全文HTML
--> --> -->为提升锂离子电池的安全性, 使用固体电解质的固态锂离子电池(solid-state lithium-ion battery, SSLIB)开始被广泛研究. 固体电解质的不可燃性, 可以解决液态锂离子电池存在的安全问题. 除了安全这一突出优势外, SSLIB可使用锂金属作为负极, 锂金属具有最低的电极电势, 理论比容量高达3860 mA·h·g–1, 可以极大地提升电池的能量密度[3,4]. 而且, SSLIB还具有宽的电化学窗口、更好的热力学稳定性、更强的机械强度和更宽的工作温度范围等优点[5,6].
但到目前为止, SSLIB离实际应用还有很大距离, 除去成本因素, 其性能不佳也是原因之一, 主要体现在: 功率密度低、循环寿命有限、倍率性能较差, 这在相当程度上归因于SSLIB的界面问题. 电池的界面会极大地影响电池的容量、循环稳定性和离子电导率等重要性质[7], 直接影响电池性能.
SSLIB的界面是影响电池性能的主要因素之一, 也是最受关注的问题. SSLIB中存在的界面、主要的界面问题以及解决界面问题的常见策略如图1所示. SSLIB中的界面有固体电解质界面、粒子间界面、以及电极与集流体之间的界面[8], 其中最为重要的是固体电解质界面, 包括电解质和正负极的界面. 而全固态锂离子电池难以实际应用, 主要归因于固体电解质界面的高电阻和差的稳定性. 固体电解质界面是固体电解质与电极之间的界面区域, 结构和成分复杂, 一般为微米或纳米尺度, 界面的性质受诸多物理、化学因素的影响, 因此界面处的各种物理和化学过程也应一并考虑.
 图 1 SSLIB中的界面、界面问题和解决策略示意图
图 1 SSLIB中的界面、界面问题和解决策略示意图Figure1. Illustration of the interfaces, interface issues, and solving strategies in SSLIB.
理想情况下, SSLIB界面应该具有高的离子电导率, 使电池具有好的倍率性能, 但固体电解质界面中固体电解质和电极为固/固接触, 这使得锂离子的活性传输位点较少, 导致界面电阻较高, 所以尽管部分固体电解质的离子电导率已经达到甚至超过液体电解质的离子电导率, 依然无法实现较好的电池性能. 此外, 充放电循环中的接触失效、空间电荷层和界面晶格失配等界面现象也会增加界面电阻[8].
固体电解质界面的不稳定性, 一方面是由于固体电解质与电极之间的化学不稳定性, 接触时发生化学反应; 另一方面是源于固体电解质自身的电化学不稳定性, 在充放电循环时发生分解. 这两类反应以及界面的相互扩散现象, 都会形成界面相. 若界面相为电子导体, 则界面反应会进一步发生, 使界面区域不断扩大并破坏体相, 所以界面相应该为电子绝缘体. 在电极和电解质的有效接触表面, 在首次充放电循环时发生氧化还原反应, 所形成的界面层称作固体电解质界面相(solid electrolyte interphase, SEI)[9-11]. 电子绝缘的SEI可以阻止进一步发生氧化还原反应, 对电池的循环起到稳定作用. 但是SEI的形成会消耗锂离子且界面电阻往往较大, 造成电池的内阻增加和容量衰减[8]. 为了提高电池的性能, SEI应该具有良好的离子电导率、抑制相互扩散的能力和足够的稳定性, 因此能否形成稳定的SEI对于电池的循环性能至关重要.
负极材料具有较高的化学势, 容易与固体电解质在界面处发生反应. SSLIB的理想负极是锂金属, 其化学性质活泼, 易与固体电解质发生界面反应. 此外, 充放电过程中界面处锂的不均匀沉积形成的锂枝晶倾向于沿固体电解质的晶界生长, 可能造成电解质的破坏使电池失效, 因此如何构建稳定的固体电解质/锂负极界面是SSLIB领域目前的一大难题.
为了保证正极具有足够的电子和离子电导率, 正极需要由正极材料(如钴酸锂LiCoO2、磷酸铁锂LiFePO4等)、导电剂(导电炭黑等)和固体电解质混合而成. 因此正极界面组成复杂, 包括固体电解质/正极材料界面、固体电解质/导电剂界面、正极材料/导电剂界面以及正极材料/集流体界面, 这些界面都需要在充放电循环中保持稳定. 另一个不容忽略的问题是, 正极材料在充放电循环时体积变化较为显著, 更容易造成界面处的接触失效.
综上可知, SSLIB界面的性质尤其是稳定性, 很大程度上是由界面化学决定的, 因此准确了解界面性质非常重要. 但由于SSLIB界面的尺寸小, 大部分测试方法对其难以进行有效表征. 作为重要的表面分析方法, X射线光电子能谱(X-ray photoelectron spectroscopy, XPS)非常适合于对SSLIB界面进行分析与研究, 不仅可以得到界面的化学成分, 对界面性能进行预测; 还可以得到空间上的化学分布, 从而评估界面化学结构和不规整性、横向和纵向的成分变化. 此外, 结合紫外光电子能谱(ultraviolet photoemission spectroscopy, UPS)还可以得到全电池的能带结构和界面能带弯曲情况[12].
 图 2 XPS测量的能级示意图
图 2 XPS测量的能级示意图Figure2. Schematic diagram of energy level in XPS measurement.
商业化的光电子能谱仪通常使用Al或Mg作为阳极靶, 产生的Al Kα和Mg Kα线的光子能量分别为1486.6和1253.6 eV, 所激发光电子(电子能量范围0—1486 eV)的非弹性平均自由程近似地与

而由于原子所处化学环境的不同所造成的内层电子结合能的位移, 即化学位移, 可在一定程度上反映原子的化学状态, 如氧化态、成键情况等. 在XPS谱图中, 化学状态变化主要表现在内层电子对应谱峰结合能的变化, 而振激峰、俄歇峰和多重分裂峰等伴峰可作为化学分析的辅助依据[15]. XPS的谱峰强度和样品表界面区域的元素浓度成正比, 因此XPS可进行元素的定量分析. 但由于定量分析采用灵敏度因子法, XPS不能实现对检测区域所含元素的绝对定量, 仅能确定各元素的相对含量(误差为20%左右).

2
3.1.非原位XPS在氧化物固体电解质界面研究中的应用
氧化物固体电解质按照结构可分为钙钛矿型、反钙钛矿型、石榴石型、LISICON和NASICON型[18,19]. 氧化物固体电解质普遍具有较宽的带隙和较弱的离子极化, 因此有较好的化学与电化学稳定性, 这对锂金属负极的应用尤为重要. 然而其硬度普遍较大, 弹性形变有限, 难以实现较好的固/固接触, 具有较大的界面电阻. 据报道, 大多数氧化物固体电解质的离子电导率在10–5—10–3 S·cm–1范围内[20]. 其中石榴石型(garnet)氧化物固体电解质具有较高的锂离子电导率, 立方相的锂镧锆氧Li7La3Zr2O12(c-LLZO)在常温下可以达到10–4 S·cm–1[21], 并可通过元素掺杂进一步提升离子电导率, 如Ta掺杂锂镧锆氧Li6.4La3Zr1.4Ta0.6O12(LLZTO)的离子电导率可达1.6 × 10–3 S·cm–1[22].立方相锂镧锆氧Li7La3Zr2O12(LLZO)的结构是由ZrO6八面体与LaO8十二面体组成, 锂离子分布在24d和96h空位, 如图3(a)所示. 虽然立方相LLZO的离子电导率较高, 但是其与锂金属的界面电阻大, 据报道一般在数百至数千Ω·cm2[22,23], 这一方面是由于接触性不良导致, 另一方面是由于石榴石本身在空气中形成的主要成分为氢氧化锂或/和碳酸锂的表面层[24]. LLZO首先会与H2O发生Li+/H+交换, 生成LiOH, 二者进一步与CO2反应生成Li2CO3[25]. 对于LLZO微米粉体, 我们利用XPS表征发现其表面生成了较厚的反应层, 如图3(b)所示, 当暴露空气的时间约1230 s时, 样品表面完全被Li2CO3覆盖. 而通过手套箱制样尽量减少暴露时间至30 s时, XPS结果证实表面层由Li2CO3和LiOH组成, 同样的现象也出现在Li6.4La3Zr1.4Ta0.6O12(LLZTO)上. 所以在考虑石榴石固体电解质的界面时, 不可忽略其表面层, 目前去除表面层的方法主要有抛光、酸洗和高温退火等[26], 表面层去除效果可利用XPS证实.
