全文HTML
--> --> -->实验上, Douglas和Lutz[2]首先在含硅烷的氦的空心阴极放电发射谱中观察到了SiH+离子的不同电子态









理论上, 人们也对SiH+离子开展了一系列高水平的从头算研究. 1986年, Hirst等[16]用多参考组态相互作用(MRCI)的方法[17]计算了SiH+离子














尽管已经对SiH+离子的各个电子态进行了大量的理论和实验研究, 但是精确的SiH+离子

2.1.单点能计算
SiH+

2
2.2.CBS方法
计算电子结构时通常采用1个高斯型轨道展开基组, 基组的不完备性产生基组重叠误差(BSSE)[29,30], 影响计算的准确性. Varandas[27,28]提出, BSSE误差可以通过有限基组单点能计算外推到完备基组(CBS)极限来纠正, 其基于Dunning的相关一致基组外推电子能量的方案如下.MRCI计算水平下的能量可写成
应用Karton和Martin[31]提出的KM方案可得CAS能量



根据Varandas[27,28]提出的统一的单重态和三重态外推方案, 动态相关(dc)能量为



2
2.3.势能曲线函数
本文中的势能函数曲线采用的是Aguado-Paniagua函数, 即双原子势能函数






3.1.势能函数
分别使用几种不同的基组和方法(AVQZ, AV5Z, AV6Z, CBS(Q, 5), AVQdZ, AV5dZ, AV6dZ, CBS(Qd, 5d)), 获得了SiH+(X1Σ+)离子的0.8a0—29a0范围内的112个从头算能量点. 基于这些能量点, 应用Aguado-Paniagua函数拟合得到APEF. 为了提高精确度, 使用了12个参数, 得到了几种不同基组和不同方法下的APEFs, 函数的具体参数列于表1 (表中只列出了主要讨论的AV6Z, CBS(Q, 5), AV6dZ和CBS(Qd, 5d)).| AV6Z | CBS(Q, 5) | AV6dZ | CBS(Qd, 5d) | SA-AV6dZ | SOC-AV6dZ | |
| a0 | 0.50876826×101 | 0.50552062×101 | 0.50858283×101 | 0.50691706×101 | 0.50773275×101 | 0.50772768×101 |
| a1 | –0.13804619×100 | –0.14563907×100 | –0.13801387×100 | –0.14215220×100 | –0.13720269×100 | –0.15322183×100 |
| a2 | –0.13563746×101 | –0.14531223×101 | –0.13570613×101 | –0.14215828×101 | –0.16375066×101 | 0.30937784×100 |
| a3 | –0.51802913×102 | –0.49687570×102 | –0.51738027×102 | –0.50317640×102 | –0.42586263×102 | –0.92938545×102 |
| a4 | 0.62749955×103 | 0.59645776×103 | 0.62624067×103 | 0.60422890×103 | 0.45856480×103 | 0.11152320×104 |
| a5 | –0.48068238×104 | –0.45486708×104 | –0.47942732×104 | –0.45972031×104 | –0.30344319×104 | –0.81653091×104 |
| a6 | 0.26148147×105 | 0.24858810×105 | 0.26073093×105 | 0.24994077×105 | 0.14706913×105 | 0.40426343×105 |
| a7 | –0.98935973×105 | –0.94946694×105 | –0.98651660×105 | –0.94886015×105 | –0.51962969×105 | –0.13681997×106 |
| a8 | 0.24960293×106 | 0.24200403×106 | 0.24891691×106 | 0.24047472×106 | 0.12676883×106 | 0.31028426×106 |
| a9 | –0.39722347×106 | –0.38879401×106 | –0.39620641×106 | –0.38442238×106 | –0.19927330×106 | –0.44979467×106 |
| a10 | 0.35937312×106 | 0.35468541×106 | 0.35853489×106 | 0.34921672×106 | 0.18029764×106 | 0.37617755×106 |
| a11 | –0.14069370×106 | –0.13986829×106 | –0.14040272×106 | –0.13721966×106 | –0.71149416×106 | –0.13802294×106 |
| β1 | 0.6890 | 0.6800 | 0.6890 | 0.6840 | 0.6870 | 0.6870 |
| β2 | 0.7470 | 0.7470 | 0.7470 | 0.7470 | 0.7470 | 0.7470 |
| ?Ermsd/ (kcal·mol–1) | 1.60176420×10–2 | 1.61755859×10–2 | 1.60042370×10–2 | 1.627559848×10–2 | 9.45767662×10–3 | 1.11170443×10–2 |
表1SiH+(X1Σ+) APEFs的拟合参数
Table1.Parameters of APEFs for SiH+(X1Σ+).
