全文HTML
--> --> -->由于实验手段的限制, 通过实验来确定金属离子与柿单宁相互作用的本质仍存在困难. 同时柿单宁的精确结构仍未知晓, 理论计算也难以对其进行研究. 近年来, Li等[11]鉴定出柿单宁末端结构由表儿茶素(epicatechin, EC)、表没食子儿茶素(epigallocatechin, EGC)、表儿茶素没食子酸酯(epicatechin gallate, ECG)、表没食子儿茶素没食子酸酯(epigallocatechin gallate, EGCG)构成, 如图1所示. 由于柿单宁末端结构中EGCG的含量最多, 因此通过量子化学方法来研究EGCG与金属离子的相互关系, 成为研究柿单宁吸附机理的桥梁. 江腾等[12]研究了EGCG与Zn2+的相互作用, 通过实验和理论计算最终确定了反应位点. 王晓巍等[13]则通过密度泛函分析了儿茶素及其相关金属化合物的结构和反应活性. 以上的研究仅仅探索了形成的金属复合物构型, 缺乏对相互作用本质的阐述. 当金属离子与EGCG相互作用时, 到底是螯合机理占优势(形成螯合键)还是主要靠静电作用相互吸引, 到目前为止还缺乏系统的理论研究.
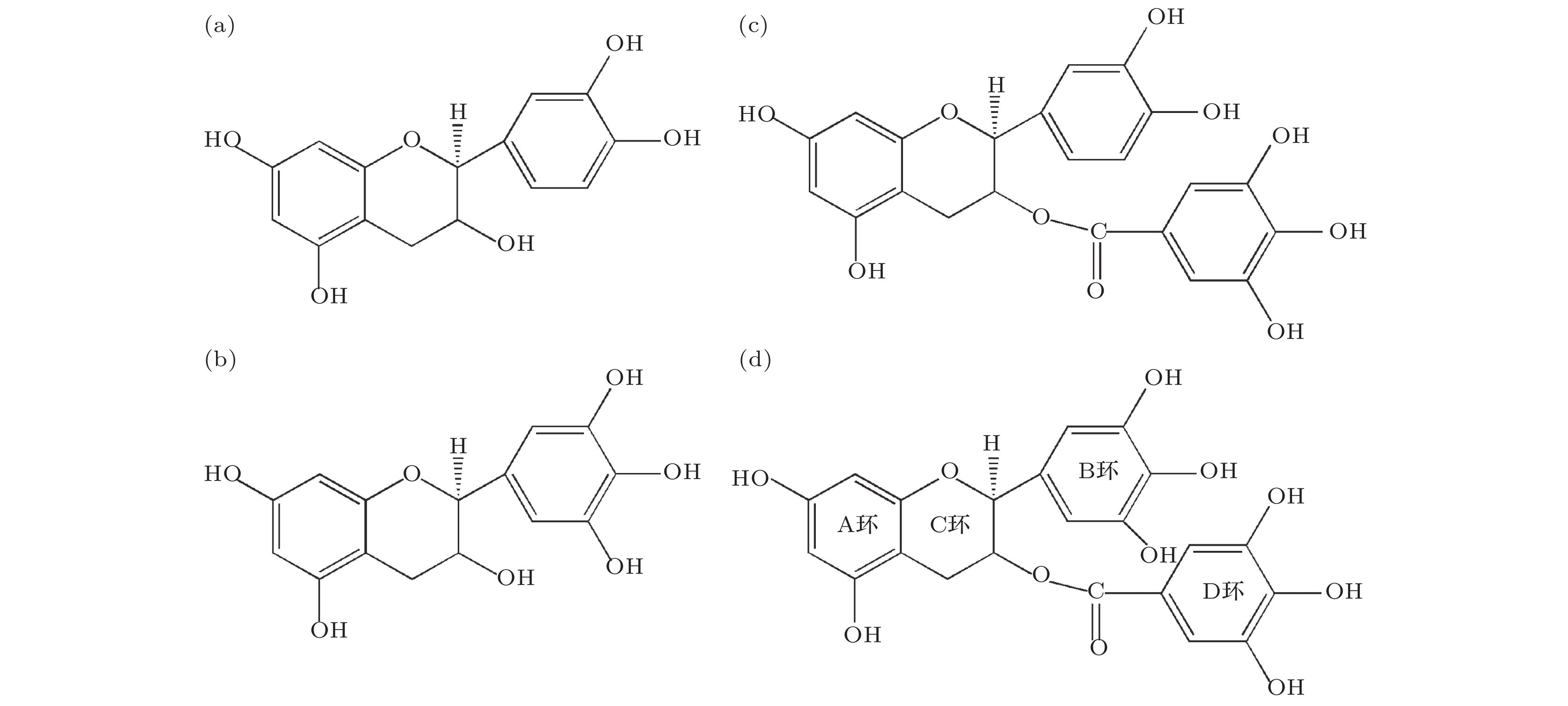 图 1 柿单宁末端结构四种单体结构示意图 (a) 表儿茶素; (b) 表没食子儿茶素; (c) 表儿茶素没食子酸酯; (d) 表没食子儿茶素没食子酸酯
图 1 柿单宁末端结构四种单体结构示意图 (a) 表儿茶素; (b) 表没食子儿茶素; (c) 表儿茶素没食子酸酯; (d) 表没食子儿茶素没食子酸酯Figure1. Structures of four monomers in the terminal structure of persimmon tannin: (a) EC; (b) EGC; (c) ECG; (d) EGCG.
密度泛函理论[14](density functional thoery, DFT)是将能量看作体系粒子密度的泛函, 它把求解N个粒子系统的3N自由度问题, 简化为3个自由度的密度问题, 从而简化计算. 经过几十年的发展, 密度泛函理论在有机分子以及更复杂的体系方面都有着广泛的应用, 其方法适用于过渡金属络合物以及金属化合物的计算. 密度泛函理论有许多种方法, 其中最常用的方法就是B3LYP [15-17] (Becke的三参数方法[18]与Lee-Yang-Parr的相关函数[19]相结合), 它将交换泛函定义为Hartree-Fock、局域和梯度修正交换项的线性组合, 从而得到较为精确的解.
