全文HTML
--> --> -->2.1.分子光致变色的机理描述
分子光致变色的机理通常用图1所示的势能曲线来描述[3,8]. 就二芳基乙烯类分子的光致开环/闭环转换过程, 曲线1代表体系从开环构型A到闭环构型B的基态势能曲线, EA和EB分别为其热活化势垒. 按照Woodward-Hoffman原理, 含杂环的二芳基乙烯类分子体系有平行和反平行两种构象, 只有反平行的构象能够发生顺旋光环化反应. 顺旋机制要求所涉及的分子构型, 包括开环构型、环闭构型以及构型转换过程中连接两种构型的过渡态, 都应该具有C2对称性. 曲线2为构型A的激发态势能曲线, 在吸收能量为hv光子后退激发, 转换为构型B; 曲线3为构型B的激发态势能曲线, 在吸收能量为
 图 1 光致变色机理示意图
图 1 光致变色机理示意图Figure1. Schematic diagram of photochromic mechanism.
2
2.2.稳态构型、最小能量路径及激发态结构计算方法
对本文研究的DFMA分子, 如图2所示, 首先需要确定处于开环(记作O-DFMA)和闭环(记作C-DFMA)时的稳态构型. 在保持原子编号一致的条件下, 分别设置开环和闭环的初始构型, 然后采用密度泛函方法进行稳态构型的优化计算. 首先在BP/SVP水平上进行优化和频率分析, 然后在所获得的结构基础上, 加大计算基组为TZVP和def2-TZVP[26], 并用杂化密度泛函B3LYP, 再次分别进行构型优化和频率分析, 将获得的O-DFMA和C-DFMA稳态构型作为后续计算的基础.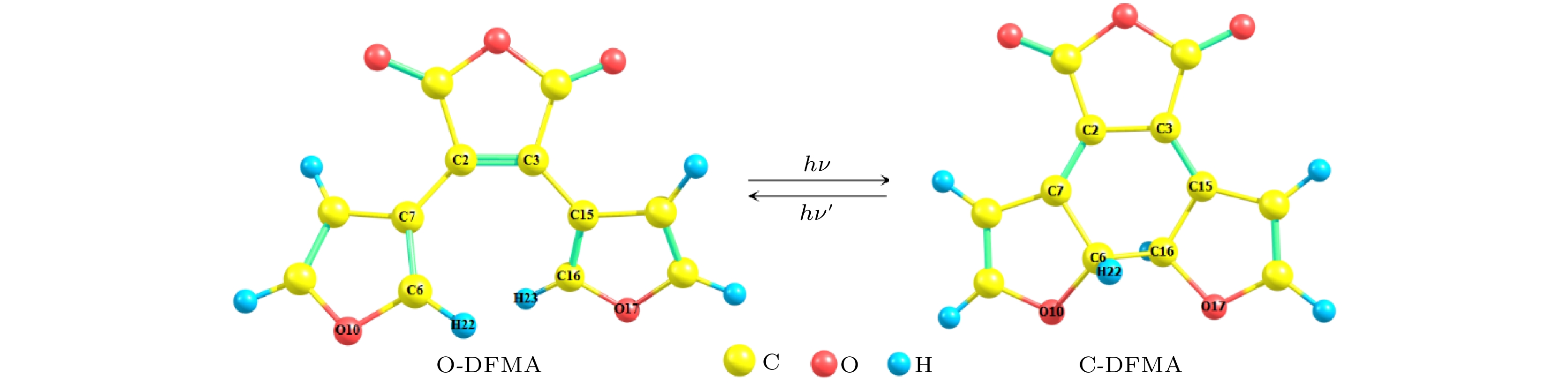 图 2 DFMA分子的开环和闭环的稳态构型
图 2 DFMA分子的开环和闭环的稳态构型Figure2. Steady-state structures of the open-ring and closed-ring of DFMA molecule.
为了寻找连接O-DFMA和C-DFMA之间的最小能量路径(minimum energy path, MEP), 并找到两稳态之间的过渡态(TS-DFMA), 以稳态的O-DFMA和C-DFMA构型为初始构型, 采用微动弹性带(nudged eleastic band, NEB)方法[27-28]进行路径寻找. 在该方法中, 初始路径由一组离散的原子配置(称为系统映像)生成和表示, 两稳态之间的分子图像数量由用户指定. 为了获得较高能量分辨率的MEP曲线并找到对应过渡态的鞍点, 同时又不加大计算的难度, 经反复尝试, 本文选择内插图像数为20, 加上两个稳态构型, 最后MEP曲线由22个分子构型组成.