图1是使用AV6Z基组和CBS(Q, 5)方法得到的SiH+(X1Σ+)离子的从头算能量点(蓝色圆点)和拟合给出的势能曲线(红色实线). 两幅图中的上部区域是短程作用范围, 由于在该范围内能量变化范围较大, 因此纵坐标采用对数标度, 横坐标的取值范围是1a0—2a0, 纵坐标的取值范围为10–2Eh—101Eh. 中间区域横坐标取值范围是2a0—10a0, 纵坐标的取值范围为0至–0.14Eh, 其内插图为长程作用范围, 相应的横坐标取值范围是6a0—26a0和纵坐标的取值范围为0至–0.005Eh. 通过将曲线纵坐标标度的减小和横坐标标度的增大, 清晰地展示出渐近区域内势能曲线和从头算能量点相吻合的特征. 底部是拟合的势能曲线与从头算能量点之间的误差, 单位为cm–1. 从整体来看, 两种方案拟合得到的APEFs不论是在短程区域还是长程区域都表现出平滑的行为特征, 从头算能量点和拟合曲线之间符合得非常好, 误差均小于10 cm–1. 从表1可以看出, 使用CBS(Q, 5)和AV6Z基组的方均根误差(RMSD)为1.61755859 × 10–2和1.60176420 × 10–2 kcal/mol.
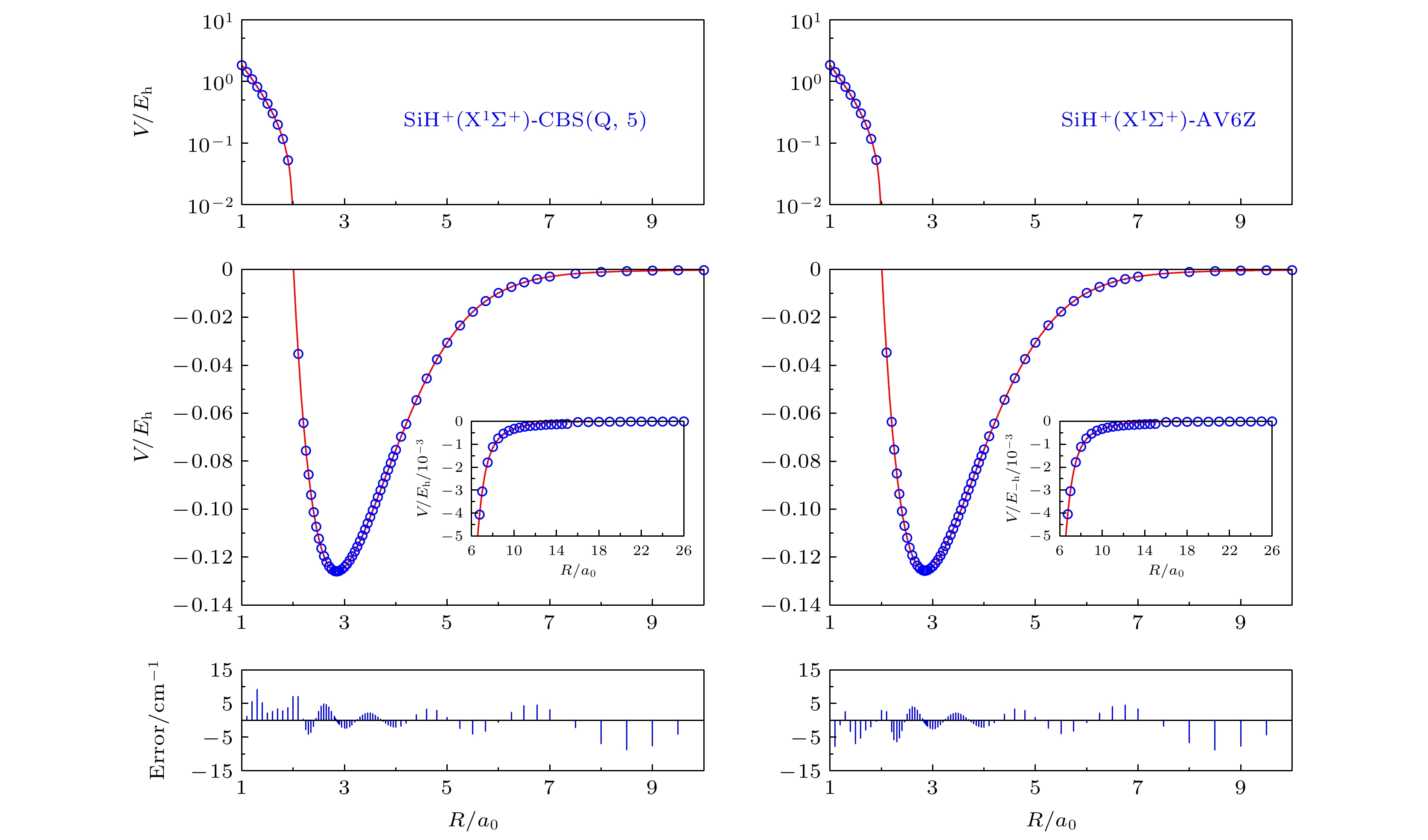 图 1 SiH+(X1Σ+)在CBS(Q, 5)和AV6Z基组下的势能曲线和从头算能量点
图 1 SiH+(X1Σ+)在CBS(Q, 5)和AV6Z基组下的势能曲线和从头算能量点Figure1. Potential energy curves and ab initio points at CBS(Q, 5) and AV6Z results.
