全文HTML
--> --> -->目前研究的LiPON具有非晶态和晶态两种类型. 非晶LiPON薄膜本质上可认为是无定形的掺氮磷酸锂. 1997年, Yu等[2]利用射频磁控溅射法, 以γ-Li3PO4为靶材, 在氮气气流下首次成功制备出LiPON. 相比于靶材γ-Li3PO4, 这样制备出LiPON的Li+电导率提升了两个数量级, 可以达到3.3 × 10–6 S/cm. 除了最常用的射频磁控溅射法制备LiPON薄膜外, 许多科研人员也探索出了其他多种制备技术, 例如脉冲激光沉积[4]、离子束溅射[5]、氧化物化学气相沉积[6]、以及原子沉积[7]等技术手段, 这不仅丰富了非晶LiPON薄膜的制备方法, 而且拓展了LiPON的应用. 2010年, Du等[8]用第一性原理的方法, 从理论上预测出了一种晶态LiPON(Li2PO2N), 空间群为Cmc21. 这种晶体结构中存在两种四面体结构, P[O2N2]四面体和Li[O3N]四面体. 由于锂离子在四面体的中心位, 而且结构中没有锂空位, 所以锂离子的扩散比较慢. 2013年, Senevirathne等[9]在实验上合成出这种材料, 并测得离子电导率约为10–7 S/cm. 虽然晶态LiPON离子电导率较低, 但其结构稳定、电化学性能稳定、晶界处不易生成锂枝晶, 非常有利于提高电池的安全性能.
用Li金属作为电池的负极材料可以提升30%左右的电池能量密度[10]. 而LiPON优异的稳定性使其能够与Li金属很好地兼容, 但由于化学反应和电化学反应, 仍然会形成很薄的一层LiPON/Li界面层[11-13]. 利用实验手段研究界面处具有何种物理化学性质比较困难, 因此本文利用从头算分子动力学(AIMD)构建和模拟非晶LiPON/Li界面和晶态LiPON/Li界面, 并研究这两类界面的结构性质和扩散性质.
2.1.模型构建
首先构造非晶LiPON与Li金属的界面. 构造a-LiPON/Li界面的首要条件是得到合理的a-LiPON结构. 实验上比较常用的做法是利用射频磁控溅射法, 通过控制氮气气流的流速可以得到不同分子式的a-LiPON, 它们之间的离子电导率存在一定的差别, 但差别不大, 都在10–6 S/cm量级[14-20]. 因此, 本文只选取其中一种实验上已经合成并报道过的非晶Li2.5PO3.5N0.5作为研究对象. 元素替换后, 通过AIMD模拟和密度泛函理论(DFT)优化后得到非晶结构. 将bcc结构的Li金属用DFT方法优化, 并截取其中比较稳定的(100)面构成表面结构, 由于a-LiPON没有特定的晶面, 因此选取三个特殊的面, 即形成N-P单桥键、双桥键和三桥键的局域结构分别暴露在表面. 将Li(100)面与a-LiPON表面搭建为界面结构, 两者的晶格失配小于5%.晶态LiPON (Li2PO2O)空间群为Cmc21, 其结构包含Li[O3N]和P[O2N2]两种四面体结构. Li2PO2N的(100)面是由-P-N-P-构成的链状结构, 是其最稳定面, 这一结论与表面能的计算结果一致[21]. 根据暴露在表面的原子的不同, 可以将Li2PO2N的(100)面分为Li, O暴露在表面和P, N暴露在表面两种情况. 因此选取Li2PO2N的(100)面与Li(100)面可以构造出两种界面.
2
2.2.计算方法
本文采用的是基于密度泛函理论的VASP程序包[22-24], 对固态电解质LiPON和金属Li做结构优化, 并对LiPON/Li界面做从头算分子动力学模拟(AIMD). 交换关联泛函采用Perdew-Burke-Ernzerhof(PBE)形式的广义梯度近似(GGA)进行描述[25,26], 电子波函数采用平面基波展开法, 截断能取为450 eV. 晶相LiPON与金属Li在结构优化时, 布里渊区积分采用的是Monkhorst-Pack特殊k点取样法[27], 其中k-网格的分辨率取为2π × 0.025 ?–1, 能量和力的收敛判据分别为10–5 eV和10–3 eV/?.对于非晶LiPON体相结构和c-LiPON/Li界面结构的AIMD计算, 为了模拟体相部分对界面的影响, 本文将界面两端的原子固定(图1中红色虚线区域). 非晶LiPON/Li界面结构模型共有320个原子, c-LiPON/Li界面结构模型共有384个原子. 计算过程采用以Γ点为中心的1 × 1 × 1网格. 在NVT系综下, 使用Nosé-Hoover热浴法[28], 时间步长设为2 fs, 能量收敛标准为10–4 eV. 由于室温原子扩散较慢, 所以将体系分别升温至1000 K加速原子扩散, 模拟了20 ps, 以研究界面层的形成及不同区域原子扩散差异.