本文通过DFT中的B3LYP方法研究EGCG与金属离子的相互作用, 选取了Ag+, Hg2+, Cu2+, Fe2+, In3+, Al3+和Au3+七种常见的金属离子与EGCG进行复合. 通过分析这些金属复合物的构型、Mayer键级、自然布居、结合能、以及弱相互作用的研究[20,21]等方面来讨论EGCG在水溶液中与金属离子复合的主要作用, 为柿单宁在吸附贵重金属的应用提供理论依据.
2.1.计算方法
本文中主要使用B3LYP结合DFT-D3色散校正的方法, 即B3LYP-D3方法[22]. 对于Ag, Hg, Cu, Fe, In, Al和Au等金属原子, 采用Lanl2dz基组; 而对于H, C和O等原子, 结构优化采用的是6-311G (d, p)基组, 计算单点能则采用的是更精确的6-311+G (d, p)基组. 计算过程中添加了溶剂为水的SMD溶剂化模型. 文中所有复合物的几何结构均通过频率计算, 确认为局域能量最小点, 即所有结构均不存在虚频. 所有计算在Gaussian 09程序包上完成[23]. 对于EGCG与金属离子弱相互作用的研究, 是利用约化密度梯度(reduced density gradient, RDG)函数方法[24,25]. RDG函数是描述均匀电子分布偏差的实空间函数, 表达式为2
2.2.EGCG模型选择
如图2(a)所示, EGCG被分为A环、B环、C环和D环, 而EGCG与金属离子反应主要发生在B环3', 4' 的酚羟基, 以及D环4", 5" 的酚羟基上[12]. 由于溶液的pH不同, 且不同的金属离子所带电荷量也不一致, 所以EGCG与金属离子的螯合方式有很多种. Navarro等[26]通过测定B和D环上1H核自旋弛豫时间鉴别了EGCG与金属的结合位点, 证实了D环比B环具有更高的配位能力, 且D环在配位过程中占主导作用. 由于吸附反应发生在溶液中, EGCG易发生去质子化反应, 即脱去质子形成阴离子. 我们统一以Inoue等在文献[27]中的方式构造模型, 即D环4"O, 5"O与金属离子1∶1发生反应, 形成EGCG-金属复合物, 其结构模型图如图2(b)所示. 图 2 (a) EGCG结构图; (b) EGCG-金属复合物结构模型图
图 2 (a) EGCG结构图; (b) EGCG-金属复合物结构模型图Figure2. (a) Structure of EGCG; (b) structure model diagram of EGCG-metal complex.