为了提高计算精度, 本文还使用了AVQdZ, AV5dZ, AV6dZ基组及CBS(Qd, 5d)方法计算和拟合了SiH+(X1Σ+)离子的势能曲线. AVX Z和AVX dZ基组二者都属于Dunning的关联一致基组, 而后者在AVX Z基础上加入了扩散函数, 得到了紧凑的扩充集. 需要说明的是, 在使用AVQdZ和AV5dZ基组计算时, 对于Si和H元素都是使用AVQdZ和AV5dZ基组; 当使用AV6dZ基组计算时, 由于Molpro程序基组使用的限定, 对于Si和H元素分别使用AV6dZ基组和AV6Z基组计算.
图2给出了分别使用CBS(Qd, 5d)方法和AV6dZ基组所得势能曲线和从头算能量点, 蓝色圆点是从头计算得到的能量点, 红色实线是拟合得到的势能曲线函数. 从图2可以看出, 不论是在长程区还是在短程区, 使用CBS(Qd, 5d)方法和AV6dZ基组所得势能曲线光滑且与从头算能量点都符合较好, RMSD较小, 分别为1.60042370 × 10–2 kcal/mol和1.627559848 × 10–2 kcal/mol. 在短程区域, 应用AV6dZ基组的拟合曲线和从头算能量点的误差上限为15 cm–1, CBS(Qd, 5d)方法的误差上限为10 cm–1, CBS(Qd, 5d)方法相对于应用较耗费机时的AV6dZ基组计算更有优势.
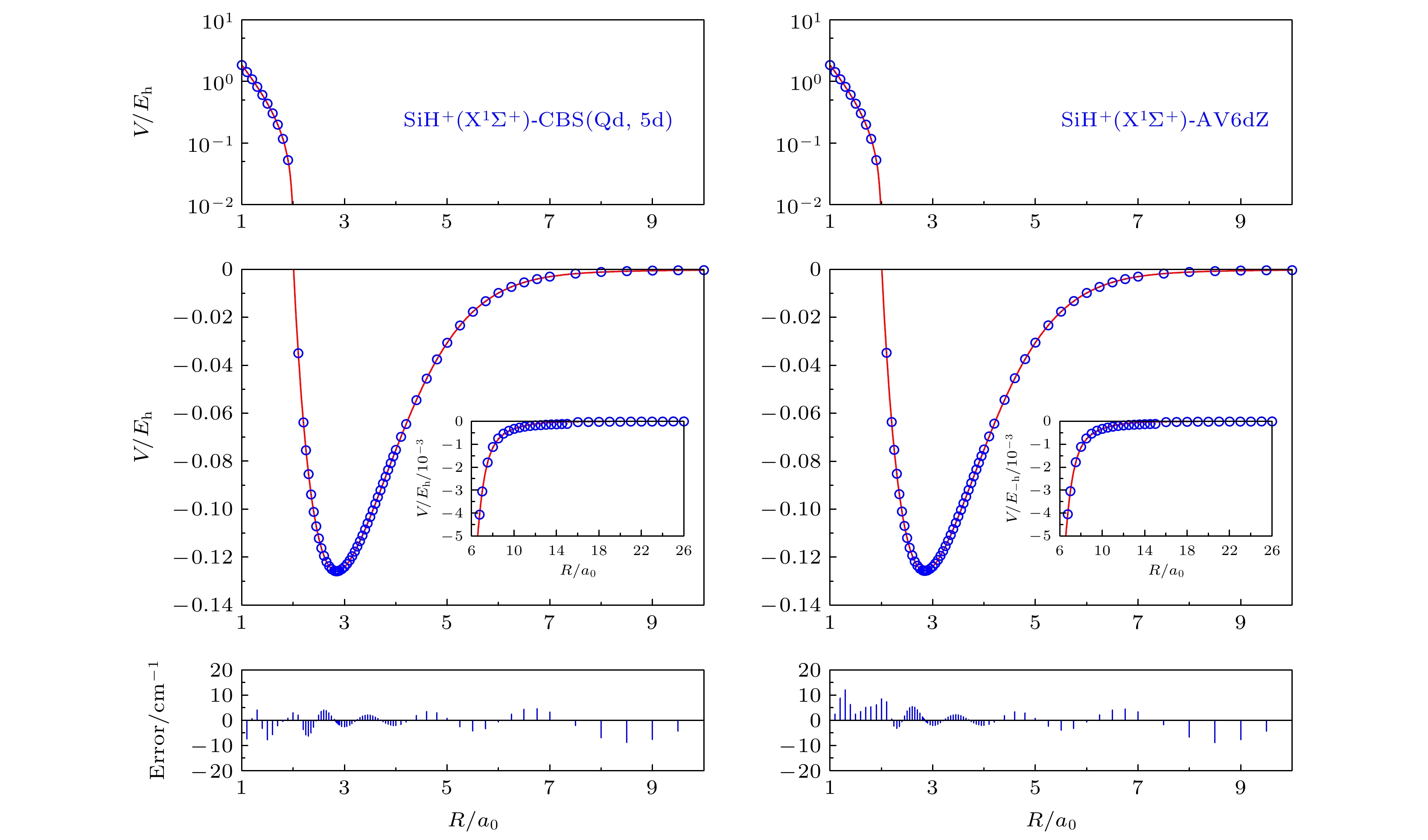 图 2 SiH+(X1Σ+)应用CBS(Qd, 5d)和AV6dZ基组的势能曲线和从头算能量点
图 2 SiH+(X1Σ+)应用CBS(Qd, 5d)和AV6dZ基组的势能曲线和从头算能量点Figure2. Potential energy curves and ab initio points at CBS(Qd, 5d) and AV6dZ results.