 图 1 (a)?(c) 三种a-LiPON/Li(100)界面沿Z方向的Li原子互扩散图; (d), (e) 两种Li2PO2N(100)/Li(100)界面沿Z方向的Li原子互扩散图. 灰色虚线表示原始的界面位置. 结构图中的红色虚线框标注的是固定的末端原子, 蓝色虚线框标注的是形成的界面区域
图 1 (a)?(c) 三种a-LiPON/Li(100)界面沿Z方向的Li原子互扩散图; (d), (e) 两种Li2PO2N(100)/Li(100)界面沿Z方向的Li原子互扩散图. 灰色虚线表示原始的界面位置. 结构图中的红色虚线框标注的是固定的末端原子, 蓝色虚线框标注的是形成的界面区域Figure1. (a)?(c) Inter-diffusion of Li atoms along the Z direction of three a-LiPON/Li(100) interfaces; (d), (e) inter-diffusion of Li atoms along the Z direction of two Li2PO2N(100)/Li(100) interfaces. The gray dotted line indicates the original interface location. The red dotted frames in the structure diagram mark fixed terminal atoms, and the blue dotted frames mark the formed interface areas.
3.1.LiPON/Li界面的原子互扩散现象
实验通过能谱仪观察到界面附近由于电化学反应发生原子互扩散, 形成界面层[11,12,29]. 为了验证这个结果, 本文通过对原始界面模型采用AIMD模拟, 来研究界面附近的原子互扩散现象, 并根据原子互扩散范围得到稳定的界面结构. 图1 (a)—图1(c)给出的是三种a-LiPON/Li(100)原始界面结构在20 ps的AIMD模拟中, Li原子沿Z方向的统计分布. 红色点线图统计的是Li金属原子的分布, 原始界面结构中Li金属的原子分布表现出晶态结构周期性的特征, 即一条条分立的峰. 随着模拟过程进行, 除了末端被固定的Li原子, 界面附近的Li原子会扩散到a-LiPON层. 蓝色点线图统计的是a-LiPON层的Li原子分布, 由于是非晶结构, 所以Li原子的分布更加无序. 可以观察到a-LiPON层的Li原子同样会扩散到Li金属层, 而且由于Li金属层对Li原子的扩散阻碍更小, 部分a-LiPON层的Li原子会扩散较快. 为了能够更好地理解元素分布和互扩散现象, 在图中添加了辅助线. 曲线交叉的部分近似认为是发生元素互扩散的范围, 这个范围作为后续研究界面性质的界面区域. 对于a-LiPON/Li(100)界面, 暴露在表面的局域结构不同, 原子的扩散会略有差异, 综合三种界面的结果可以确定出大约10 ?厚度的界面层.从图1 (d)和图1(e)可以看到, 在晶态Li2PO2N(100)/Li(100)界面同样观察到了类似的原子互扩散现象. Li2PO2N电解质和Li金属均为晶态结构, 所以原始界面中Li原子沿Z方向呈周期性分立排布. 随着MD模拟过程, 界面附近会发生原子互扩散. 除了固定的原子层外, Li2PO2N中仅界面附近一两层的Li原子会发生明显扩散, 离界面较深的Li原子仍保持晶态结构的分布特征. 因此, Li2PO2N(100)/Li(100)界面的Li原子分布没有用多项式拟合. 确定界面层的区域是结合了Li金属层中的Li原子和Li2PO2N层中Li, P, O, N原子的扩散范围, 最终得到7 ?左右的界面层用于后续研究.