3.1.EGCG-金属复合物的构型及键长
构建好的复合物模型利用Gaussian软件进行结构优化, 得到了各自最稳定的结构, 列在图3中. 通过对比不难发现, ECGC-Ag+, EGCG-Hg2+和EGCG-Fe2+复合物的形成对整体结构没有什么影响, 同时Fe2+离子与D环的4"O, 5"O形成了一个五元环, 而EGCG-Ag+与EGCG-Hg2+复合物则未形成. 不同的是, EGCG与Cu2+, In3+, Al3+和Au3+这四种离子复合后使得EGCG的D环发生折叠向B环靠近, 例如图3(c)展现的EGCG-Cu2+复合物(D环折叠与B环类平行, 而Cu2+离子在两个环之间, 形成“腔状结构”[28]). 由此, 推断这类“腔状结构”复合物中, 不仅D环的4"O, 5"O与金属离子存在相互作用关系, 由于B环与D环形成堆积, 因此两者可能也存在相互作用. 图3(e)—(g)中的EGCG-In3+, EGCG-Al3+, EGCG-Au3+复合物结构与图3(c)相似. 此外图3(c)和图3(d)中形成了明显的螯合键, 而其他金属复合物则没有. 图 3 EGCG-金属复合物的几何构型图 (a) EGCG-Ag+; (b) EGCG-Hg2+; (c) EGCG-Cu2+; (d) EGCG-Fe2+; (e) EGCG-In3+; (f) EGCG-Al3+; (g) EGCG-Au3+
图 3 EGCG-金属复合物的几何构型图 (a) EGCG-Ag+; (b) EGCG-Hg2+; (c) EGCG-Cu2+; (d) EGCG-Fe2+; (e) EGCG-In3+; (f) EGCG-Al3+; (g) EGCG-Au3+Figure3. Geometric diagrams of EGCG-metal complexes: (a) EGCG-Ag+; (b) EGCG-Hg2+; (c) EGCG-Cu2+; (d) EGCG-Fe2+; (e) EGCG-In3+; (f) EGCG-Al3+; (g) EGCG-Au3+.
还计算了金属离子与EGCG中D环4"O, 5"O形成的复合键键长. 通过表1中的数据看出, Cu2+, Fe2+离子与EGCG之间形成的键长较短(分别为复合键4"O—Cu: 1.941 ?; 5"O—Cu: 1.916 ?; 4"O—Fe: 1.926 ?; 5"O—Fe: 1.893 ?), 因此Cu2+, Fe2+离子与EGCG形成的复合键较稳定(螯合作用较强). 而像Ag+, Hg2+离子和最近的酚羟基氧原子距离分别为2.302 ?和2.333 ?, 虽然Hg2+离子与EGCG之间可能存在较强的相互作用关系, 但由于二者的距离较远, 螯合作用较弱. 对于EGCG-In3+, EGCG-Al3+和EGCG-Au3+复合物, 与EGCG- Fe2+复合物对比, 可以明显地看出这类的“腔状结构”复合物的复合键更长, 螯合作用自然也是最弱的.
| EGCG-金属复合物 | 复合键 | 键长/? |
| ECGC-Ag+ | 4"O—Ag | 2.363 |
| 5"O—Ag | 2.302 | |
| EGCG-Hg2+ | 4"O—Hg | 2.366 |
| 5"O—Hg | 2.333 | |
| EGCG-Cu2+ | 4"O—Cu | 1.941 |
| 5"O—Cu | 1.916 | |
| EGCG-Fe2+ | 4"O—Fe | 1.926 |
| 5"O—Fe | 1.893 | |
| EGCG-In3+ | 4"O—In | 2.929 |
| 5"O—In | 2.683 | |
| EGCG-Al3+ | 4"O—Al | 2.418 |
| 5"O—Al | 2.008 | |
| EGCG-Au3+ | 4"O—Au | 2.979 |
| 5"O—Au | 2.194 |
表1EGCG-金属复合物的复合键键长
Table1.Composite bond lengths of the EGCG-metal complexes.