2
3.2.自旋-轨道耦合计算
关于SiH+(X1Σ+)离子的SOC效应的讨论涉及到对应同一解离极限




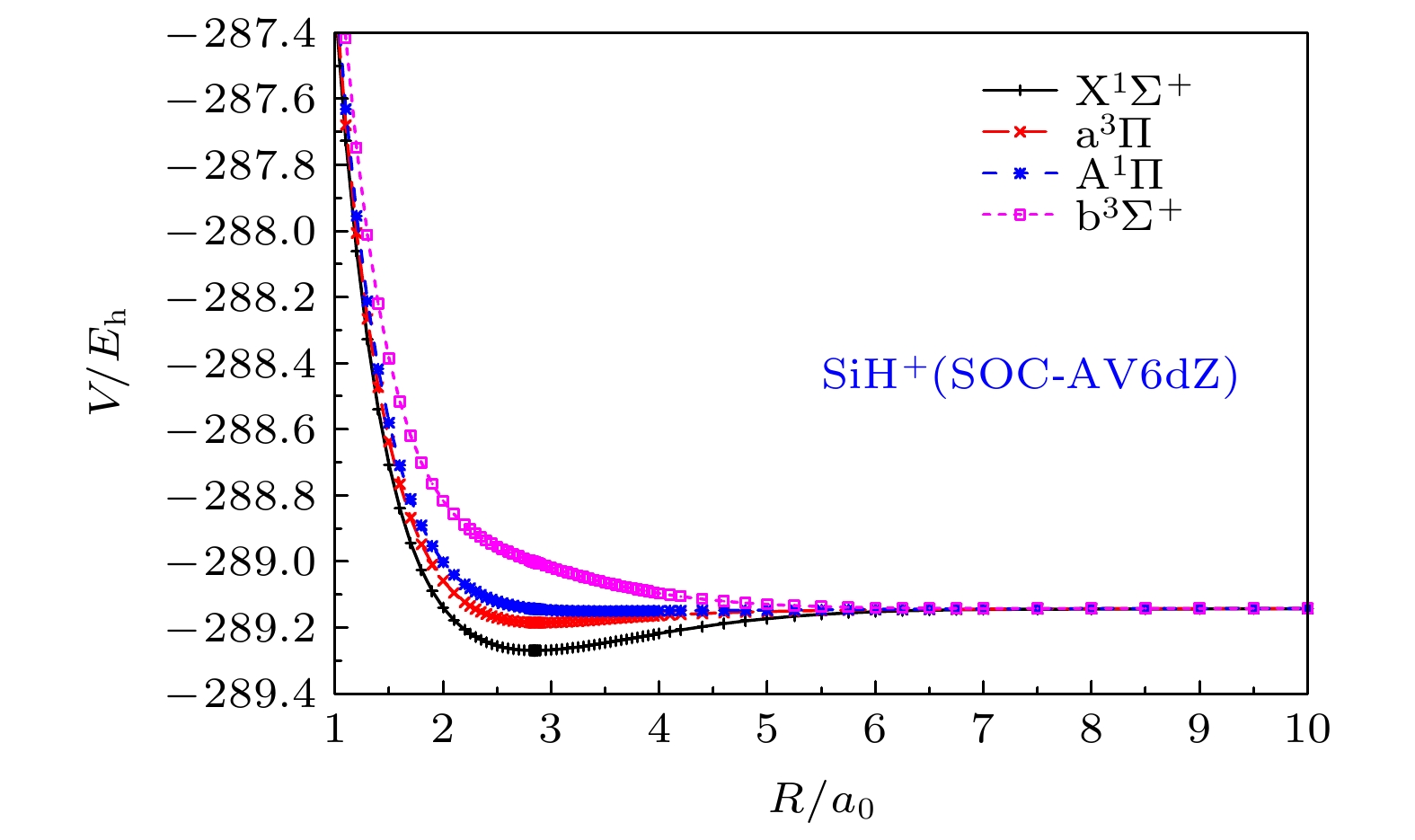 图 3 SiH+离子的
图 3 SiH+离子的



Figure3. The ab initio points of




基于SA-AV6dZ和SOC-AV6dZ两种方法得到的单点能, 应用Aguado-Paniagua函数进行拟合, 拟合的参数如表1所列. 从表1可以看出, SA-AV6dZ和SOC-AV6dZ两种方法的RMSD分别为9.45767662 × 10–3和1.11170443 × 10–2 kcal/mol, 是略小于使用基组AV6Z, AV6dZ以及应用CBS (Q, 5), CBS(Qd, 5d)方法的结果. 图4为相应的从头算单点能和拟合势能曲线, 可以看出, 在长程区和短程区域, 使用SA-AV6dZ和SOC-AV6dZ方法所得的势能曲线光滑且与单点能量点符合都比较好, 误差上限分别为10 cm–1和20 cm–1, 其中单纯的态平均计算结果(SA-AV6dZ)更优.
