷烯吸附Si原子体系能带结构Figure11. (a)?(e) Band structure of black phosphorene with 2%?10% tensile deformation; (f)?(j) band structure of Si adsorbed on black phosphorene with 2%?10% tensile deformation.
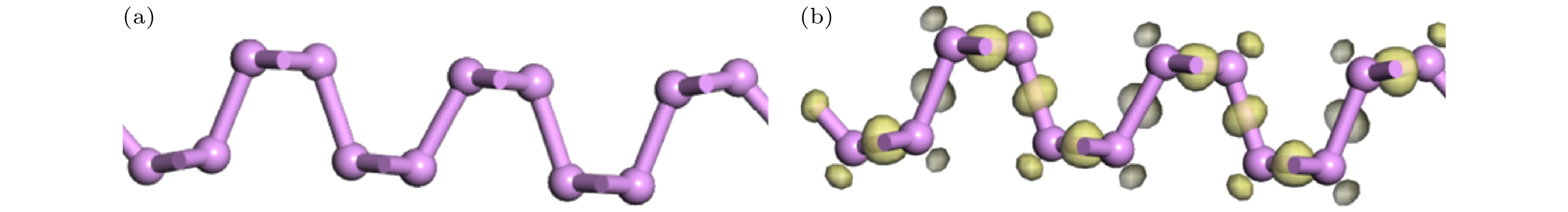 图 12 (a)形变为8%的黑磷烯结构俯视图; (b)形变为8%的黑磷烯电荷差分密度
图 12 (a)形变为8%的黑磷烯结构俯视图; (b)形变为8%的黑磷烯电荷差分密度Figure12. (a) Top view of the structure of 8% tensile deformation black phosphorene; (b) differential charge density of 8% tensile deformation phosphorene.
图11(f)—(j)为黑磷烯吸附Si原子拉伸体系能带结构, 可以看出变形较小时, 带隙打开较小, 形变量为2%时带隙为0.460 eV, 且直接带隙出现在价带内. 当形变量增加至4%时, 带隙骤增至1.750 eV, 但此时VBM和CBM分别移动到点X和L处, 对应直接带隙. 之后带隙随形变量增大而增加, 但增幅较缓. 形变量为8%和10%的体系的VBM出现在Γ点, 电子跃迁的所需动量增加. 在当前研究范围, 纯黑磷烯带隙的范围在1.578—1.771 eV, 而黑磷烯吸附Si原子带隙在0—1.924 eV范围变化, 远宽于纯黑磷烯的变化范围. 这意味着吸附Si原子的黑磷烯体系的间隙调谐比纯黑磷烯的更有效. 观察形变量为8%的吸附体系的能带结构发现, 尽管几何结构也出现了波浪状的形变, 但带隙并未出现骤减的情况. 这说明Si原子的吸附体系电子结构稳定性较强, 容易实现带隙的稳定调控.
图13为纯黑磷烯和黑磷烯吸附体系在拉伸应变作用下的态密度结构. 对比纯黑磷烯的PDOS图, 发现只有形变量为8%的结构在能量为–7和–12 eV的价带内有局域内小波峰的减少与增加. 其余体系DOS形状大体相同, 仅在费米能级处为零的区域范围有所不同, 显现出的带隙大小与能带图相对应. 观察黑磷烯吸附Si原子体系的DOS发现, 除了形变量为2%, 其余体系在费米能级附近的–0.5—0 eV处, Si原子p轨道的PDOS与P原子s轨道的PDOS重合, 说明两者轨道能量相近且发生杂化, 对应着吸附后体系内存在共价键的特征. 这也对应着带隙在形变量为4%时的带隙骤增.