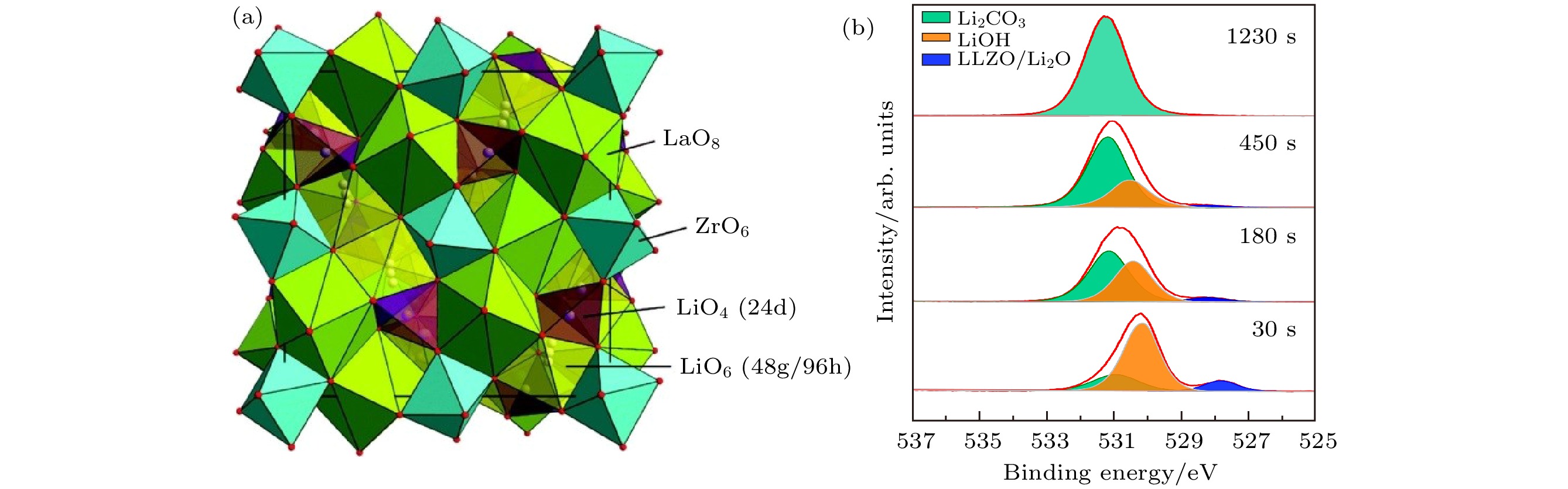 图 3 (a)立方相Li7La3Zr2O12(c-LLZO)晶体结构示意图[27]; (b)空气中不同暴露时间的LLZO微米粉体表面的O1s谱图变化
图 3 (a)立方相Li7La3Zr2O12(c-LLZO)晶体结构示意图[27]; (b)空气中不同暴露时间的LLZO微米粉体表面的O1s谱图变化Figure3. (a) Crystal structure of cubic Li7La3Zr2O12(c-LLZO)[27]; (b) O1s spectra of LLZO micro particles exposed to air for different times.
LLZO等石榴石型固体电解质与锂负极的界面被认为是稳定的, 此结论是基于循环伏安法(cyclic voltammetry, CV)等电化学表征的结果进行判断的[28,29], 而对界面直接的观察则较少. 由于实验条件不一, 对锂负极的稳定性仍没有确切的结论. 根据第一性原理计算, c-LLZO会与锂金属反应并形成含Zr, La2O3和Li2O的反应层[30], 而XPS证实循环后的Li/LLZO界面有Zr3O生成[31](如图4(a)). 为了去除表面层, Wolfenstine等[32]将LLZO加热至800 ℃并保持2—4 h, 再将其浸渍于300 ℃熔融态的锂196 h后, 对LLZO表面进行XPS测试, 发现表面的锂离子含量有所增加, 而La谱峰和Zr谱峰没有发生位移, 即LLZO的结构并未破坏, 说明LLZO对锂金属的化学稳定性.
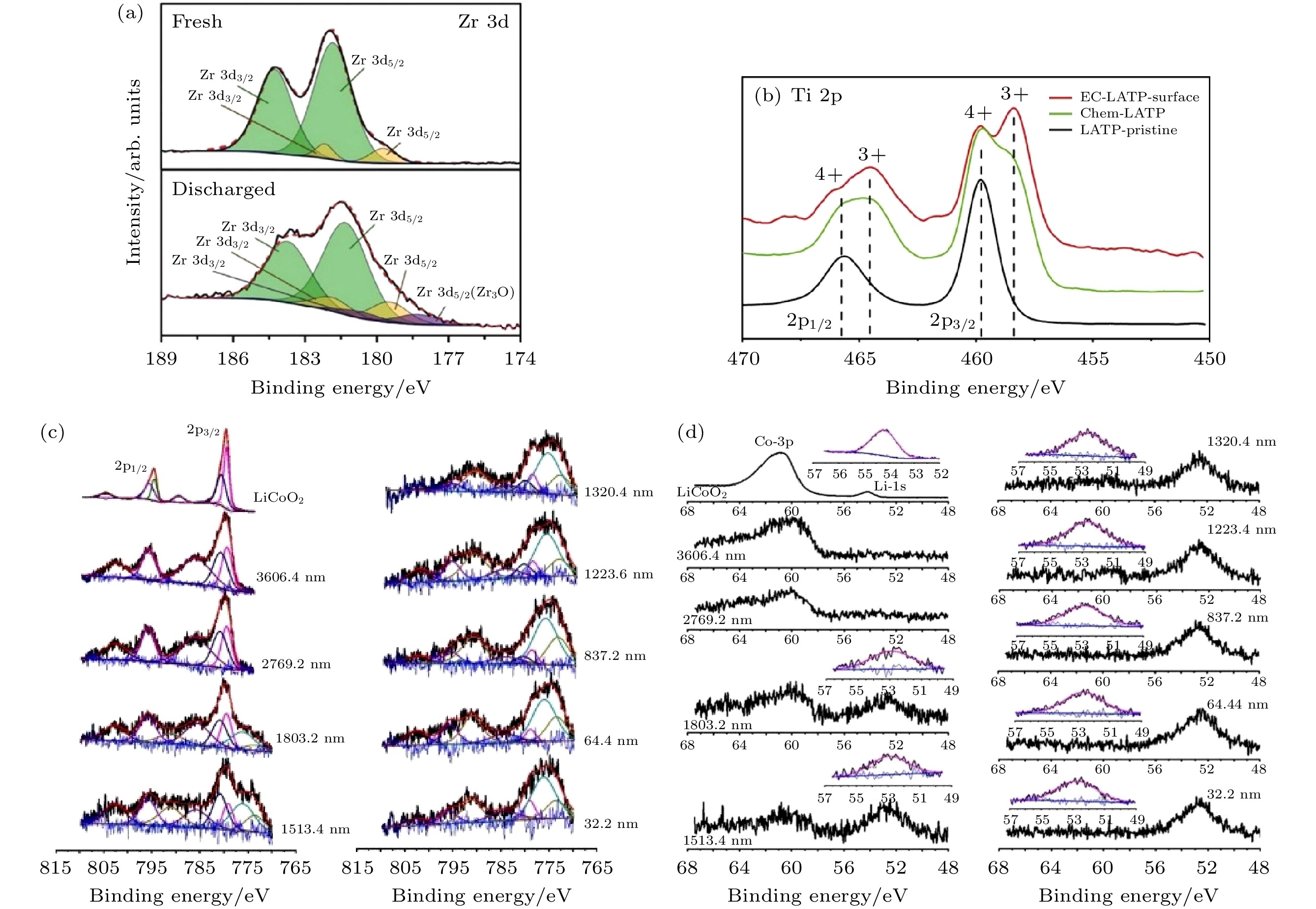 图 4 (a) 原始和放电后的Li7La3Zr2O12(LLZO)表面的Zr 3d分峰拟合结果[31]; (b)原始Li1.3Al0.3Ti1.7(PO4)3(LATP)、化学失效LATP、电化学失效LATP的Ti 2p谱图[35]; LiCoO2衬底上LLZO薄膜不同刻蚀深度的(c)Co 2p峰以及(d)Li 1s和Co 3p峰的XPS深度剖析谱图[41]
图 4 (a) 原始和放电后的Li7La3Zr2O12(LLZO)表面的Zr 3d分峰拟合结果[31]; (b)原始Li1.3Al0.3Ti1.7(PO4)3(LATP)、化学失效LATP、电化学失效LATP的Ti 2p谱图[35]; LiCoO2衬底上LLZO薄膜不同刻蚀深度的(c)Co 2p峰以及(d)Li 1s和Co 3p峰的XPS深度剖析谱图[41]Figure4. (a) Peak deconvolution of Zr 3d spectra of fresh and discharged Li7La3Zr2O12 (LLZO)[31]; (b) Ti 2p XPS spectra of the Li1.3Al0.3Ti1.7(PO4)3 (LATP)-pristine, chem-LATP(chemical failure), and EC-LATP(electrochemical failure) surface[35]; XPS depth profiles of (c) Co 2p spectra, (d) Li 1s and Co 3p spectra of LLZO thin film on LiCoO2 at different depth[41].
含Ti4+, Sn4+, Gd3+等阳离子的氧化物固体电解质, 容易与锂金属发生还原反应形成不稳定的混合导电界面相[33,34]. Zhu等[35]用XPS研究了NASICON型Li1.3Al0.3Ti1.7(PO4)3(LATP)固体电解质与锂负极的界面(图4(b)), 发现界面处的Ti4+在循环中被还原为Ti3+, 该还原反应不但使界面处形成富锂相, 而且使电子不断注入到Li1.3Al0.3Ti1.7(PO4)3, 导致局部区域电势降低, 作为锂离子沉积的位点生成锂枝晶, 加速电池的失效.
大多数固体电解质具有足够的机械强度而不会被锂枝晶刺穿, 因此可以抑制锂枝晶的生长, 尤其是对于硬度较高的氧化物固体电解质. 但Sharafi等[36]发现Li/LLZO对称电池进行充放电循环后, LLZO表面有黑色区域产生, XPS结果表明黑色区域为金属锂, 即界面生成了锂枝晶. 许多研究表明, 锂枝晶倾向于在界面处的空隙、晶界中进行沉积并生长[22,37-39]. 固体电解质内部残留的电子和负离子也可能使锂离子还原[39], 所以即使对于SSLIB, 也需要合适的方法解决锂枝晶生长的问题.