 图 4 SiH+(X1Σ+)在SA-AV6dZ和SOC-AV6dZ基组下的从头算能量点与拟合势能曲线
图 4 SiH+(X1Σ+)在SA-AV6dZ和SOC-AV6dZ基组下的从头算能量点与拟合势能曲线Figure4. Potential energy curves and ab initio points at SA-AV6dZ and SOC-AV6dZ results.
2
3.3.光谱常数
基于不同的基组和拟合方法(AVQZ, AV5Z, AV6Z, CBS(Q, 5), AVQdZ, AV5dZ, AV6dZ, CBS(Qd, 5d), SA-AV6dZ和SOC-AV6dZ)得到的APEFs, 本文计算了SiH+(X1Σ+)离子的平衡键长Re, 解离能De, 振动频率ωe, 光谱常数ωeχe, αe, βe. 各个光谱常数的公式如下:SiH+ (X1Σ+)离子的光谱常数列于表2, 表中同时列出了实验[12,34]和其他一些理论研究结果[16,18,21,23]. 通过对比发现, 本文的计算结果与其他理论计算[16,18,21,23]符合得较好, 与实验数值[12,34]也基本吻合. 需要说明的是, 就解离能De而言, 与实验最接近的是使用AVQZ基组得到的结果, 实验值[12,34]De为0.123203Eh, 用AVQZ基组得到的De为0.124640Eh, 与实验值的误差为1.166367%, 理论计算值和实验值差别较大. Zhang等[21]计算结果是0.125317Eh, 与实验值的误差是1.715867%; Biglari等[23]的数值是0.124980Eh, 与实验值的误差为1.442335%, 与实验同样偏差较大, 而与本工作的计算结果符合较好. 另外, 应用更巨大的基组、更高级的方法和考虑SOC效应(AV5Z, AV6Z, CBS(Q, 5), AVQdZ, AV5dZ, AV6dZ, CBS(Qd, 5d), SA-AV6dZ和SOC-AV6dZ), 本应得到更精确的结果, 却发现其理论计算值与实验值的偏差反而增大. 基于此, 有理由对实验结果提出疑问, 建议实验工作者重新测定SiH+离子的包含解离能在内的各项光谱常数.
| 基组 | De(Eh) | Re(a0) | ωe/cm–1 | βe/cm–1 | αe/cm–1 | ωeχe/cm–1 |
| AVQZ | 0.124640 | 2.851925 | 2153.245 | 7.607440 | 0.219327 | 42.373 |
| AV5Z | 0.125388 | 2.848589 | 2155.792 | 7.625272 | 0.218960 | 42.220 |
| AV6Z | 0.125584 | 2.848001 | 2156.686 | 7.628410 | 0.218833 | 42.189 |
| CBS(Q, 5) | 0.125856 | 2.847053 | 2157.189 | 7.633502 | 0.218622 | 42.117 |
| AVQdZ | 0.125067 | 2.848877 | 2156.187 | 7.623729 | 0.219427 | 42.343 |
| AV5dZ | 0.125461 | 2.848067 | 2156.737 | 7.628067 | 0.219011 | 42.232 |
| AV6dZ | 0.125622 | 2.847728 | 2157.046 | 7.629881 | 0.218849 | 42.190 |
| CBS(Qd, 5d) | 0.125801 | 2.847782 | 2156.602 | 7.629593 | 0.218534 | 42.112 |
| SA-AV6dZ | 0.127264 | 2.848531 | 2163.448 | 7.625581 | 0.216725 | 41.893 |
| SOC-AV6dZ | 0.126533 | 2.848382 | 2164.033 | 7.626378 | 0.217885 | 42.158 |
| Expe[12,34] | 0.123203 | 2.842338 | 2157.17 | 7.6603 | 0.2096 | 34.24 |
| Theory[16] | 0.118665 | 2.834590 | 2155.4 | 7.6786 | 0.2082 | 38.8 |
| Theory[18] | 0.123982 | 2.844039 | 2172.0 | — | — | — |
| Theory[21] | 0.125317 | 2.834590 | 2177.9 | 7.6984 | — | 36.7 |
| Theory[23] | 0.124980 | 2.842149 | 2154.3 | 7.6609 | 0.2032 | 35.0 |
表2SiH+(X1Σ+)的平衡键长Re, 解离能De, 振动频率ωe, 光谱常数ωeχe, αe和βe
Table2.Spectroscopic constants compared with the experimental values and other theoretical results for SiH+(X1Σ+).