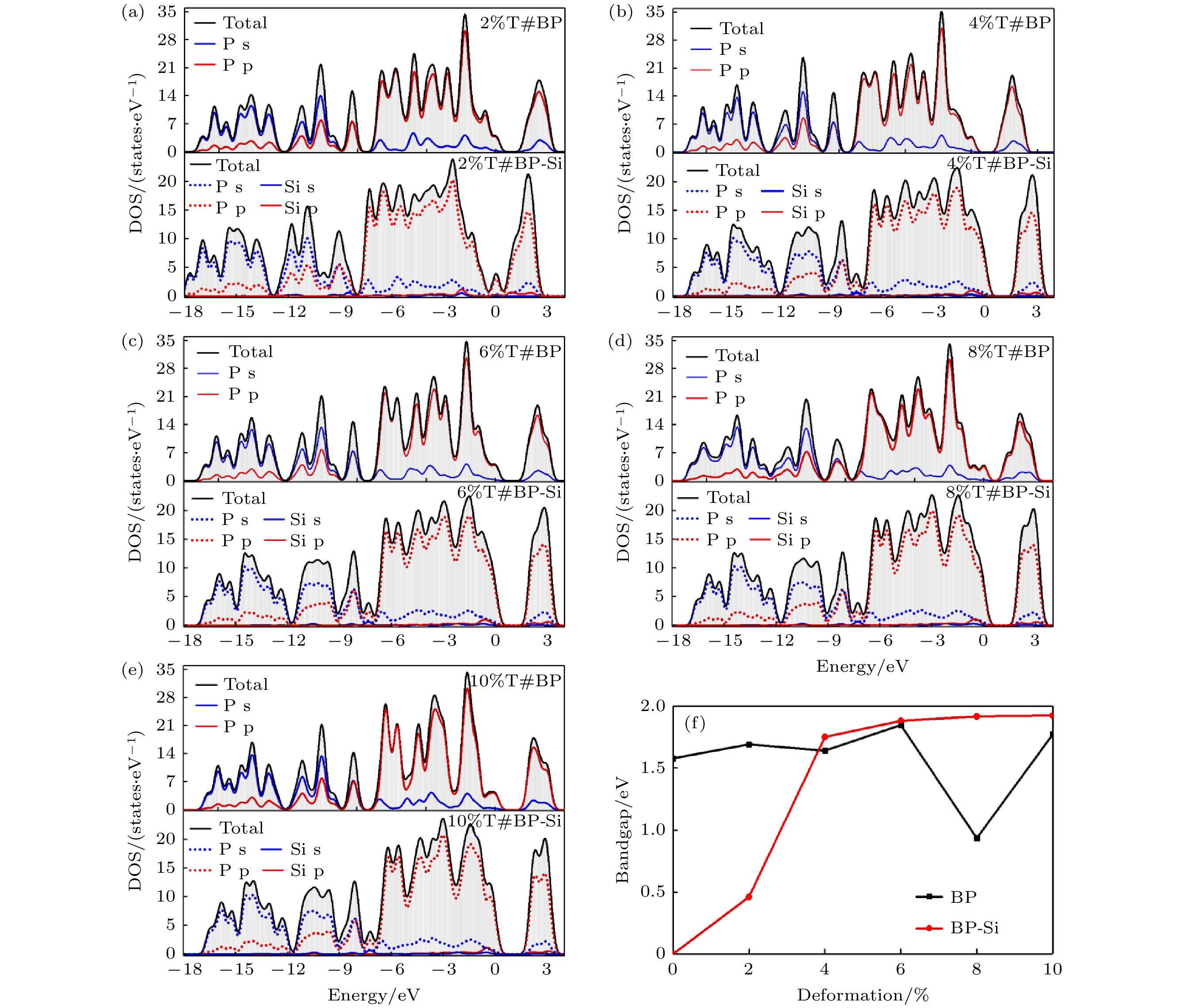 图 13 (a)—(e)拉伸形变量为2%—10%的纯黑磷烯(BP)以及黑磷烯吸附Si原子体系(BP-Si)态密度结构; (f)黑磷烯带隙变化曲线
图 13 (a)—(e)拉伸形变量为2%—10%的纯黑磷烯(BP)以及黑磷烯吸附Si原子体系(BP-Si)态密度结构; (f)黑磷烯带隙变化曲线Figure13. (a)?(e) The DOS of black phosphorene (BP) and Si adsorbed on black phosphorene (BP-Si) with 2%?10% tensile deformation; (f) band gap curves of black phosphorene.