在正极界面, 即使氧化物固体电解质对正极具有足够的稳定性而不发生反应, 也可能会有界面相形成, 这是由界面处的相互扩散导致的, 常见的是过渡金属离子和固体电解质阳离子之间的相互扩散[33,40]. Zarabian等[41]以XPS深度剖析研究了400 ℃下制备的LLZO固体电解质薄膜/LiCoO2(LCO)正极之间的界面, 如图4(c)和图4(d)所示, 发现循环后的界面生成了Co3O4以及不同的含Co相, 距离正极区域约1803.2 nm范围内没有Li1s的信号, 说明该区域锂离子脱嵌形成Li1–xCoO2. Liu等[42]利用同步辐射可变能量硬X射线光电子能谱(variable energy hard X-ray photoelectron spectroscopy, VE-HAXPES)观察到LiCoO2正极与Li1.3Al0.3Ti1.7(PO4)3(LATP)界面处强的氧元素扩散现象, 对应LATP的O1s峰在LiCoO2正极仍保持高的强度, 而在选用Li3PO4作为界面缓冲层后, LiCoO2正极中Li1.3Al0.3Ti1.7(PO4)3相关的峰有相当程度的降低, 说明缓冲层可以有效抑制O元素的扩散.
2
3.2.非原位XPS在硫化物固体电解质界面研究中的应用
硫化物固体电解质可分为Argyrodite型、Li2S-P2S5体系、thio-LISICON型和Li10GeP2S12型[19]. 由于硫离子的半径大于氧离子, 硫化物固体电解质的极化率比氧化物固体电解质的更高, 削弱了材料骨架结构与锂离子之间的相互作用, 拓宽了锂离子的迁移通道; 且由于硫元素较低的电负性, 硫离子与锂离子之间的结合强度小于氧离子与锂离子之间的结合强度, 因此硫化物固体电解质具有更高的离子电导率[43]. 其中thio-LISICON型固体电解质(典型化学式为Li4–xGe1–xPxS4)和Li2S-P2S5体系玻璃态固体电解质离子电导率可达10–3 S·cm–1, 快锂离子导体Li10GeP2S12的离子电导率更是可达到10–2 S·cm–1[44-47]. 如图5(a)所示, Li10GeP2S12结构在c轴方向上由LiS6八面体与Ge/PS4四面体共边构成的一维离子通道, 由16h和8f位点的LiS4组成, 并由PS4四面体相互联接组成三维离子传输网络[47]. Li2S-P2S5体系通过改变Li2S与P2S5的比例可以得到不同成分的材料[48](图5(b)), 如Li7P3S11[49], β-Li3PS4[50]和Li2P2S6[51]. 由于结构不同, 材料离子电导率的差异可达几个数量级, 其中280 ℃下退火的Li7P3S11的离子电导率可达1.4 × 10–3 S·cm–1[52]. 这一方面是由于结构中有较多孤立的PS4和P2S7四面体提供锂离子传输的间隙, 另一方面是由于退火使玻璃相部分晶化. 与氧化物固体电解质相比, 硫化物固体电解质的硬度较低, 可实现较好的固/固接触从而降低界面电阻, 但是在空气中容易与水反应形成H2S气体[53], 给实际应用造成很大的困难. 图 5 (a) Li10GeP2S12的晶体结构示意图[47]; (b) Li2S-P2S5体系中不同成分的部分材料的晶体结构示意图[48]
图 5 (a) Li10GeP2S12的晶体结构示意图[47]; (b) Li2S-P2S5体系中不同成分的部分材料的晶体结构示意图[48]Figure5. (a) Crystal structure of Li10GeP2S12[47]; (b) crystal structures of some materials within Li2S-P2S5 binary system[48].
硫化物固体电解质的电化学窗口有一些互相矛盾的报道. 循环伏安法测试表明硫化物固体电解质的电化学窗口可达5 V (vs. Li+/Li)或更宽[47, 54,55], 从而在电化学循环中保持稳定. 而另有报道其电化学窗口仅在1.5—2.5 V (vs. Li+/Li)范围内[56], 这可能是由于硫化物固体电解质与电极的接触面积有限, 其分解反应的法拉第电流过小而被忽略. 许多XPS研究也证实了硫化物固体电解质对电极的不稳定性[57-59].
Jérémie等[57]对以Argyrodite型Li6PS5Cl为电解质的SSLIB进行了非原位XPS研究, LiCoO2/Li6PS5Cl/Li4Ti5O12(LTO) SSLIB在充放电循环后出现了极大的容量衰减, 这可能是由Li6PS5Cl与正极的界面反应造成的. XPS结果表明充电时部分Li6PS5Cl在正极界面上发生氧化分解, 生成LiCl, P2S5和多硫化物Li2Sn, 其中LiCl的生成被认为是界面电阻增加的主要原因; 在多次循环后, Li6PS5Cl还与LiCoO2反应生成磷酸盐, 而负极界面在循环过程中保持稳定. Gao等[58]对充放电循环后的Li/Li10GeP2S12界面进行了深度剖析, 如图6(a), 发现Li10GeP2S12在循环过程中还原, 还原产物为Li2S, Ge以及低价态P的化合物. 由于生成金属相, 界面具有电子导电性可使界面反应进一步发生, 不能形成稳定的界面层. 此外, 硫化物固体电解质的不稳定性并不仅限于电极界面. Koerver等[59]利用深度剖析研究了结构为Li-In||β-Li3PS4||NCM-811-β-Li3PS4的SSLIB在不同截止电压下经过25次充放电循环后的集流体/正极界面, 如图6(b)所示, β-Li3PS4的S 2p中有两个峰对应PS4四面体中的P—S键和P—[S]n—P结构. 经过充放电循环后, 更高结合能处出现了新峰, 对应热力学稳定的β-Li3PS4氧化产物, 其中4.0和4.3 V的谱图大致相同, 而4.6和5.0 V下β-Li3PS4的氧化更加剧烈, 氧化产物即使在远离集流体/正极界面处也有少量存在. 所以正极中的电解质与活性材料(NCM-811)在体相几乎是稳定的, 而在集流体表面区域形成具有一定厚度的界面层.
 图 6 (a) 循环后的Li/Li10GeP2S12界面的XPS深度剖析谱图, S 2p, Ge 3d, P 2p, Li 1s[58]; (b) 在不同截止电压(4.0—5.0 V)循环25次后的集流体/正极界面随时间变化的S 2p深度剖析谱图[59]
图 6 (a) 循环后的Li/Li10GeP2S12界面的XPS深度剖析谱图, S 2p, Ge 3d, P 2p, Li 1s[58]; (b) 在不同截止电压(4.0—5.0 V)循环25次后的集流体/正极界面随时间变化的S 2p深度剖析谱图[59]Figure6. (a) XPS depth profiles of cycled Li/Li10GeP2S12 interface: XPS spectra of S 2p, Ge 3d, P 2p, Li 1s[58]; (b) S 2p XPS depth profiles after 25 cycles for different upper cut-off voltage (4.0–5.0 V) as a function of different etching time[59].