DFMA激发态的计算是采用含时密度泛函方法, 在B3LYP/def2-TZVP水平上完成的. 为了获得尽可能多的激发态结构信息, 基于MEP所对应的22个分子图像, 计算每个图像下最低的8个单重激发态及其从基态到激发态的跃迁偶极矩, 最后将各激发态能量按照不同分子构型画出曲线, 即得到不同电子激发态的势能曲线, 结合跃迁偶极矩数值, 可用于光致变色分子开关机理的讨论及光吸收效率的分析.
2
2.3.稳态分子吸收速率的计算方法
分子的电子激发量子效率是光致变色分子开关的关键指标. 本文对两稳态O-DFMA和C-DFMA的光吸收速率计算是基于量子化分子体系与经典光场相互作用的理论, 采用路径积分近似的方法进行的[29]. 分子体系在初态










上述分子动力学计算跃迁速率的过程, 可归结为以下步骤: 1)读取初、末态的几何构型参数和力常数矩阵(Hessians); 2)计算初、末态坐标之间的Duschinsky转动矩阵及位移矢量; 3)计算跃迁偶极矩






本文的所有计算是用ORCA程序包[30-31] (版本4.2.1)在计算服务器上完成的.
3.1.DFMA分子开环和闭环的稳态构型
为了整体了解分子的空间结构, 我们选择乙烯桥的键长(RC2-C3)、两个芳香环上O原子的距离(RO10-O17)和两个O原子与乙烯桥构成的二面角(DO10-C2-C3-O17)来近似描述分子结构. 图2和表1分别给出了优化后的稳态结构及相应的几何参数. 从图2可以发现: 开环和闭环稳定构型中作为乙烯桥的马来酸酐环均保持近平面结构, 说明马来酸酐环的确起到了“稳定器”的作用; 两个芳香杂环在O-DFMA稳定构型中也各自保持了近平面结构(参与构成闭环的H22, H23除外), 而在C-DFMA稳定构型中参与构成闭环的C6和C16明显偏离杂环平面.| Methods | Structure | RC2-C3/nm | RO10-O17/nm | DO10-C2-C3-O17/(°) | Total thermal energy (hartree) | Energy difference/cm–1 |
| BP/SVP | O-DFMA | 0.1382 | 0.5552 | 20.24 | –836.23050 | 1232 |
| C-DFMA | 0.1450 | 0.3156 | 0.92 | –836.22488 | ||
| B3LYP/TZVP | O-DFMA | 0.1359 | 0.5482 | 21.33 | –836.63680 | 1815 |
| C-DFMA | 0.1447 | 0.3153 | 0.98 | –836.62853 | ||
| B3LYP/def2-TZVP | O-DFMA | 0.1359 | 0.5480 | 19.82 | –836.68342 | 2055 |
| C-DFMA | 0.1445 | 0.3150 | 0.75 | –836.67406 | ||
| NEB/def2-SVP | TS-DFMA | 0.1380 | 0.3365 | 8.45 |
表1O-DFMA和C-DFMA分子稳态构型参数
Table1.Stable state configuration parameters of O-DFMA and C-DFMA.
比较开环/闭环的几何结构参数(如表1所示)发现: 1)乙烯桥的键长变化不大, 闭环比开环仅拉长了0.01 nm; 2)两个芳香环间O原子距离RO10-O17变化达0.2 nm以上, 说明开环/闭环转换时两个芳香环整体有明显位移, 由此可推测, 如果芳香环通过配位延伸, 或者芳香环被其他配体所“定位”, 可能影响开环/闭环的有效转换; 3)两个O原子与乙烯桥构成的二面角(DO10-C2-C3-O17)变化达到了近似20°, 说明分子从开环时的整体近似“平面”向闭环转换时, 两个芳香环在相互靠近的同时发生了明显的反式扭转, 根据对O-DFMA分子的振动模式的计算发现, 最符合这种扭转的振动模式对应一种分子整体的“骨架”振动, 谐振频率仅为100 cm–1, 红外谱强度很小(仅为0.18, 最强振动模强度达729), 说明其红外活性不强; 4)比较总热能(电子能、零点振动能、常温下的转动和平动能之和), 发现在3种计算水平下所得结果都显示O-DFMA的能量低于C-DFMA, 说明开环更稳定, 但开环/闭环的总热能差并不大, B3LYP/def2-TZVP计算给出的总热能差仅为2055 cm–1, 预示着DFMA分子可能是开环/闭环双稳态分子, 这一点从下面的最小能量路径(MEP)计算中得到了证明.