2
3.4.拉伸及电场共作用对黑磷烯吸附Si原子体系影响
33.4.1.外加电场对黑磷烯吸附Si原子体系影响
图14为电场作用下纯黑磷烯与黑磷烯吸附Si原子体系的能带与态密度结构. 在能带图中, 蓝色虚线代表未加电场的黑磷烯体系. 从图14可以看出, 电场使本征黑磷烯带隙减小, 且由直接带隙转变为间接带隙. 对于黑磷烯吸附Si原子体系来说, 电场作用没有改变其零带隙, 但导带中接近费米能级附近的能级变得稀疏. DOS结构可以看出, Si原子的DOS贡献与P原子的DOS曲线较为离散, 未见强烈的杂化作用.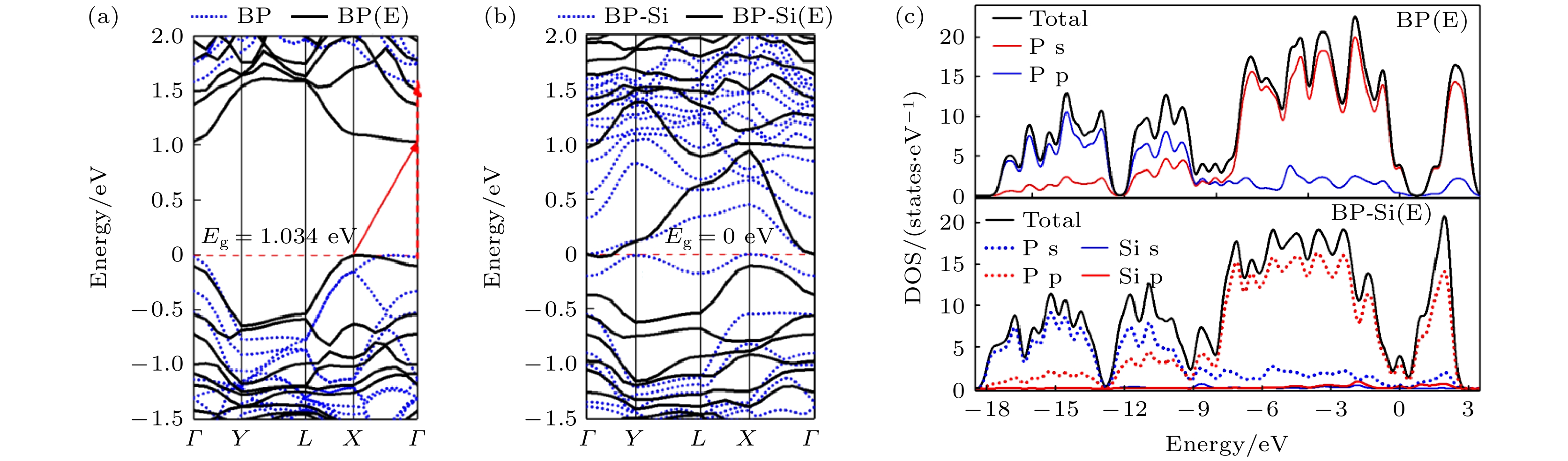 图 14 电场作用下纯黑磷烯与黑磷烯吸附Si原子体系 (a), (b)能带结构, 其中蓝色虚线代表黑磷及黑磷烯吸附体系的能带结构, 黑色实现代表电场作用下黑磷烯及其吸附体系的能带结构; (c) DOS结构
图 14 电场作用下纯黑磷烯与黑磷烯吸附Si原子体系 (a), (b)能带结构, 其中蓝色虚线代表黑磷及黑磷烯吸附体系的能带结构, 黑色实现代表电场作用下黑磷烯及其吸附体系的能带结构; (c) DOS结构Figure14. Si adsorbed on black phosphorene system and pure black phosphorene under electric field: (a), (b) Band structure, the blue dotted line represents the energy band structure of black phosphorus and black phosphorene adsorption system, and the black realization represents the energy band structure of black phosphorus and its adsorption system under the action of electric field; (c) DOS.