2
3.2.LiPON/Li界面的锂离子扩散性质
对LiPON/Li界面进行模拟, 验证了电解质与电极之间会由于化学反应或电化学反应发生原子互扩散, 形成界面层. 在实际电池体系中, 界面层的Li+电导率(σLi)会直接影响到锂离子电池的性能. 基于构造的LiPON/Li界面, 本节采用AIMD模拟, 探究界面发生反应后形成的界面层结构对离子电导率的影响.由于LiPON电解质的Li+扩散性能较差, 且AIMD难以做长时间尺度的模拟, 因此本节对a-LiPON体相结构, 以及三种a-LiPON/Li(100)界面分别在800, 1000, 1200, 1400 K温度下模拟来加速扩散, 再利用高温的扩散数据推导出室温的扩散系数. 根据模拟得到的Li+运动轨迹, 利用以下公式可以求得Li+的均方位移(mean square displacement, MSD):



通常来说, 如果统计的样本足够多, 体系足够稳定, Li+的MSD与时间间隔

 图 2 (a)?(c)三种a-LiPON/Li(100)界面和(d) a-LiPON在不同高温下Li+的均方位移(MSD); (e) a-LiPON和三种a-LiPON/Li(100)界面体系中温度与Li+扩散系数(DLi)的关系. 图中灰色虚线为300 K对应的位置
图 2 (a)?(c)三种a-LiPON/Li(100)界面和(d) a-LiPON在不同高温下Li+的均方位移(MSD); (e) a-LiPON和三种a-LiPON/Li(100)界面体系中温度与Li+扩散系数(DLi)的关系. 图中灰色虚线为300 K对应的位置Figure2. (a)?(c) the three a-LiPON /Li(100) and (d) a-LiPON MSD of Li+ at different temperatures, (e) Arrhenius plot of Li diffusivity (DLi) as a function of temperature in a-LiPON and three kinds of a-LiPON/Li (100). The corresponding positions of 300 K are presented by dotted lines.





利用推导出的室温Li+扩散系数, 可以进一步根据Nernst–Einstein关系计算出室温Li+电导率. 公式的具体形式如下:







| Structure | DLi/(cm2·s–1) | σLi/(S·cm–1) | σLi/(S·cm–1) exp. |
| a-LiPON | 2.18×10–9 | 5.56×10–5 | 1.8×10–6 exp.[30] |
| a-Interface-1 | 4.67×10–8 | 9.69×10–3 | — |
| a-Interface-2 | 3.44×10–6 | 7.14×10–3 | — |
| a-Interface-3 | 2.23×10–6 | 4.63×10–3 | — |
| Li2PO2N | — | — | 8.8×10–7 exp.[10] |
| c-Interface-1 | 1.19×10–8 | 3.19×10–3 | — |
| c-Interface-2 | 3.57×10–8 | 7.78×10–3 | — |
表1a-LiPON、三种a-LiPON/Li(100)界面和两种Li2PO2N(100)/Li(100)界面的室温Li+扩散系数(DLi)与电导率(σLi)
Table1.Li+ diffusion coefficient (DLi) and electrical conductivity (σLi) of a-LiPON, three a-LiPON/Li(100) interfaces and two Li2PO2N(100)/Li(100) interfaces at room temperature.
用同样的方法计算Li2PO2N及Li2PO2N(100)/Li(100)界面的DLi和σLi, 但是从图3 (a)可以看出Li2PO2N体相结构的MSD并没有明显的线性关系. 这是因为晶态Li2PO2N中所有Li都是以Li[O3N]四面体结构稳定存在的, 且模拟的晶体是有限大小的完美晶体, 所以在有限的模拟时间内很难观察到明显的扩散, 在实验上测得Li2PO2N的σLi约为8.8 × 10–7 S/cm[10]. 而由电化学反应形成Li2PO2N(100)/Li(100)界面层在相同的高温下可以发生显著的Li+扩散, 其MSD具有明显的线性关系. 根据图3 (d)可以推导出两种Li2PO2N(100)/Li(100)界面室温的DLi, 并进一步计算出电导率. 在实验中测量电化学阻抗谱发现[31], LiPON/Li界面的阻抗小于LiPON体相的阻抗, 而本文计算得到两种界面的σLi约在10–3 S/cm量级, 比Li2PO2N的σLi (实验数据10–7 S/cm量级)大得多. 理论结果与实验现象符合良好.
 图 3 (a) Li2PO2N、(b), (c) Li2PO2N(100)/Li(100)界面在不同高温下的MSD; (d)两种Li2PO2N(100)/Li(100)界面体系中温度与Li+扩散系数(DLi)的关系. 图中灰色虚线为300 K对应的位置
图 3 (a) Li2PO2N、(b), (c) Li2PO2N(100)/Li(100)界面在不同高温下的MSD; (d)两种Li2PO2N(100)/Li(100)界面体系中温度与Li+扩散系数(DLi)的关系. 图中灰色虚线为300 K对应的位置Figure3. (a) Li2PO2N and (b), (c) Li2PO2N(100)/Li(100) MSD of Li+ at different temperatures. Arrhenius plot of Li diffusivity (DLi) as a function of temperature in two kinds of Li2PO2N(100)/Li(100). The corresponding positions of 300 K are presented by dotted lines.