2
3.3.非原位XPS在人工SEI修饰层研究中的应用
针对各种界面问题, 尤其是为了提高界面锂离子电导率和界面稳定性, 需要进行界面修饰以提高界面的性能, 其中一种方式是构建人工SEI, 也称之为修饰层. 通过对界面元素化学状态的分析, XPS可以直接证明修饰层对界面稳定性的作用. 人工SEI可分为有机SEI、无机SEI和混合型SEI, 其中有机SEI一般是聚合物固体电解质, 如聚氧化乙烯-双三氟甲基磺酰亚胺锂(PEO-LiTFSI); 无机SEI包括锂合金、无机物薄膜、氧化物、锂酸盐和锂化合物; 混合型SEI则是有机SEI与无机SEI的复合物[60]. 氟化锂(LiF)是负极修饰层的常用材料, 尽管LiF的离子电导率不高, 但是作为人工SEI不仅可以提高固体电解质对锂负极的稳定性, 还可以保持锂离子的快速传输[61-63]. Fan等[64]提出将双氟磺酰亚胺锂(LiFSI)涂覆在Li3PS4电解质上进行渗透, 再将电解质与锂负极接触反应形成含LiF的SEI. 他们对Li/Li3PS4/不锈钢半电池进行循环后, 取出Li3PS4对其表面进行非原位XPS表征(图7(a)), 出现对应Li3P的新峰, 是Li3PS4被锂还原的产物; 作为对比, LiFSI@Li3PS4表面只有对应Li3PS4和LiF的峰, 表明LiF层成功包覆Li3PS4, 并可以提高Li/Li3PS4界面的电化学稳定性. Chang等[65]设计了一种氧化锌改性的氟化三维泡沫铜作为锂负极的表面层, XPS证实其在循环过程中产生LiF, 提高了界面的稳定性. 在室温下, Li2.9B0.9S0.1O3.1玻璃陶瓷固体电解质有良好的延展性, Nagao等[66]以其作为缓冲层提升Li/LLZO的界面接触性, 同时该缓冲层可有效阻止界面反应. 电化学阻抗谱(electrochemical impedance spectroscopy, EIS)显示Li/Li2.9B0.9S0.1O3.1对称电池的界面电阻(约1 Ω)在恒电流充放电循环前后无明显变化(图7(b)). XPS显示界面产物为Li2O, Li2S和LixB, 且相应谱峰的强度在界面刻蚀后迅速下降(如图7(c)所示), 表明界面层厚度小, 且有足够的电子绝缘性阻止反应的进一步发生. 该界面层具有循环稳定性, 同时对锂离子的快速迁移没有影响. Liang等[67]在LiNixMnyCo1–x–yO2正极表面原位生成了非晶态LixBOyFz包覆层, XPS显示在100次循环后, 包覆正极表面的Ni2+含量与初次循环相比并无明显增加, 表明非晶态LixBOyFz包覆层具有良好的化学稳定性.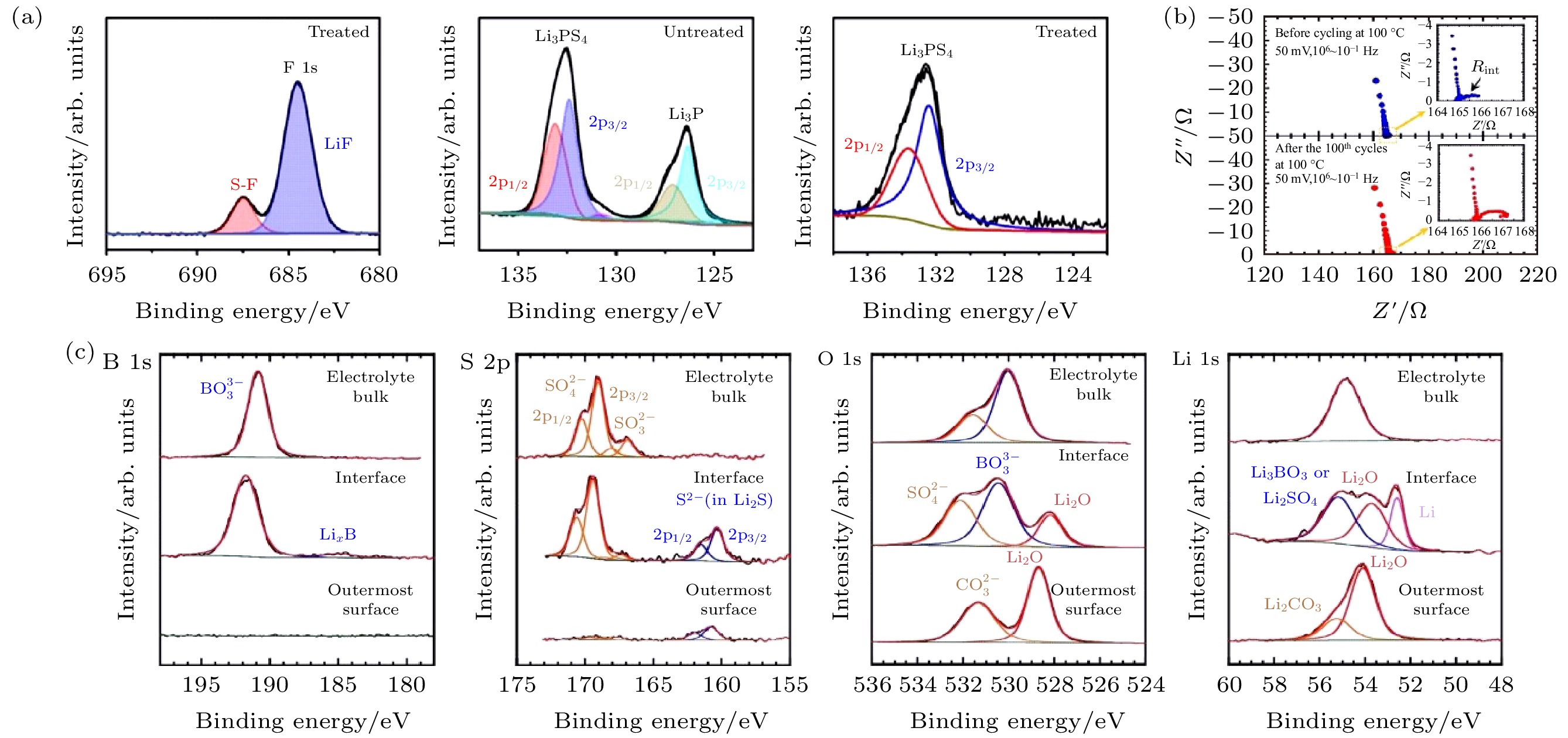 图 7 (a) 循环后LiFSI修饰的Li3PS4的F 1s和P 2p(右)谱图, 循环后未处理的Li3PS4的P 2p(左)谱图[64]; (b) Li/Li2.9B0.9S0.1O3.1对称电池恒流充放电循环前后的阻抗图[66]; (c)刻蚀得到的Li/Li2.9B0.9S0.1O3.1区域表面/界面/体相的B 1s, S 2p, O 1s, Li 1s谱图[66]
图 7 (a) 循环后LiFSI修饰的Li3PS4的F 1s和P 2p(右)谱图, 循环后未处理的Li3PS4的P 2p(左)谱图[64]; (b) Li/Li2.9B0.9S0.1O3.1对称电池恒流充放电循环前后的阻抗图[66]; (c)刻蚀得到的Li/Li2.9B0.9S0.1O3.1区域表面/界面/体相的B 1s, S 2p, O 1s, Li 1s谱图[66]Figure7. (a) F 1s and P 2p (right) spectra of LiFSI-treated Li3PS4 from cycled cell, P 2p (left) spectra of untreated Li3PS4 from cycled cell[64]; (b) impedance plots of the Li/ Li2.9B0.9S0.1O3.1 symmetric cell before and after the galvanostatic test for cycles[66]; (c) XPS spectra of B 1s, S 2p, O 1s, Li 1s for the outermost surface, interface, and electrolyte bulk regions uncovered by etching of Li/Li2.9B0.9S0.1O3.1 interface area[66].
4.1.原位XPS简介
通常, 分析对象的真实反应情况难以用非原位分析方法得到, 在非原位分析表征中, 对同一分析对象, 其分析与反应的环境、时间不同, 测试结果往往是反应已经完成的“状态”, 无法监测分析对象与其原始环境的相互作用, 无法排除外界环境的影响, 导致测试结果的非实时性. 为了对样品进行更具真实性、准确性、实时性的分析表征, 得到样品在反应过程中的真实状况, 需要采用原位分析方法.原位分析方法不需要将分析对象脱离其原始体系环境, 而是在原始体系环境中, 对反应过程中的样品进行分析表征[68]. 原位分析表征方法包含operando方法(进行原位实时动态分析), 该方法是在原位条件基础上又有进一步的限定, 故本文将原位XPS分为常规原位XPS和operando XPS进行介绍. 由于各方面条件的限制, 特别是光电子能谱仪需要超高真空的条件, XPS难以实现严格意义上的原位分析. 为使制备方法与分析环境达成一定的妥协, 目前可行且较为成熟的原位XPS系统其实是准原位的, 即实现制备与表征过程都维持在超高真空环境下, 样品不暴露于大气环境, 本文中也同样称为原位XPS. 原位XPS系统中通过真空互联传输装置实现样品在制备设备和分析仪器之间的真空传递, 如darmstadt integrated system for battery research系统(DAISY-BAT, 图8(a))[69], 集成了多个制备腔室, 包括碱金属蒸发源(dispenser)、三个溅射沉积室(正极、负极和电解质)、金属有机化合物气相外延室(metal-organic chemical vapour deposition, MOCVD)、化学气相合成室(chemical vapor synthesis, CVS)、脉冲激光沉积室(pulsed laser deposition, PLD)以及加热台, 居中的“转盘”将各个腔室与光电子能谱仪连接. 由此, 原位XPS对逐层生长的薄膜进行分析表征, 获得动态的界面变化, 反映更为真实的界面信息. 此外, 通过对分析室配置相关的附件或进行合适的改造, 如配置加热冷却样品台、充放电样品台、导入光纤和安装溅射靶材支架(图8(b))等[70,71], 可使样品的反应与表征同时进行, 实现原位XPS表征.
 图 8 (a) DAISY-BAT系统的示意图[69]; (b)利用离子枪溅射金属靶材构筑界面以实现原位XPS的过程示意图[70]
图 8 (a) DAISY-BAT系统的示意图[69]; (b)利用离子枪溅射金属靶材构筑界面以实现原位XPS的过程示意图[70]Figure8. (a) Scheme of the DAISY-BAT system[69]; (b) scheme of performing in situ XPS using ion gun to sputter the fixed metal target to build the interface[70].