从表2还可知, 基于态平均技术的SA-AV6dZ方法的平衡键长Re、解离能De分别是2.848531a0和0.127264Eh, 考虑SOC效应后的(SOC-AV6dZ)的Re和De分别是2.848382a0和0.126533Eh, 平衡位置基本吻合, 误差为0.005231%, 解离能误差为0.577715%, 偏差较小. 基于SA-AV6dZ方法的振动频率ωe, 光谱常数ωeχe, αe, βe分别为2163.448 cm–1, 7.625581 cm–1, 0.216725 cm–1和41.893 cm–1, 考虑SOC效应后的数值分别为2164.033 cm–1, 7.626378 cm–1, 0.217885 cm–1和42.158 cm–1, 误差分别为0.027033%, 0.010451%, 0.532391%和0.628588%, 偏差均较小. 根据以上数值对比, 发现SOC效应对SiH+离子光谱常数各项的影响较小.
2
3.4.振动能级
基于SiH+(X1Σ+)离子的APEF, 核运动的径向薛定谔方程[35]为表3和表4列出了SiH+(X1Σ+)离子在SOC-AV6dZ方法下, 转动量子数j = 0时的前23个振动能级G(v), 经典拐点, 转动常数Bv和6个离心畸变常数Dv, Hv, Lv, Mv, Nv和Ov. 理论上说, SOC-AV6dZ方法的计算结果由于采用更巨大的基组(AV6dZ)、更高级的方法(SA-MRCI(Q))和考虑SOC效应可以得到最高精度的APEF、光谱常数和振动能级. 图5给出了SiH+(X1Σ+)离子在SOC-AV6dZ方法下, j = 0时的前23个振动能级图.
| v | G(v)/ cm–1 | Rmin(a0) | Rmax(a0) | Bv/ cm–1 |
| 0 | 1074.467 | 2.62981 | 3.11141 | 7.530487 |
| 1 | 3171.224 | 2.49347 | 3.33862 | 7.336827 |
| 2 | 5200.543 | 2.40984 | 3.51594 | 7.142529 |
| 3 | 7162.231 | 2.34756 | 3.67498 | 6.947281 |
| 4 | 9055.742 | 2.29761 | 3.82512 | 6.750433 |
| 5 | 10880.124 | 2.25595 | 3.97091 | 6.551101 |
| 6 | 12634.005 | 2.22031 | 3.91505 | 6.348250 |
| 7 | 14315.600 | 2.18934 | 4.26009 | 6.140735 |
| 8 | 15922.726 | 2.16213 | 4.40742 | 5.927312 |
| 9 | 17452.802 | 2.13803 | 4.55893 | 5.706603 |
| 10 | 18902.835 | 2.11661 | 4.71650 | 5.477032 |
| 11 | 20269.364 | 2.09752 | 4.88227 | 5.236702 |
| 12 | 21548.363 | 2.08051 | 5.05888 | 4.983201 |
| 13 | 22735.066 | 2.06540 | 5.24986 | 4.713296 |
| 14 | 23823.694 | 2.05206 | 5.46016 | 4.422419 |
| 15 | 24807.010 | 2.04041 | 5.69731 | 4.103776 |
| 16 | 25675.599 | 2.03041 | 5.97379 | 3.746622 |
| 17 | 26416.625 | 2.02208 | 6.31278 | 3.332604 |
| 18 | 27011.579 | 2.01553 | 6.76545 | 2.826988 |
| 19 | 27432.546 | 2.01096 | 7.48310 | 2.158867 |
| 20 | 27650.956 | 2.00861 | 9.05975 | 1.323582 |
| 21 | 27734.462 | 2.00771 | 11.47553 | 0.838944 |
| 22 | 27769.256 | 2.00734 | 16.60576 | 0.360244 |
表3SiH+(X1Σ+)离子在SOC-AV6dZ方法下, j = 0时的前23个振动能级G(v)、经典拐点和惯性转动常数Bv
Table3.Vibrational levels G(v), classical turn point androtational constant Bv for SiH+(X1Σ+) when j = 0 at SOC-AV6dZ result.