2
3.3.LiPON/Li界面的局域结构特征
计算结果表明a-LiPON/Li(100)界面和Li2PO2N(100)/Li(100)界面附近Li+扩散要比LiPON体相部分Li+扩散快得多, 而DLi与材料结构息息相关. 因此统计界面附近局域结构有何种特征, 能够更好地理解界面Li+扩散性质变化的原因. 图4表示的是a-LiPON中各类元素之间的径向分布函数(RDF). 由于原子之间的成键作用, 在键长附近的粒子数密度会比较大, 所以对于非晶体系的RDF, 第一条峰的位置可以近似认为是原子之间的键长. 界面由于化学反应而非晶化, 所以可以根据图4中的数据, 将第一条峰谷的位置作为判断界面各类原子之间成键的截断距离, P-O之间和P-N之间的截断距离取1.8 ?, Li-O之间和Li-N之间的截断距离取2.5 ?.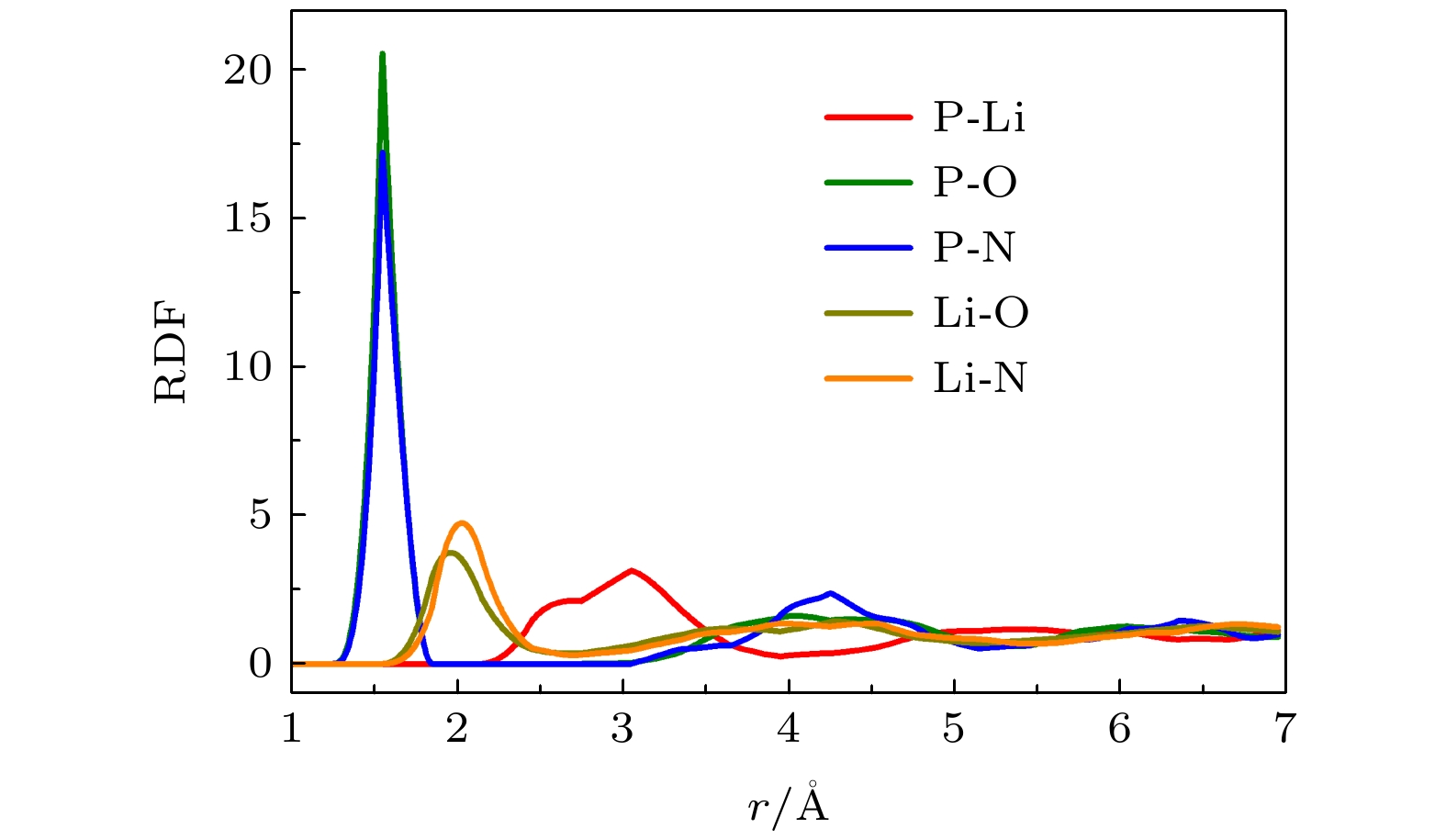 图 4 a-LiPON体系中各类原子之间的径向分布函数(RDF)
图 4 a-LiPON体系中各类原子之间的径向分布函数(RDF)Figure4. Radial Distribution Functions of a-LiPON.