2
4.2.原位构筑界面
利用原位XPS可有效地研究电解质和电极之间发生化学反应的过程, 关键的前提是能够可控地原位构筑电解质/电极界面. Wenzel等[70]通过原位XPS研究了Li/Li0.35La0.55TiO3(LLTO)界面层随金属锂沉积时间的变化情况. 金属锂在Li0.35La0.55TiO3(LLTO)表面的沉积是在光电子能谱仪样品台上安装锂金属靶材支架, 利用氩离子枪溅射锂金属靶使锂沉积在Li0.35La0.55TiO3(LLTO)样品表面, 并在每一次沉积后记录XPS数据, 结果如图9(a)所示. La 3d谱图随着Li沉积的进行未出现明显变化, 说明La元素价态在金属锂沉积过程中没有发生改变, 证实La3+不会被锂金属还原. O 1s谱在初始状态有两个峰, 随着沉积的进行逐渐变为单峰, 约531 eV处的高结合能峰可能对应于碳酸盐或氢氧化物组分, 峰强度因锂的沉积而减少, 说明Li0.35La0.55TiO3(LLTO)中碳酸盐或氢氧化物相应地减少. Ti 2p中随着锂沉积的进行出现三个额外的峰, 说明Ti4+被还原至Ti3+, Ti2+和Ti0, 证实LLTO与锂金属之间的化学不稳定性主要来自Ti. 在1000 min的沉积时间后, Ti4+的还原仍然在进行, 表明Li/Li0.35La0.55TiO3(LLTO)的界面层具有足够的电子电导率, 易于传导电子从而使界面反应不断进行.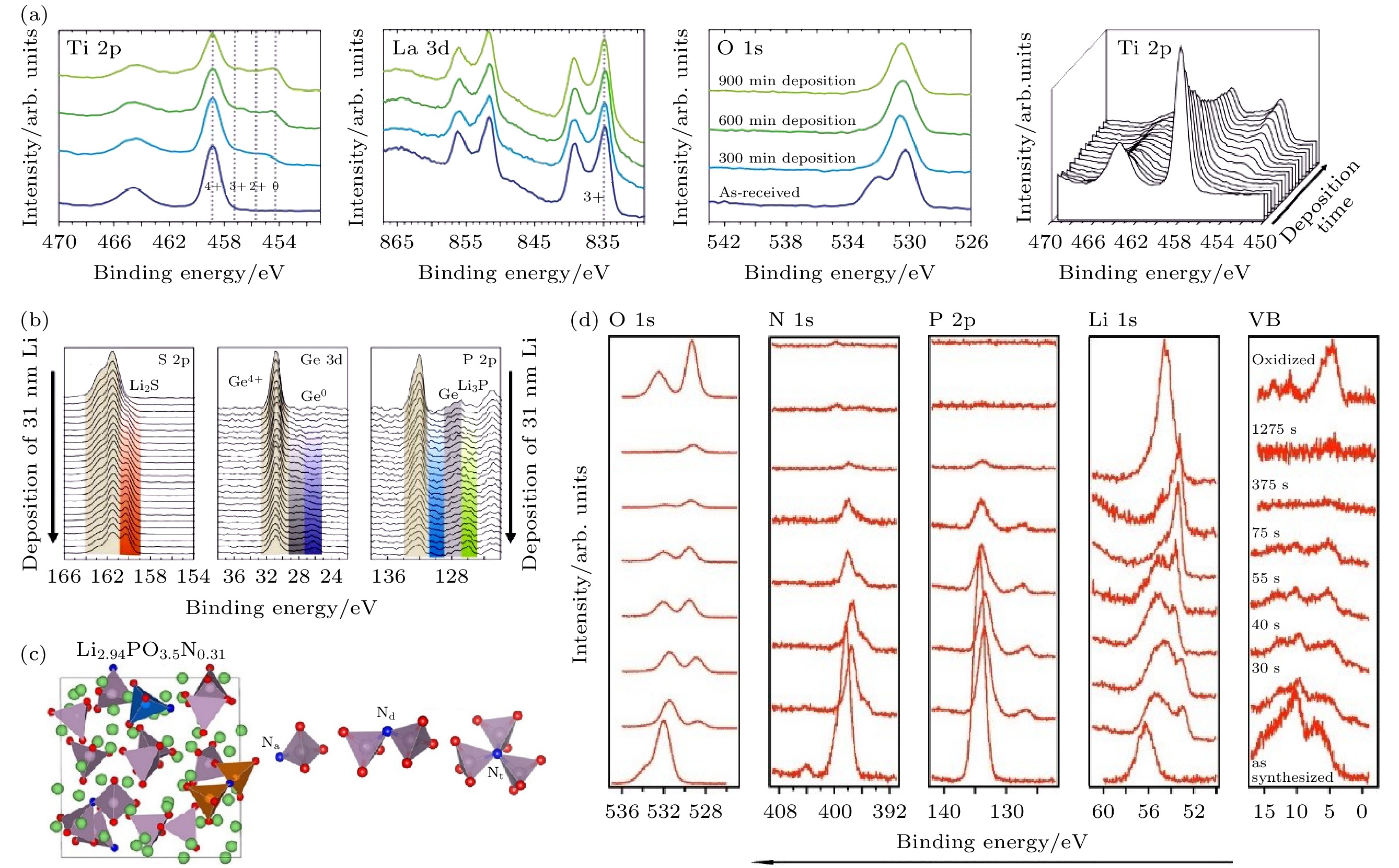 图 9 (a) LLTO表面不同锂沉积时间的Ti 2p, La 3d, O 1s谱以及Ti 2p瀑布图[70]; (b) 31 nm金属锂沉积过程中Li10GeP2S12表面的S 2p, Ge 3d和P 2p-Ge 3p谱图[53]; (c) Li2.94PO3.5N0.31的结构和磷酸盐结构中N的结合情况示意图[73,74]; (d)金属锂沉积过程中LiPON表面的O 1s, N 1s, P 2p, Li 1s和价带图[76]
图 9 (a) LLTO表面不同锂沉积时间的Ti 2p, La 3d, O 1s谱以及Ti 2p瀑布图[70]; (b) 31 nm金属锂沉积过程中Li10GeP2S12表面的S 2p, Ge 3d和P 2p-Ge 3p谱图[53]; (c) Li2.94PO3.5N0.31的结构和磷酸盐结构中N的结合情况示意图[73,74]; (d)金属锂沉积过程中LiPON表面的O 1s, N 1s, P 2p, Li 1s和价带图[76]Figure9. (a) Ti 2p, La 3d, O 1s detail spectra and Ti 2p waterfall plot for different Li metal deposition times on LLTO surface[70]; (b) S 2p, Ge 3d, and P 2p-Ge 3p detail spectra during 31 nm Li metal deposition on Li10GeP2S12 surface[53]; (c) scheme of Li2.94PO3.5N0.31 structure and possible N configurations in phosphate structures[73,74]; (d) O 1s, N 1s, P 2p, Li 1s, and valence band spectra of LiPON surface during Li metal deposition[76].
Wenzel等[53]以同样的溅射沉积方式对Li/ Li10GeP2S12界面的变化进行研究, 如图9(b)所示, 在31 nm锂金属薄膜的沉积过程中, XPS谱图显示Li10GeP2S12与锂反应发生分解, 产生低价Ge化合物, Ge0, P的还原组分, Li3P以及大量的Li2S. 由于金属导电相的形成, 电子流向界面使分解反应随着锂的沉积持续进行.
利用功能强大的DAISY-BAT原位XPS系统, 可以实现部分电解质的原位制备, 利用原位制备的电解质作为基底, 实现金属锂的原位沉积, 研究电解质和金属锂的界面变化. LiPON是一种非晶态固体电解质, 离子电导率大约为10–6 S·cm–1, 与电极的界面稳定性较好, 可用于制备薄膜电池[72]. 研究表明, LiPON的结构可能是N取代Li3PO4中的O, 作为PO4四面体的“桥”构成非晶态网络(图9(c))[73,74]. Yu等[75]对Li与LiPON界面电阻进行电化学阻抗测试, 表明没有明显的界面反应发生, 但是否产生界面层仅依据电阻的变化难以证实. 因此, Schw?bel等[76]以原位XPS研究了LiPON与Li的负极界面, 实验设备为DAISY-BAT系统, 以射频磁控溅射制备LiPON基底, 通过碱金属释放剂(dispenser)使金属锂逐步蒸发沉积到LiPON表面, 于两次沉积之间进行XPS表征. 如图9(d)所示, XPS谱图显示原始LiPON表面有对应P—O—P键、P—O—Li键和P=O键的O 1s峰, 对应三键N(连接三个PO4四面体)和双键N的N 1s峰, 对应Li—O键的Li 1s峰以及对应磷酸盐的P 2p峰. 随着金属锂沉积的进行, O 1s中出现新峰, 与氧化锂样品的O 1s对比, 确定该峰对应氧化锂的O 1s, 同时P—O—P峰的减弱更为明显, 表明金属锂倾向于与桥氧发生反应. N 1s中三键N峰明显减弱, 并且出现了对应Li3N的峰, 证实锂更倾向与三键N反应. P 2p出现一个新的峰, 对应Li3P. Li 1s出现肩峰, 但由于各种可能组分的Li 1s结合能十分接近, 难以分峰进行定性分析. XPS结果证实了LiPON的分解, 确定的产物有Li2O, Li3N和Li3P, 其中Li3N和Li3P的离子电导率在10–4 S·cm–1左右[77,78], 这可能是界面电阻无明显变化的原因, 同时, 锂蒸发后各谱峰位置有一定的偏移, 这是界面处能带结构发生变化导致的.