| v | Dv (× 10–4) | Hv (× 10–8) | Lv | Mv | Nv | Ov |
| 0 | –3.7712102 | 1.4824556 | –9.1045 × 10–13 | 4.1069 × 10–17 | –2.4011 × 10–21 | 6.9631 × 10–26 |
| 1 | –3.7362090 | 1.4243863 | –8.8032 × 10–13 | 3.5297 × 10–17 | –4.1863 × 10–21 | 1.8363 × 10–25 |
| 2 | –3.7039865 | 1.3618539 | –8.9866 × 10–13 | 2.9262 × 10–17 | –5.1444 × 10–21 | 1.9171 × 10–25 |
| 3 | –3.6778687 | 1.2884893 | –9.4914 × 10–13 | 2.3397 × 10–17 | –5.8593 × 10–21 | 9.6796 × 10–26 |
| 4 | –3.6608093 | 1.2011917 | –1.0216 × 10–12 | 1.6648 × 10–17 | –6.8904 × 10–21 | –1.1678 × 10–25 |
| 5 | –3.6553038 | 1.0982022 | –1.1140 × 10–12 | 6.8522 × 10–18 | –8.7371 × 10–21 | –4.8697 × 10–25 |
| 6 | –3.6635199 | 0.9774540 | –1.2320 × 10–12 | –9.1302 × 10–18 | –1.2003 × 10–20 | –1.1229 × 10–24 |
| 7 | –3.6875661 | 0.8351772 | –1.3901 × 10–12 | –3.5900 × 10–17 | –1.7702 × 10–20 | –2.2781 × 10–24 |
| 8 | –3.7298613 | 0.6645057 | –1.6143 × 10–12 | –8.0925 × 10–17 | –2.7799 × 10–20 | –4.5064 × 10–24 |
| 9 | –3.7936180 | 0.4536483 | –1.9481 × 10–12 | –1.5748 × 10–16 | –4.6321 × 10–20 | –9.0505 × 10–24 |
| 10 | –3.8835055 | 1.8293995 | –2.4663 × 10–12 | –2.9088 × 10–16 | –8.1928 × 10–20 | –1.8869 × 10–23 |
| 11 | –4.0066389 | –0.1804552 | –3.3026 × 10–12 | –5.3251 × 10–16 | –1.5445 × 10–19 | –4.1577 × 10–23 |
| 12 | –4.1741837 | –0.6927552 | –4.7113 × 10–12 | –9.9426 × 10–16 | –3.1311 × 10–19 | –9.8710 × 10–23 |
| 13 | –4.4041727 | –1.4546951 | –7.2135 × 10–12 | –1.9417 × 10–15 | –6.9300 × 10–19 | –2.5871 × 10–22 |
| 14 | –4.7268845 | –2.6591579 | –1.1978 × 10–11 | –4.0783 × 10–15 | –1.7160 × 10–18 | –7.7417 × 10–22 |
| 15 | –5.1961278 | –4.7106022 | –2.1959 × 10–11 | –9.5527 × 10–15 | –4.9431 × 10–18 | –2.7809 × 10–21 |
| 16 | –5.9157772 | –8.5688303 | –4.5904 × 10–11 | –2.6336 × 10–14 | –1.7665 × 10–17 | –1.2991 × 10–20 |
| 17 | –7.1117968 | –16.938203 | –1.1615 × 10–10 | –9.3420 × 10–14 | –8.7454 × 10–17 | –9.0301 × 10–20 |
| 18 | –9.3643712 | –39.467832 | –3.9578 × 10–10 | –4.9264 × 10–13 | –7.1396 × 10–16 | –1.1444 × 10–18 |
| 19 | –14.323856 | –114.68916 | –1.7949 × 10–9 | –3.4005 × 10–12 | –7.1696 × 10–15 | –1.6154 × 10–17 |
| 20 | –18.399913 | –47.324191 | 1.5501 × 10–9 | –7.5768 × 10–13 | –3.7548 × 10–14 | –1.9274 × 10–17 |
| 21 | –14.637253 | –291.40012 | –2.0665 × 10–8 | –1.4452 × 10–10 | –1.3084 × 10–12 | –1.3806 × 10–14 |
| 22 | –57.213156 | –20756.133 | –1.6655 × 10–5 | –1.7512 × 10–6 | –2.1241 × 10–7 | –2.8187 × 10–8 |
表4SiH+(X1Σ+)离子在SOC-AV6dZ方法下, j = 0时的前23个振动能级的6个离心畸变常数Dv, Hv, Lv, Mv, Nv和Ov
Table4.Six centrifugal distortion constants Dv, Hv, Lv, Mv, Nv和Ov for the top 23 vibrational states of SiH+(X1Σ+) when j = 0 at SOC-AV6dZ result.