2
3.2.O-DFMA与C-DFMA之间最小能量路径
图3是采用NEB法找到的最小能量路径(MEP), 即基态S0的势能曲线. 图3中横坐标0和21分别对应于O-DFMA和C-DFMA的稳态构型, 14对应TS-DFMA构型. 由MEP曲线可以得出O-DFMA和C-DFMA的热活化势垒分别是24959 cm–1 (3.0945 eV)和23328 cm–1 (2.8923 eV). 考虑到DFMA分子的开环/闭环反应是分子骨架的整体变化, 而分子骨架振动模的谐振频率处于分子振动模谐振频率(28—3300 cm–1)的低频段, 即分子骨架振动的谐振频率远小于热活性势垒, 预示着O-DFMA和C-DFMA均具有良好的热稳定性, 即DFMA可能为热稳定的双稳态分子. 从NEB计算中提取出TS-DFMA(image14)结构参数(见表1)与开环/闭环稳态结构对比发现, TS在结构上更接近闭环的稳态构型.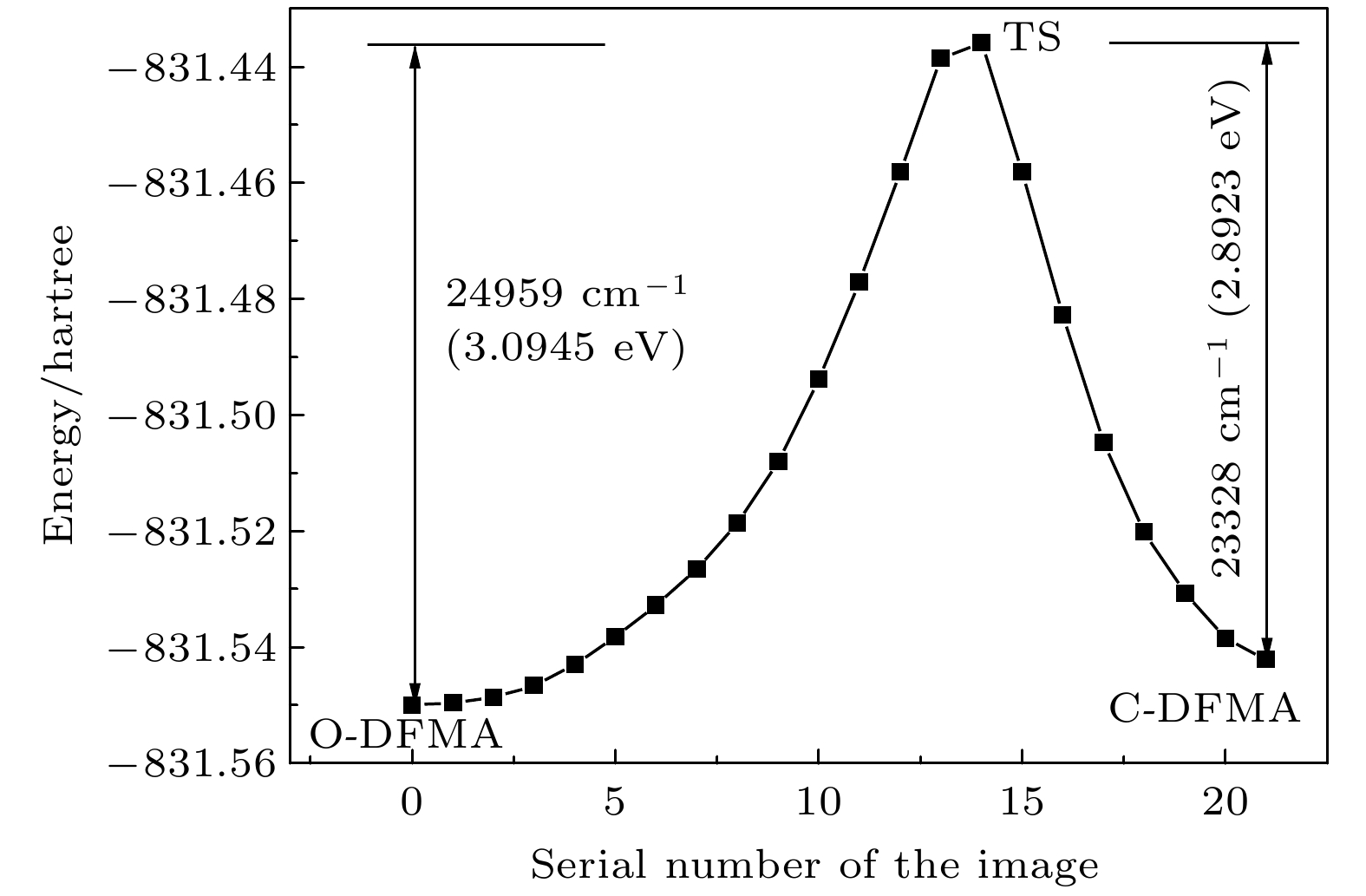 图 3 NEB法计算的开环/闭环之间的最小能量路径(MEP)