2
4.3.电化学原位XPS
电化学原位XPS研究的优势之一在于能直接研究界面在电化学条件下的化学变化. Liu等[71]设计了三明治结构的Li薄膜负极/Ga掺杂LLZO/ LiFeO4+Ga-LLZO+导电炭黑混合正极的原位电化学池, 并将其通过XPS能谱仪分析室内的样品操纵杆与外部电化学工作站连接, 在恒电位电化学测试(电池的开路电压为2.1 V, 恒电位电压分别选择为2.1, 2.5, 3.6 V)的同时对负极界面进行原位XPS测试, 其中Li薄膜负极是通过气相沉积法得到的(图10(a)). 如图10(b)所示, 当形成3 nm的Li金属薄膜后, 施加正电压导致负极界面各XPS谱峰结合能发生负偏移(单位为eV), 结合能位移值与所施加电压相近(如施加电压为2.1 V时, 负极界面所有谱峰结合能都会负移约2 eV). 开路电压(2.1 V)时, O 1s在526.3 eV结合能处产生新峰, 证明生成了Li2O; Li 1s在52.8 eV结合能处也产生新峰(对应金属锂), 而在电压增加至3.6 V时, 金属锂峰强度增大, 这是由于正极迁移过来的锂离子沉积导致; Li2O峰强度增加, 表明界面反应加剧; La 4d, Zr 3d峰信号减弱, 说明界面层具有一定厚度, 几乎不含La与Zr元素.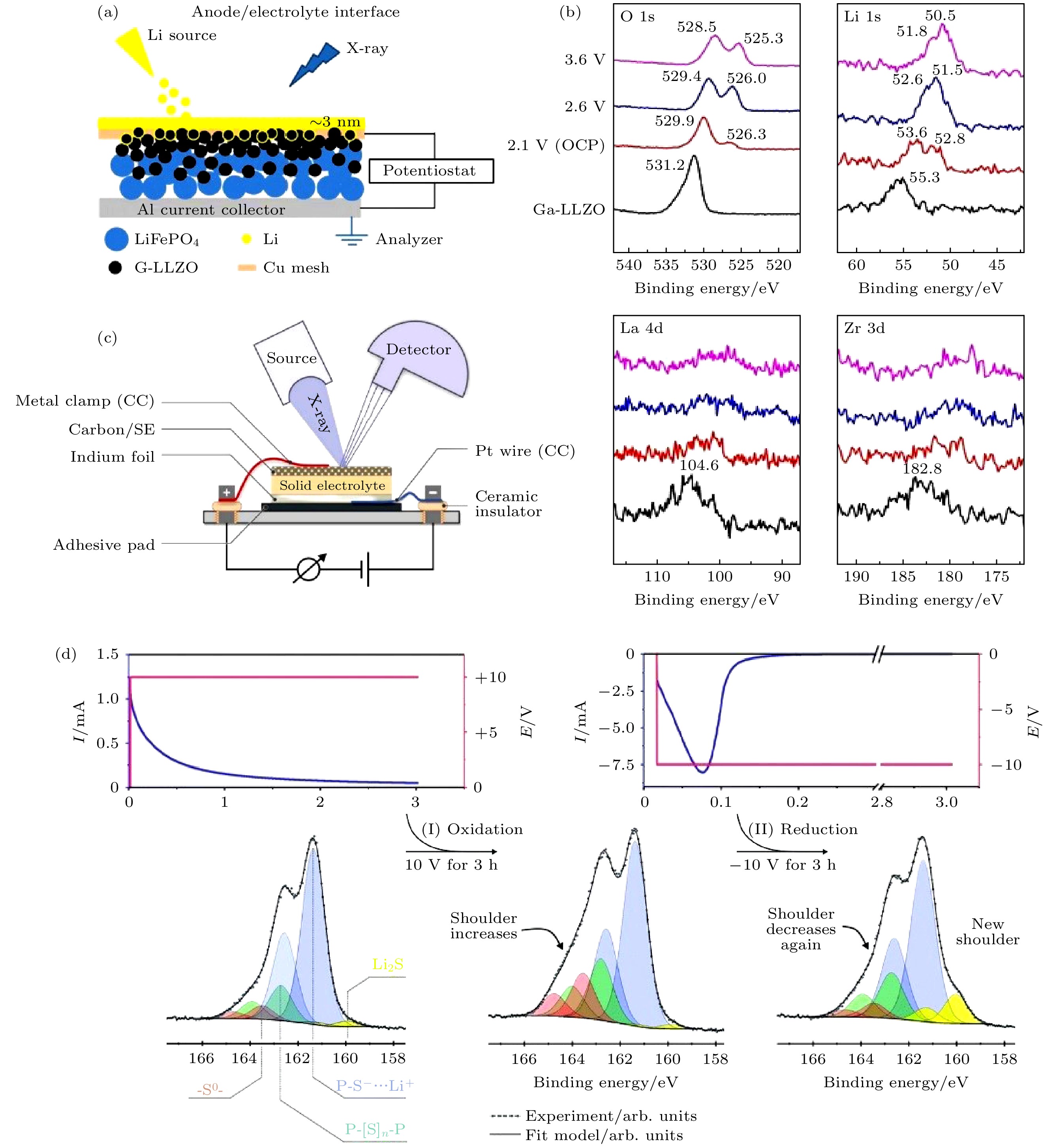 图 10 (a)电化学电池的负极界面原位XPS测试示意图; (b)不同电势下Li/Ga-LLZO界面的O 1s, Li 1s, La 4d, Zr 3d谱图[71]; (c)原位XPS装置示意图; (d)电化学极化中混合正极的S 2p谱图[59]
图 10 (a)电化学电池的负极界面原位XPS测试示意图; (b)不同电势下Li/Ga-LLZO界面的O 1s, Li 1s, La 4d, Zr 3d谱图[71]; (c)原位XPS装置示意图; (d)电化学极化中混合正极的S 2p谱图[59]Figure10. (a) Scheme of the configuration of the anode surface of electrochemical cell for in situ XPS; (b) in situ XPS spectra of O 1s, Li 1s, La 4d, and Zr 3d at the Li/Ga-doped LLZO interphase as a function of potential[71]; (c) scheme of the measurement setup for in situ XPS; (d) S 2p spectra of composite cathode during electrochemical polarization[59].
Koerver等[59]运用电化学原位XPS方法研究了硫化物电解质β-Li3PS4在电化学条件下的局部变化, SSLIB为三明治结构: In负极/β-Li3PS4固体电解质/C65-β-Li3PS4混合正极, 电池在初始、充电后、放电后的开路电压分别为0.61, 2.11, 0.20 V. 电化学原位XPS测试使用恒电位法, 利用电化学工作站分别对电池在10和–10 V的电位下进行3 h的充/放电, 并在过程中持续记录电流的变化, 对正极表面进行XPS表征(图10(c)). 由实验结果(图10(d))所示, β-Li3PS4与C65在制备过程会形成界面层, 主要成分为S0以及少量的Li2S. 在电化学氧化过程中, 高结合能的S0含量增大, 电流从初始最大值1.2 mA逐渐降低, 意味着电阻在逐渐增加, 说明界面层的生长. 在电化学还原过程中, S0峰下降, 而Li2S峰增加, 电流在初始的几分钟最大可达8 mA, 说明氧化过程中生成的高电阻组分在还原过程中转变为电阻更低的组分, 之后电流迅速减小甚至低于氧化态, 可能进一步发生了还原反应, 生成高电阻的组分和Li2S.
2
4.4.原位XPS/UPS研究SSLIB能带排列
当两种材料接触时, 由于化学势的不同, 界面处发生载流子的重新分布, 形成空间电荷层并建立内建电场[79]. 内建电场导致界面能带弯曲, 从而改变界面的导电性质. SSLIB中, 锂离子倾向于流向低化学势的正极, 平衡时界面的固体电解质侧形成锂耗尽层, 正极侧积累间隙锂离子. 由于正极一般具有较好的离子/电子混合导电性, 间隙锂离子可被电子消除, 使空间电荷层进一步扩大, 锂离子迁移势垒增大, 导致界面电阻的增加[80]. 通过沉积薄膜进行原位XPS/UPS表征, 可证实在固体电解质界面处形成空间电荷层, 能带发生弯曲, 能带弯曲程度表现在界面处光电子结合能发生偏移. 在沉积过程中能带的变化情况可由界面光电子结合能的位移情况和界面功函数的变化确定. Schw?bel等[12]以原位XPS/UPS研究了Li/LiPON/LiCoO2电池的正负极界面并得出全电池的能带图, 其中负极界面的分析方法如前所述, LiPON/LiCoO2正极界面的构建是通过在LiCoO2薄膜上以射频磁控溅射法沉积LiPON, 并于每一次沉积后进行XPS/UPS表征, 采用的实验装置是SoLiAs(solid liquid analysis system, Helmholtz centre Berlin for materials and energy), 由超高真空系统和同步辐射光电子能谱仪组成, 并与射频磁控溅射室相连接.对于LiPON/LiCoO2正极界面, 如图11(a)所示, LiCoO2价带顶相对费米能级的位置为0.3 eV. 沉积较长时间得到LiPON体相表面, 其荷电效应导致的谱峰位移非常明显, 故仅考虑较短沉积时间(12 s)内的结果. 同时LiCoO2的内层电子的谱峰都往高结合能移动了0.3 eV并趋于稳定, 这表明界面具有向下0.3 eV的能带弯曲. LiPON为电子绝缘体, 因此能带弯曲主要是锂离子由LiCoO2衬底迁移至LiPON造成的. LiCoO2和LiPON的ΔEVBM (价带顶之差)可由沉积前后Co 2p3/2和P 2p相对VBM (价带顶)的位置计算得到, ΔEVBM为1.26 eV.