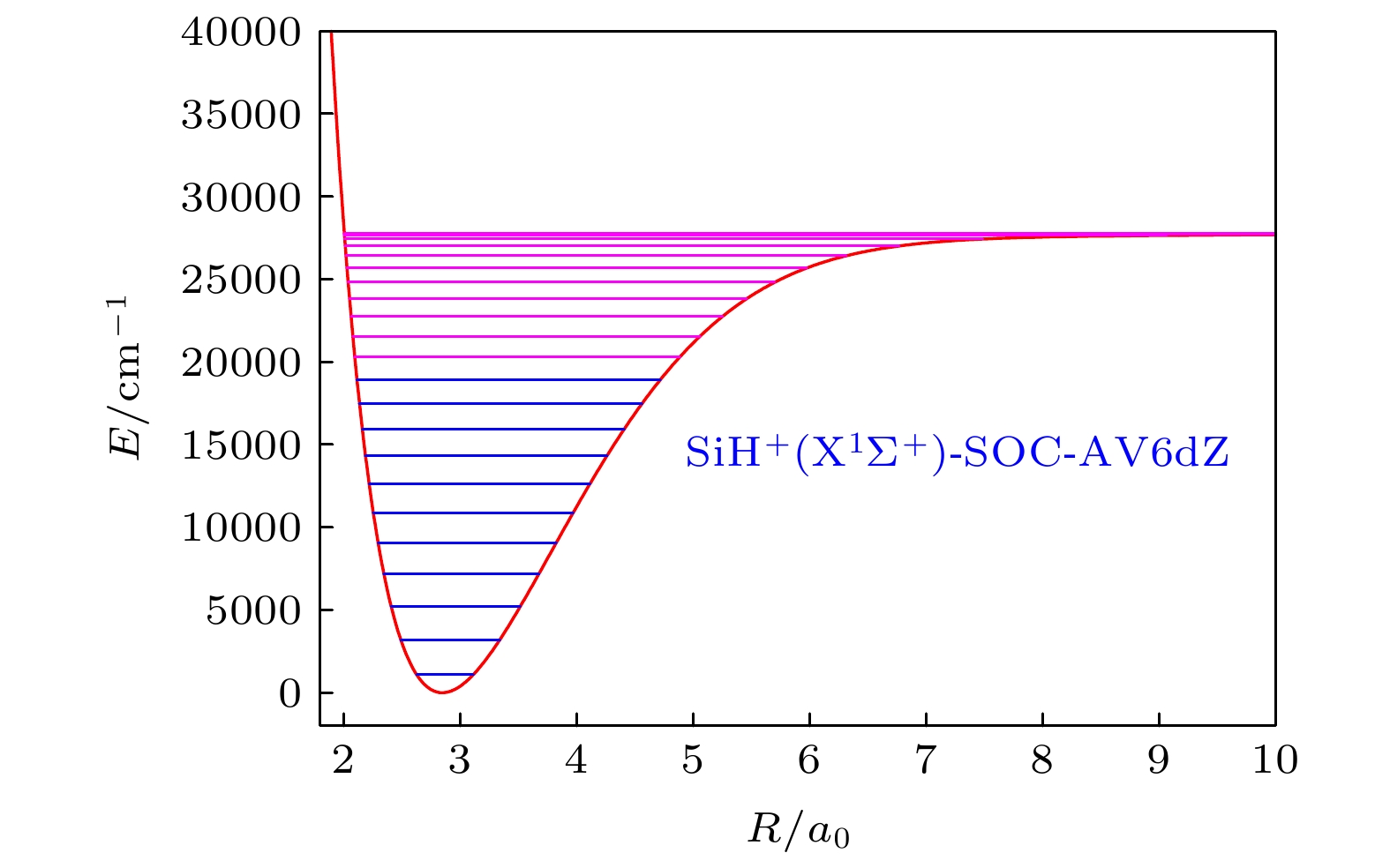 图 5 SiH+(X1Σ+)离子在SOC-AV6 dZ方法下, j = 0时的前23个振动能级
图 5 SiH+(X1Σ+)离子在SOC-AV6 dZ方法下, j = 0时的前23个振动能级Figure5. Top 23 vibrational energy levels of SiH+(X1Σ+) when j = 0 at SOC-AV6 dZ result.
从表3可以看出, 对于j = 0的前23个能级, 其经典拐点的取值范围较大, 从v = 0的Rmin = 2.62981a0, Rmax = 3.11141a0到v = 22的Rmin = 2.00734a0, Rmax = 16.60576a0, 这与能级较多相关. v = 22的振动能级(27769.256 cm–1)与解离极限(27770.8 cm–1)非常接近, 二者差值仅为1.544 cm–1. 并且v = 22和v = 21的振动能级差值非常小, 为34.794 cm–1, 仅为v = 22的振动能级的0.125%, 以至于在图5的能级图中几乎区别不出这两个振动能级.
由表4可以看出, 在这6个离心畸变常数中, 除Dv范围变化较小外(–3.7712102 × 10–4— –5.721315 × 10–3), 其余的5个数值变化范围非常大, 例如, Ov的范围为6.9631 × 10–26— –2.8187 × 10–8, 横跨了18个数量级. 所以在表4中只有Dv和Hv把数量级标注在第1行, 其余各常数的数量级都附在数值后.