3
3.4.2.拉伸及电场共作用对黑磷烯吸附Si原子体系影响
根据(1)式和(2)式, 计算了纯黑磷在不同拉伸量和外加电场共作用下的单原子结合能和黑磷烯吸附体系在不同拉伸量和外加电场共作用下黑磷烯的吸附能, 具体结果列于表5.| 形变量/% | 0 | 2 | 4 | 6 | 8 | 10 |
| 结合能/eV | –2.507 | –2.485 | –2.457 | –2.439 | –2.433 | –2.421 |
| 吸附能/eV | 3.461 | 3.420 | 3.366 | 3.254 | 3.214 | 3.210 |
表5电场与拉伸共作用下纯黑磷烯单原子结合能和黑磷烯吸附Si原子吸附能
Table5.Single atom binding energy of black phosphorene and adsorption energy of Si adsorbed on black phosphorene system under the action of electric field and tensile.
在当前研究范围内, 电场的作用对黑磷烯及其吸附体系影响较大. 比较表4和表5可得, 外加电场使纯黑磷烯体系的结合能大幅降低, 相比于未加电场体系大约降低了3 eV. 对有电场作用的纯黑磷烯来说, 体系结合能的绝对值随拉伸变形量增加而减小, 但变化较为微小. 说明拉伸对电场作用的黑磷烯体系能量影响较小. 外加电场对黑磷烯吸附Si原子体系的吸附稳定性影响较小, 吸附能变化不大. 电场作用的吸附Si原子黑磷烯体系的吸附能随形变量的增强而减小.
图15(a)—(e)显示了纯黑磷烯在拉伸应变和电场共同作用下的能带结构. 从图15(a)—(e)可以看出, 引入电场后所有体系变为间接带隙. 与未加电场的拉伸体系相比, 电场的作用使体系带隙大约减少了0.5 eV. 图15(f)—(j)为拉伸应变和电场共同作用下吸附体系的能带结构, 带隙与未加电场的体系相比减少量在0.3—0.7 eV不等. 形变量增加至4%之后, 带隙几乎无太大改变. 说明此时形变量对于体系带结构影响微弱. 值得一提的是, 在电场作用下形变量为10%的吸附体系由间接带隙转变为直接带隙, VBM与CBM均出现在X点. 引入电场后, 黑磷烯体系的带隙随形变量的变化趋势与未加电场的相一致, 只是整体数值有所减小. 说明电场会使体系的带隙整体变窄. 形变与电场共作用的黑磷烯体系态密度如图16所示. 同未加电场的一样, Si原子的p轨道与P原子的s轨道有剧烈的杂化, 其成键具有共价性.