 图 11 (a) LiPON沉积过程中LiCoO2表面的Co 2p, O 1s, P 2p和价带谱图; (b) LiCoO2内层电子结合能, LiPON P 2p电子结合能和表面功函数在沉积过程中的变化; (c)金属锂沉积过程中LiPON内层电子结合能的变化; (d)Li/LiPON/LiCoO2 SSLIB的能带图[12]
图 11 (a) LiPON沉积过程中LiCoO2表面的Co 2p, O 1s, P 2p和价带谱图; (b) LiCoO2内层电子结合能, LiPON P 2p电子结合能和表面功函数在沉积过程中的变化; (c)金属锂沉积过程中LiPON内层电子结合能的变化; (d)Li/LiPON/LiCoO2 SSLIB的能带图[12]Figure11. (a) Co 2p, O 1s, P 2p and valence band spectra of LiCoO2 surface during the deposition of LiPON; (b) evolution of the core level binding energies of the LiCoO2 substrate and the P 2p binding energy of the covering LiPON layer as a function of deposition time; (c) evolution of the binding energies during the stepwise evaporation of lithium on top of LiPON; (d) energy band diagram of Li/LiPON/LiCoO2 SSLIB[12].
对于Li/LiPON负极界面(图9(d)), 原始衬底LiPON的各谱峰都往高结合能方向发生约1.5 eV的位移, 这是因为LiPON本身的电子绝缘性而产生荷电效应[68], 当锂金属沉积达到一定厚度时, 可使界面与样品台实现电子传导消除荷电效应. 锂金属薄膜的Li 1s峰的结合能仅为53.4 eV, 而其参考结合能为54.9 eV, 说明界面能带发生弯曲, 存在空间电荷层, 由于锂金属和固体电解质导电性的差异, 电压降主要集中在界面区域. 当空间电荷层接近稳定时, LiPON的价带顶位置结合能负移0.5 eV, 说明能带向上弯曲0.5 eV. Li和LiPON的ΔEVBM同样由内层电子结合能与价带顶的相对位移求得, 为4.1 eV. 再根据参考数据(如LiPON的带隙、Li的功函数)可得到Li/LiPON/LiCoO2固态薄膜电池的能带图(图11(d)).
Operando电池应该同时满足工作状态和原位表征的要求[83]. 对operando XPS电池, 需要一定的压力维持电池循环, 以及供X射线与光电子通过的窗口, 目前常见的设计是通过两端对夹层施加压力的三明治结构的电池, 于电极侧开孔使X射线透过[84]. Wu等[83]在SSLIB界面的operando XPS研究中使用了自行设计的operando XPS电池(图12(a)), 产生电池正常工作需要的机械压力, 其中③为顶部电池盖, 由螺钉⑤固定产生机械压力, 中心有狭缝, 在最大机械压力时使X射线依然可以无阻碍地入射; ②是聚甲醛(polyoxymethylene, POM)环, 工作电极/固体电解质/对电极结构的电池安装于POM环中; ①为电池底座, ⑥与顶部电池盖接触构成回路; ④为塑料垫圈, 防止内部短路. 电池的工作电极由质量分数为50% 的 Li2S-P2S5, 质量分数为45% 的LiCoO2和质量分数为5% 的气相生长碳纤维VGCF混合制得, 固体电解质为Li2S-P2S5, 对电极由铟锂合金(InLix)和In/ Li2S-P2S5/C缓冲层组成. 利用循环伏安法(扫描速度为5 μV·s–1), 比较了operando XPS电池在超高真空(真空度 < 2 × 10–9 mbar (1 mbar = 100 Pa))下与在150 MPa下工作的标准SSLIB的循环性能(图12(b)). 在1.9—3.6 V (vs. InLix)电位区间内, 循环伏安法结果显示工作电极在充电过程出现了两个氧化峰, 分别位于2.6和3.4 V附近, 对应Li2S-P2S5的氧化分解和LiCoO2的电化学反应. Operando XPS电池中没有测量到额外的过电位, 显示电池设计的可靠性. 采用恒电位法作为operando XPS的电化学表征方法, 在不同电位下operando XPS S 2p, P 2p和Co 3p-Li 1s-Fe 3p芯能级谱变化情况如图12(c)所示. 在充电过程对应的XPS结果中(图12(c)), S 2p和P 2p中高结合能出现肩峰并逐渐增强, 归因于PS

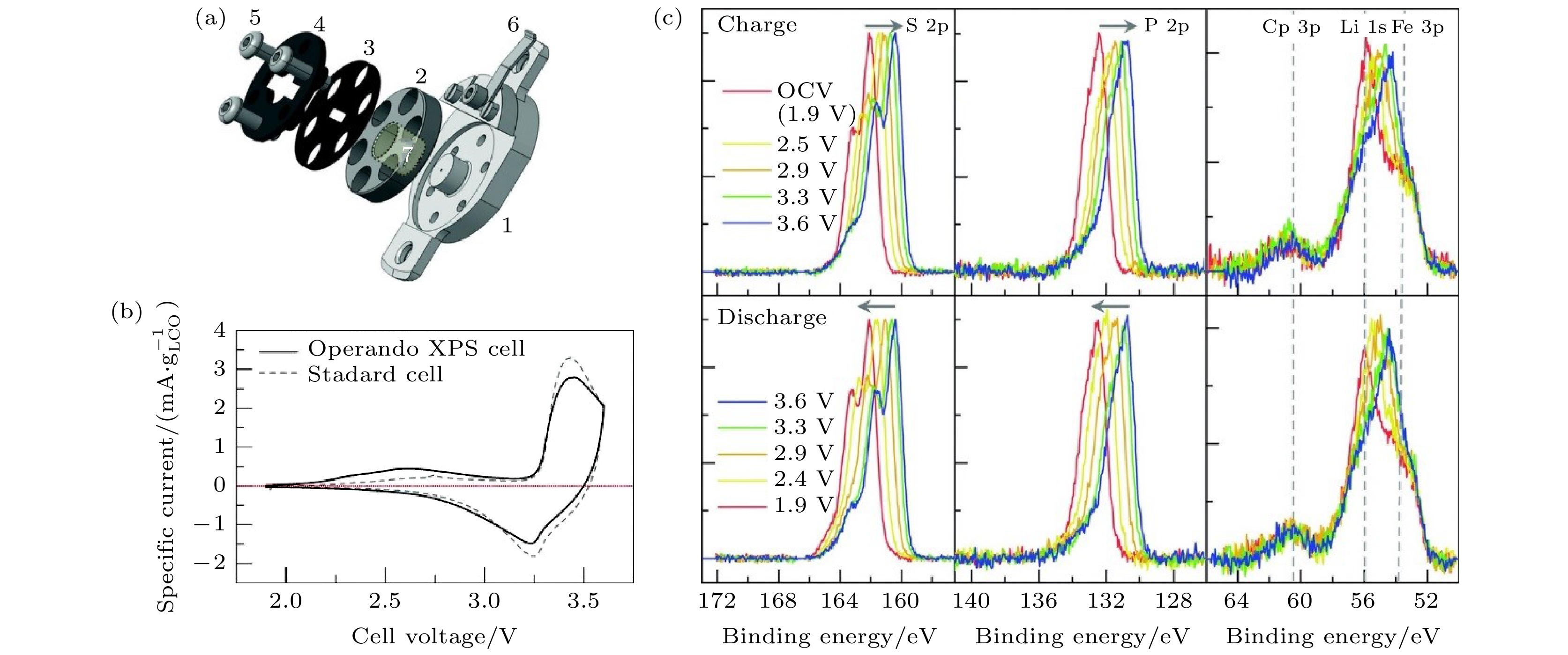 图 12 (a)全固态operando XPS电池的结构示意图[83]; (b) Operando XPS电池和标准电池工作电极的循环伏安测试结果[83]; (c) S 2p, P 2p, Co 3p-Li 1s-Fe 3p谱图在operando XPS测试过程中的变化[83]
图 12 (a)全固态operando XPS电池的结构示意图[83]; (b) Operando XPS电池和标准电池工作电极的循环伏安测试结果[83]; (c) S 2p, P 2p, Co 3p-Li 1s-Fe 3p谱图在operando XPS测试过程中的变化[83]Figure12. (a) An operando XPS cell design for all-solid-state batteries[83]; (b) cyclic voltammetry measurements of the work electrodes of the operando XPS cell and a standard test cell[83]; (c) evolution of S 2p, P 2p, and Co 3p-Li 1s-Fe 3p core level spectra recorded during the operando XPS measurements[83].
Wood等[85]通过一种虚拟电极法实现电池的充放电循环, 利用光电子能谱仪自带的电子枪对Li/L3PS4半电池的电解质表面实现充电, XPS分析结果证实了界面层和锂金属的产生, 实现了Li/L3PS4对称电池的原位构建. 其后电子照射持续进行使锂离子向锂金属表面迁移模拟充电过程, 以紫外光辐照表面激发光电子使锂离子从锂金属表面脱出模拟放电过程, 同时利用XPS监测表面的变化情况(图13(a)). 如图13(b)所示, XPS观察到原始表面有氧元素, 其晶格内存在替代的氧原子, 表面成分为Li3POxS4–x. 充电过程中控制电子枪的电子动能为11.5 eV, 法拉第杯测得对应的入射电流密度约为0.17 mA/cm2. 充电0.5 h时界面层成分为Li2S, Li3–xP, Li3PO4和Li3PS4. 充电2 h时界面层已经达到一定厚度, 进一步产生Li2O和Li0, 同时Li3–xP部分转变为Li3P. 放电过程使用波长为405 nm、光子能量为3.06 eV的紫外光, 并施加–45 V的样品偏压提高离子电流, 在紫外光照射和外加偏压的条件下, 得到了约150 nA的净光电发射电流, 表明该方法有足够的电化学驱动力驱使Li+离子离开表面并通过Li2S-P2S5迁移. 放电过程中表面P 2p中重新产生Li3–xP峰, 且Li3P峰强度减少, Li3PO4峰增强; O 1s中出现代表Li2O2的峰并且逐渐增强, 同时Li2O强度减弱, 说明界面层的Li-P相具有一定的反应可逆性, 放电过程中Li2O与Li3P反应生成Li3PO4和Li2O2.