Li+的扩散性质与其邻域的结构特征有很大关系. 图5 (a)是以Li为中心原子, 统计a-LiPON体系和a-LiPON/Li(100)界面在截断距离内形成的Li[OxNy]局域结构以及相应的占比, 其中a-LiPON/Li(100)界面的统计结果是对之前构造的三种界面局域结构取统计平均. 统计结果表明在体相a-LiPON中有20%以上的Li是在Li[O2N2], Li[O3N]或者Li[O4]四面体的中心位, 这些四面体中心的Li较难发生扩散, 而在a-LiPON/Li(100)界面受负极Li原子的作用, 这三种以Li为中心的四面体结构占比明显减小, 而受O, N影响比较小的局域结构占比明显更高, 所以计算得到界面附近的Li+扩散要比在体相a-LiPON中快得多. 由于在体相Li2PO2N中所有Li都处于Li[O3N]四面体中心位, 所以Li+扩散比较困难, Li+电导率较差, 而Li2PO2N/Li界面附近Li+电导率变大必然也与Li邻域的结构变化有关. 图5 (b)统计了以Li为中心原子Li2PO2N(100)/Li(100)界面中Li[OxNy]局域结构的占比. 根据暴露在表面的不同原子, 将界面分成了两种, 图5 (b)中表示的是对这两种界面局域结构做统计平均的结果. 可以看出, 界面范围内Li[O3N]四面体的占比非常小, 由于Li负极对界面的作用使得四面体结构会被分解为更小的局域结构, 因此Li+在界面扩散受到的束缚要小很多.
 图 5 (a) a-LiPON体系与a-LiPON/Li(100)界面、(b) Li2PO2N(100)/Li(100)界面, 以Li原子为中心的Li[OxNy]局域结构统计图
图 5 (a) a-LiPON体系与a-LiPON/Li(100)界面、(b) Li2PO2N(100)/Li(100)界面, 以Li原子为中心的Li[OxNy]局域结构统计图Figure5. Statistics of local structures (Li[OxNy]) of (a) a-LiPON and (b) a-LiPON/Li(100) interfaces.
实验上通过Cryo-TEM对Li/LiPON界面进行研究[29], 发现LiPON和Li之间存在互扩散现象形成界面层, Li/LiPON界面从Li到LiPON之间结构成分存在一个渐变的过程, 在Li负极这一侧的界面主要成分为Li金属和Li2O, 接着会出现Li3N, 到LiPON这一侧界面主要成分为Li2O, Li3N和Li3PO4, 这些物质均为LiPON的分解产物. LiPON/Li界面形成界面层后四面体结构均被分解成了更小结构, 与我们的计算结果一致.
另外在表2中统计了体相a-LiPON, a-LiPON/Li(100)界面、体相Li2PO2N以及Li2PO2N(100)/Li(100)界面中阳离子和阴离子之间的平均配位数. 由于电解质与Li负极之间原子互扩散, 使得界面Li+含量较大. 在a-LiPON/Li(100)界面中Li-O, Li-N平均配位数分别为1.75和0.34, 小于对应a-LiPON电解质的Li-O平均配位数2.49和Li-N平均配位数0.50. 在Li2PO2N(100)/Li(100)界面中Li-O, Li-N平均配位数分别为1.63和0.71, 同样小于对应Li2PO2N电解质的Li-O平均配位数3和Li-N平均配位数1. 界面处Li-O, Li-N的平均配位数更小表明界面附近的Li与O, N之间的离子键作用更弱, 所以Li+扩散更快. 此外, 界面处的P-O, P-N配位数均小于对应的LiPON电解质中P-O, P-N配位数, 表明P-O-N原子构成的网状结构随着Li原子的互扩散会被分解为更小的局域结构, 使得Li+扩散过程受到的阻碍更小.
| Structure | Li-O | Li-N | P-O | P-N |
| a-LiPON | 2.49 | 0.50 | 2.43 | 0.51 |
| a-LiPON/Li (100) | 1.75 | 0.34 | 2.07 | 0.42 |
| Li2PO2N | 3 | 1 | 2 | 2 |
| Li2PO2N (100)/Li (100) | 1.63 | 0.71 | 1.04 | 1.72 |
表2LiPON体相与LiPON/Li界面中原子间的平均配位数
Table2.The average coordination number between atoms in LiPON bulk and LiPON/Li interface