图 3 NEB法计算的开环/闭环之间的最小能量路径(MEP)Figure3. Minimum energy path (MEP) between O-DFMA and C-DFMA calculated by NEB method.
2
3.3.激发态势能曲线
基于NEB计算获得的开环与闭环之间最小能量路径上22个分子结构, 计算了8个单重激发态的势能曲线. 结果见图4, 在最低的8个激发态中, 仅第1激发态S1的势能曲线在对应TS-DFMA构型时出现极小, 而其余7个激发态的势能曲线与基态S0相似, 在TS构型处存在势垒. 由此特征可推断, 分子处于O-DFMA或C-DFMA稳态时, 最有可能实现光致开关的途径是从基态S0到S1态的光吸收, 而其余激发态的光激发不具备这样的可能. 为了了解S0态到S1态的光激发效率, 图5给出了S0态到S1态跃迁偶极矩的平方值随MEP的变化曲线. 从图5可以看出, 处于基态时的双稳态O-DFMA和C-DFMA到S1态的跃迁偶极矩平方分别是1.2 arb.units2和3.1 arb.units2, 说明开环/闭环的激发跃迁均为允许跃迁, 且跃迁偶极矩相对最小能量路径上的其他构型而言数值较大, 意味着开环和闭环均有相对较高的激发量子效率, 符合作为光致开关分子的基本要求. 从图4可知, O-DFMA和C-DFMA向S1态电子激发跃迁的波长分别为387.4 nm和392.4 nm.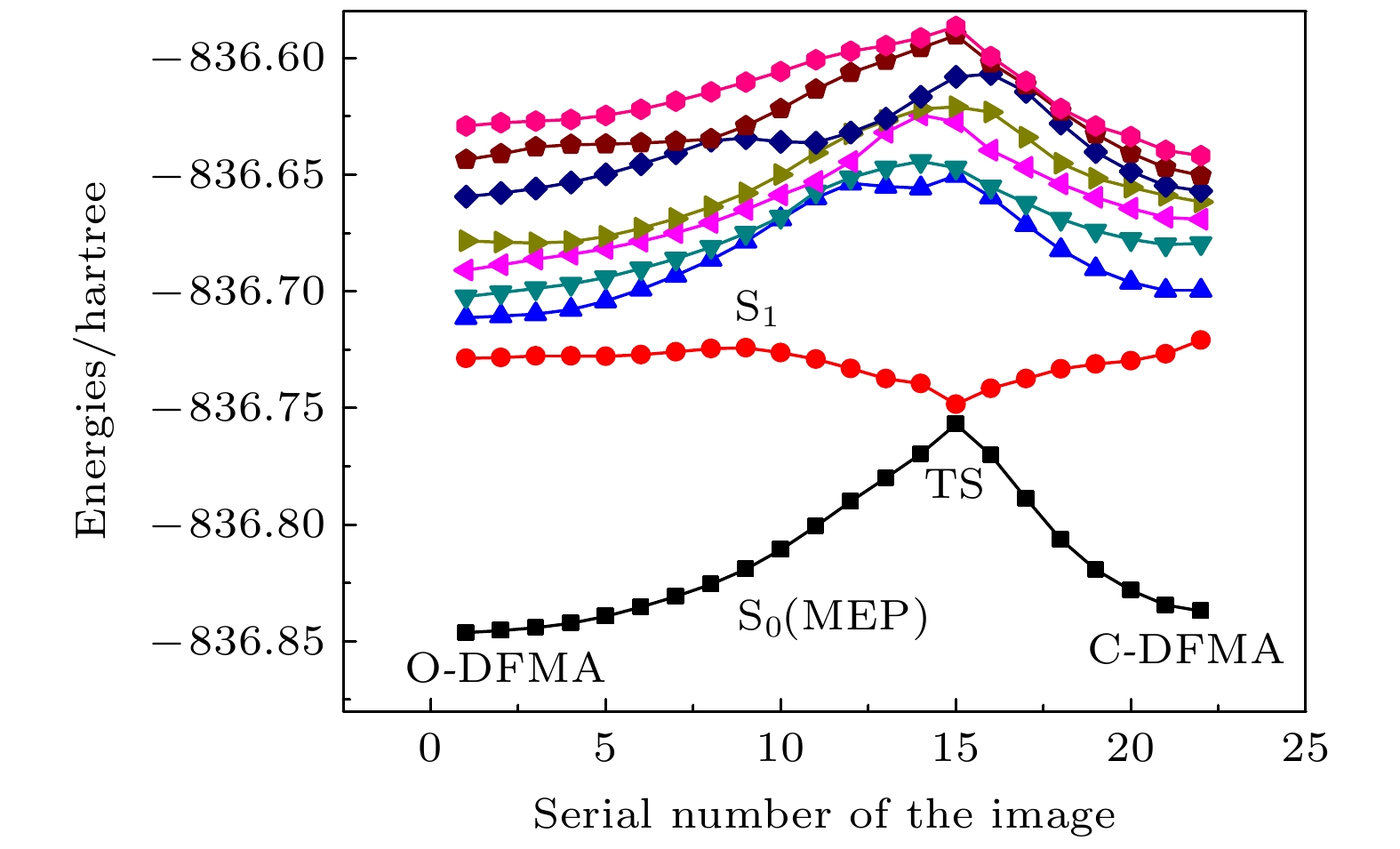 图 4 TD-DFT/B3LYP/def2-TZVP水平上计算的DFMA分子势能曲线
图 4 TD-DFT/B3LYP/def2-TZVP水平上计算的DFMA分子势能曲线Figure4. DFMA molecular potential energy curves calculated with TD-DFT/B3LYP/def2-TZVP.
 图 5 S0态到S1态激发跃迁偶极矩平方随MEP的变化
图 5 S0态到S1态激发跃迁偶极矩平方随MEP的变化Figure5. Square of the transition dipole moment from S0 to S1 state along the MEP.