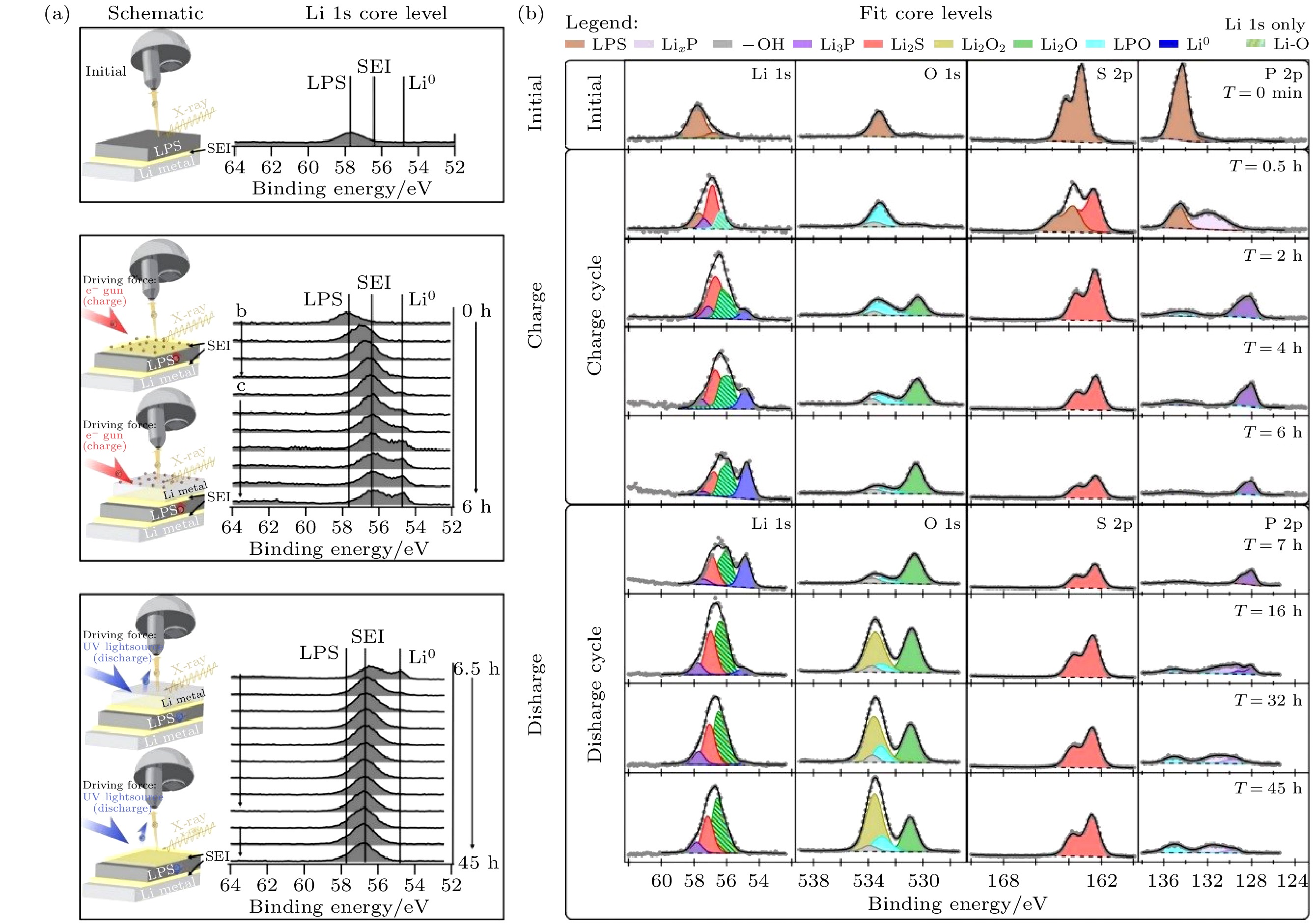 图 13 (a)虚拟电极法实现operando XPS测试的原理图和原始、充电、放电状态下Li2S-P2S5表面的Li 1s谱图变化[85]; (b)各XPS谱图及拟合结果在operando XPS过程中的变化[85]
图 13 (a)虚拟电极法实现operando XPS测试的原理图和原始、充电、放电状态下Li2S-P2S5表面的Li 1s谱图变化[85]; (b)各XPS谱图及拟合结果在operando XPS过程中的变化[85]Figure13. (a) Schematic of operando XPS measurements via virtual electrode cycling and the evolution of Li 1s spectra on Li2S-P2S5 surface during the cycling process[85]; (b) evolution of XPS spectra showing peak deconvolution during operando XPS measurements[85].
界面层各组分的过电位η表现为其光电子结合能的位移





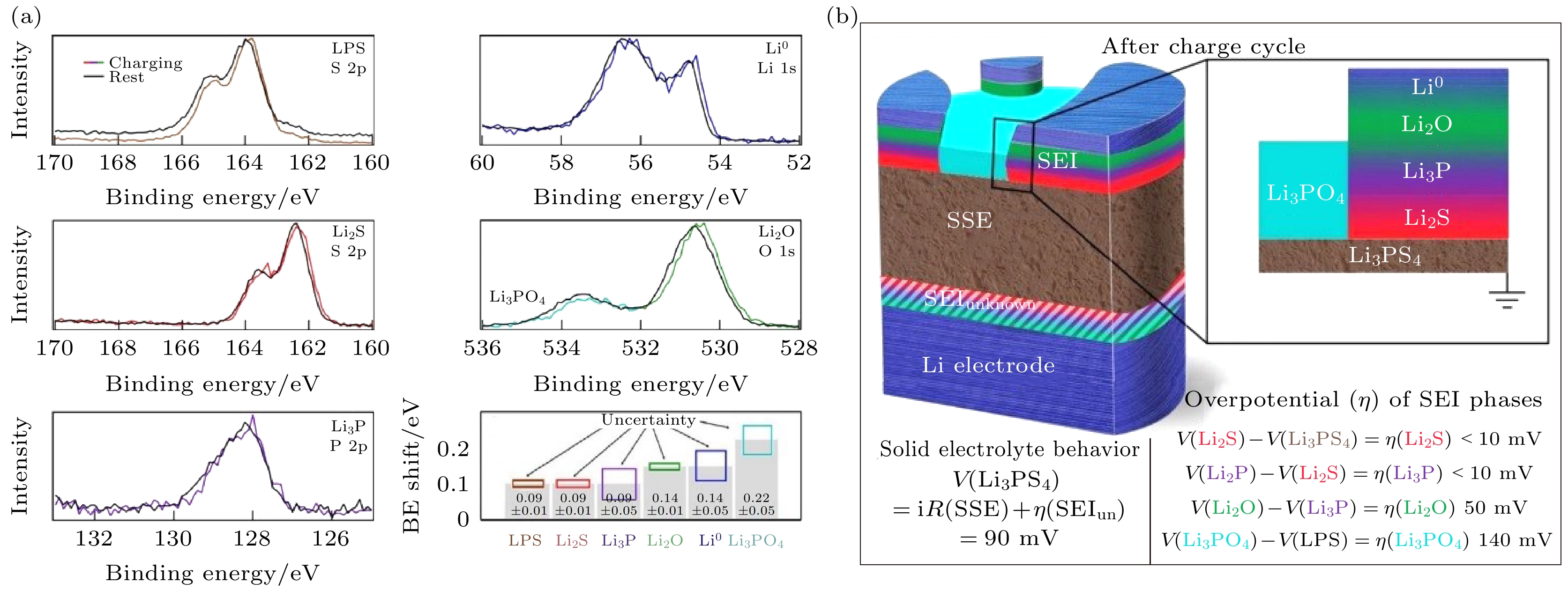 图 14 (a) SEI各组分充电过程中结合能的位移[85]; (b) SEI各组分的过电位及得到的SEI结构示意图[85]
图 14 (a) SEI各组分充电过程中结合能的位移[85]; (b) SEI各组分的过电位及得到的SEI结构示意图[85]Figure14. (a) Binding energy shifts of each SEI phase composition during charging process[85]; (b) overpotential of each SEI phase composition during charging process and scheme of SEI structure after charging based on the overpotential[85].
由于实现条件的困难, 目前operando XPS的研究实例并不多, 研究方法也还有很大的发展空间. 电池在常压环境下工作, 而XPS需要超高真空环境, 所以需要使用近常压XPS(near-ambient pressure XPS)进一步接近SSLIB实际工作状况. Operando XPS与其他表征方法的联用也是一个具有前景的方向, 比如与质谱、拉曼光谱等技术联用, 实现对电池较全面的综合分析. 此外, 对operando XPS电池进一步的改进与全新的设计, 对于实现更好的原位XPS表征也非常重要.
XPS作为表面灵敏的分析方法, 实验结果难免会受到各种因素的影响, 比如表面形貌、荷电效应和离子溅射效应等, 因此在构筑界面和实验过程中, 特别是对于原位实验, 需要考虑如何减少干扰因素的影响. Operando XPS方法结合了实时电化学表征, 是未来研究的趋势, 但满足其实验条件是一大难题. 如何设计operando电池实现真实电池工作条件下的XPS分析和电化学表征, 以及将电池电化学性能变化与XPS反映的界面变化联系起来, 还需要进一步研究和探索.