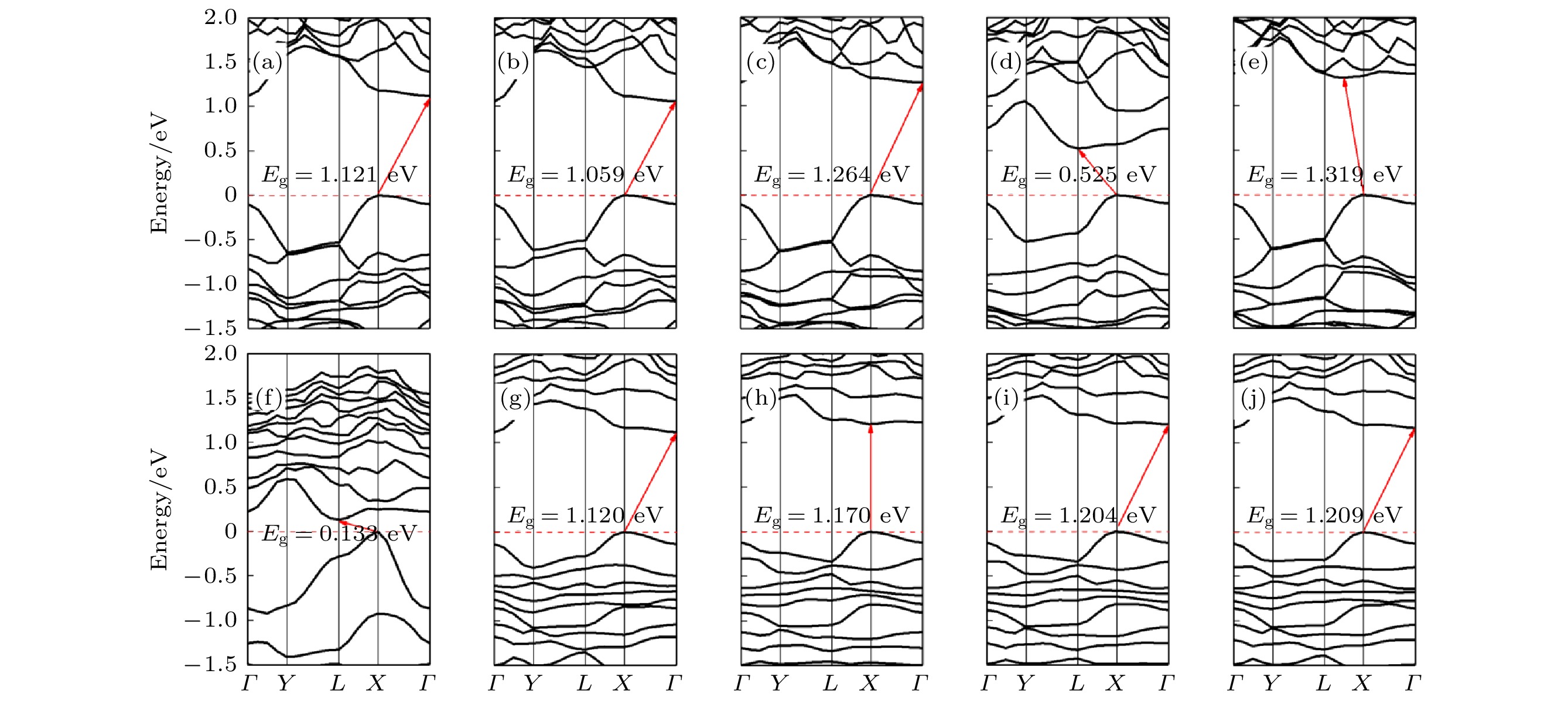 图 15 (a)—(e)电场作用下拉伸形变量为2%—10%的黑磷烯能带结构; (f)—(j)电场作用下拉伸形变量为2%—10%的黑磷烯吸附Si原子体系能带结构
图 15 (a)—(e)电场作用下拉伸形变量为2%—10%的黑磷烯能带结构; (f)—(j)电场作用下拉伸形变量为2%—10%的黑磷烯吸附Si原子体系能带结构Figure15. (a)?(e) Band structure of black phosphorene with 2%?10% tensile deformation under electric field; (f)?(j) band structure of Si adsorbed on black phosphorene system with 2%?10% tensile deformation under electric field.