2
3.4.O-DFMA和C-DFMA光致变色开关机理分析
沿着MEP, 分析基态S0到S1态跃迁所对应的主要分子轨道, 发现该跃迁主要发生在最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)之间. 为此, 从MEP的22个分子构型下提取出这2个分子轨道的图像. 根据分子轨道的图像特征, 可将MEP中的22个点大致分为5组, 其中0–11为开环组, 12–13为开环→TS过渡组, 14为TS组, 15–18为TS→闭环过渡组, 19–21为闭环组.将O-DFMA, TS和C-DFMA的HOMO和LUMO的空间分布图与S0态和S1态势能曲线结合来看(如图6所示), 可将开环/闭环的激发及相互转换做如下分析: 1)O-DFMA时: HOMO电子主要分布于由乙烯桥和2个呋喃环构成的大“C”形C链上, 且呈现出由C6-C7-C2-C3-C15-C16(见图2)构成的3个“局域”π轨道电子的特征; LUMO电子则主要分布于乙烯桥的马来酸酐环, 也呈现出2个局域π轨道的特征. 由此可认为, O-DFMA时的激发过程对应为电子从“C”形环到乙烯桥马来酸酐环的π–π跃迁. 2)TS时: 随着两个芳香环逐渐靠近, HOMO轨道上的电子逐渐从三个“局域”π轨道转移至“C”形环上分属原子C7, C3, C16的垂直各自所属环的p轨道上; 而LUMO上电子则从乙烯桥马来酸酐环的2个局域π轨道也转移至“C”形环上分属C6, C2, C15的p轨道上. 可见, TS构型时HOMO和LUMO上的电子布局恰好是在“C”形环上6个C原子p轨道的交错分布, 所以HOMO和LUMO的轨道能接近, 这也是S0和S1态势能曲线在TS构型时出现“重合”的原因. 3)C-DFMA时: HOMO电子主要分布于靠近乙烯桥的分属2个呋喃环的“局域”π轨道上; LUMO电子则分布于乙烯桥上C2–C3之间的局域π轨道和芳香环的C7和C15的p轨道上. 可见, 闭环时C-DFMA的激发过程仍可认为是一种π–π跃迁.
 图 6 S0态和S1态势能曲线, 其中插图为HOMO和LUMO空间分布图
图 6 S0态和S1态势能曲线, 其中插图为HOMO和LUMO空间分布图Figure6. Potential energy curves of S0 state and S1 state, where the inset is the spatial distribution diagram of HOMO and LUMO.
根据上面的分析, 结合图3的MEP曲线, 可将DFMA光致变色开关的机理描述为: 1) C-DFMA分子在S1–S0共振跃迁的波长激光作用下从S0态跃迁至S1态, 然后分子沿S1态势能曲线退激发, 在TS-DFMA构型处发生从S1态到S0态的交叉跳变, 然后回到O-DFMA构型, 完成由闭环→开环的开关动作. 因为在这区间S1态势能曲线单调下降, 预示着开环动作很难有荧光辐射发生. 2) O-DFMA分子在S1–S0共振跃迁的波长激光作用下从S0态跃迁至S1态, 由于从O-DFMA构型到TS-DFMA构型, S1态势能曲线存在一个相对“平坦区”, 直到接近TS-DFMA构型时, S1态的势能曲线才明显单调下降, 这意味着O-DFMA分子可能需要借助分子某些模的振动激发才能完成由开环→闭环的开关动作, 同时有荧光辐射发生. 开环到闭环转换完成, 也意味着荧光辐射结束. 所以作为潜在的光致变色荧光分子开关, 荧光的持续时间及强度, 与分子在光激励下完成闭环动作的效率密切相关.
2
3.5.稳态分子吸收光谱的模拟
为了更细致地了解O-DFMA和C-DFMA稳态分子从基态S0到第一激发态S1之间的光致吸收效率, 用ORCA程序包提供的分子激发态动力学(ESD)分析模块模拟计算了O-DFMA和C-DFMA的吸收光谱(图7), 激发态势能面构建选择了垂直梯度(VG)模型. 理论上, 吸收强度与实验上的消光系数ε相对应, 并且取决于光谱线型. 图7的模拟是选择了均匀加宽的Lorentz线型, 半高全宽(FWHM)参数为100 cm–1. 吸收总速率由弗兰克-康顿因子决定的强度(FC强度)和电子-振动耦合效应强度(HT强度)之和组成. 但比较发现, HT强度不到FC强度的1%, 说明电子-振动耦合效应不强, 几乎可以忽略. 由图7模拟结果可见, 两稳态分子的最强吸收峰均出现在S1–S0电子跃迁的0–0带, C-DFMA的吸收强度峰值大约是O-DFMA吸收强度峰值的5倍, 再次说明C-DFMA具有更高的吸收效率. 和图6所示的纯电子跃迁激发能相比, 0–0带相应的吸收波长明显增大, 分别是448.37 nm (C-DFMA)和442.73 nm (O-DFMA). 图 7 ESD模拟的O-DFMA和C-DFMA吸收光谱
图 7 ESD模拟的O-DFMA和C-DFMA吸收光谱Figure7. O-DFMA and C-DFMA absorption spectra simulated by ESD.