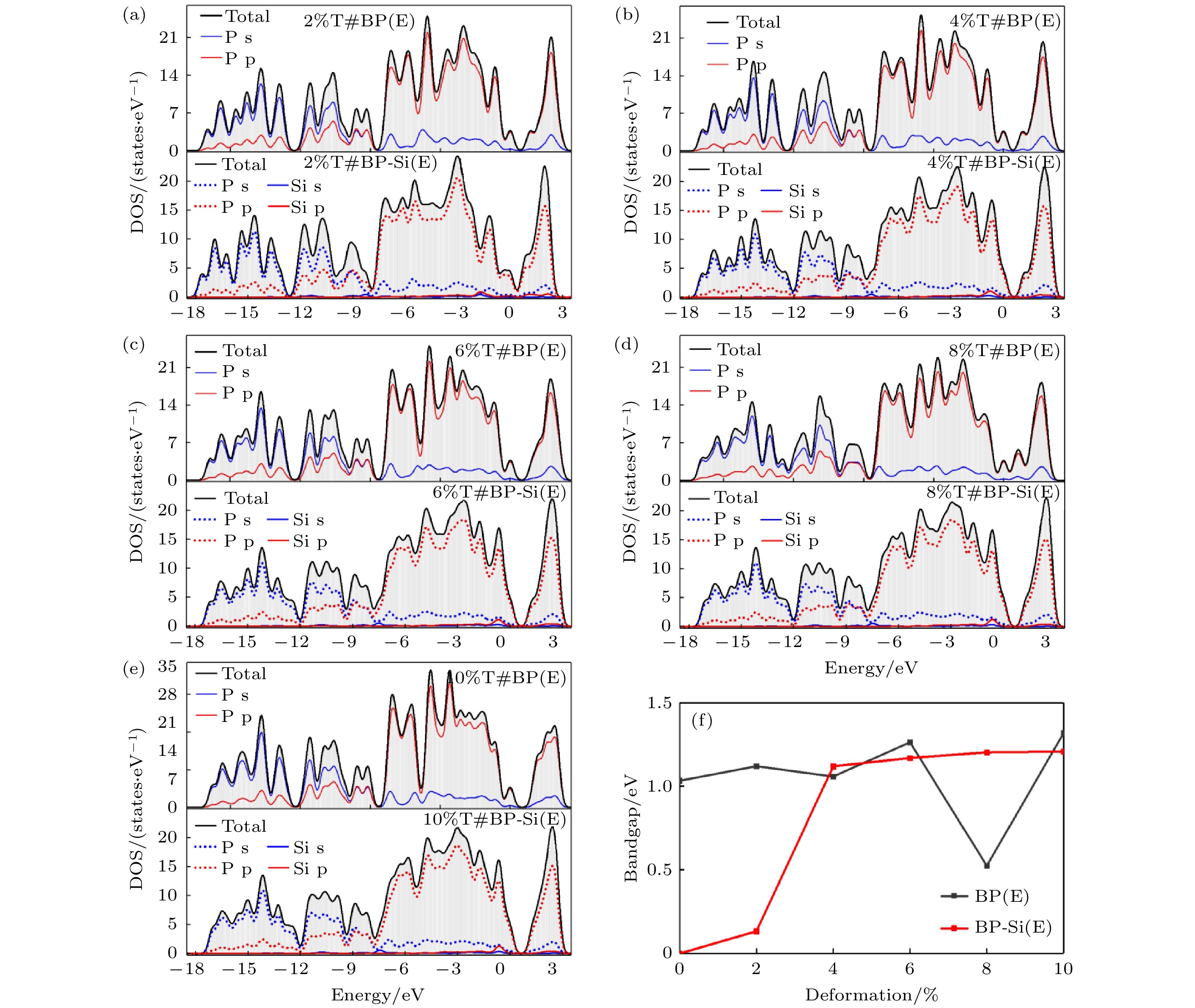 图 16 (a)—(e)电场作用下拉伸形变量为2%—10%的黑磷烯吸附Si原子体系能带结构; (f)黑磷烯带隙变化曲线
图 16 (a)—(e)电场作用下拉伸形变量为2%—10%的黑磷烯吸附Si原子体系能带结构; (f)黑磷烯带隙变化曲线Figure16. (a)?(e) Band structure of Si adsorbed on black phosphorene system with 2%?10% tensile deformation under the action of electric field; (f) band gap curves of black phosphorene.