2
3.2.EGCG-金属复合物的Mayer键级
判断原子间是否成键, 光从Gaussview中观察几何构型和计算相连键长是远远不够的, 因此我们还计算了金属复合物的Mayer键级[29]. Mayer键级是基于量子化学计算产生的波函数计算的, 从物理意义上可以理解为原子间共享的电子对数. 因此对于单/双/三重键, Mayer键级应比较接近1.0/2.0/3.0, 而没有或几乎没有成键的原子间Mayer键级应当接近0. 一般情况下, Mayer键级小于0.5, 都被称为弱成键. 对应下面的表2中, 唯有EGCG-Cu2+和EGCG-Fe2+复合物形成的Mayer键级超过了0.5, 即形成的螯合键作用较强. 尤其是EGCG与Fe2+离子的复合, 形成的复合键Mayer键级分别为4"O—Fe键级0.594, 5"O—Fe键级0.748. 而其他金属离子与EGCG中D环4"O, 5"O形成的复合键键级则较小, 可能仅仅形成了弱的复合键, 即螯合作用较弱. 通过表2还可以发现, In3+离子与EGCG复合形成的键级是最小的(4"O—In的Mayer键级仅为0.0742, 5"O—In的Mayer键级为0.1183), 可能与复合物中金属离子与EGCG距离较远有关, 这也导致了EGCG-In3+复合物的形成几乎不存在螯合作用.| EGCG-金属复合物 | 复合键 | Mayer键级 |
| ECGC-Ag+ | 4"O—Ag | 0.3462 |
| 5"O—Ag | 0.4304 | |
| EGCG-Hg2+ | 4"O—Hg | 0.3870 |
| 5"O—Hg | 0.4697 | |
| EGCG-Cu2+ | 4"O—Cu | 0.5089 |
| 5"O—Cu | 0.5458 | |
| EGCG-Fe2+ | 4"O—Fe | 0.5940 |
| 5"O—Fe | 0.7480 | |
| EGCG-In3+ | 4"O—In | 0.0742 |
| 5"O—In | 0.1183 | |
| EGCG-Al3+ | 4"O—Al | 0.1635 |
| 5"O—Al | 0.3022 | |
| EGCG-Au3+ | 4"O—Au | 0.1058 |
| 5"O—Au | 0.4331 |
表2EGCG-金属复合物中复合键的Mayer键级
Table2.The Mayer bond orders of composite bond in the EGCG-metal complexes.
2
3.3.EGCG-金属复合物的自然布居分析
既然说金属离子与EGCG在溶液中存在静电吸引作用, 那么自然就要观察它反应前后的电荷变化. 因此, 考察复合物部分原子的自然布居分析 (natural population analysis, NPA)十分必要[30]. NPA是NBO分析中的核心组成部分, 复合物形成前后原子电荷的变化只能反映整个原子的电子得失, 而利用NPA分析则可以得知每个原子轨道的电子得失. Ag+和Hg2+离子在复合前分别带1, 2个单位正电荷; 与EGCG的复合后, 通过表3可以看出Ag+和Hg2+离子分别转移0.22和0.29个单位正电荷, 这仅仅有少量的电荷转移, 归结其原因是螯合作用较弱, 主要依靠静电作用吸附. 而Fe2+和Cu2+离子与EGCG复合后, 构型中金属离子与EGCG距离较近且形成了螯合键, 由于静电作用依然存在, Fe2+和Cu2+离子上的电子仍然有部分的转移. EGCG-In3+, EGCG-Al3+和EGCG-Au3+复合物的自然电荷分布则较为特别, 由于它们形成了“腔状结构”, EGCG的B环与D环对In3+, Al3+和Au3+离子都有较强的相互作用. 这导致这些金属离子都转移了大部分的电荷, 分别是In3+离子转移了1.92个单位正电荷, Al3+离子转移了2.13个单位正电荷, 而Au3+离子也转移了2.13个单位正电荷, 因此可以判断EGCG对In3+, Al3+和Au3+这三种金属离子的吸附明显是静电吸引作用主导的.| EGCG-金属复合物 | 复合原子 | 自然电荷分布 |
| ECGC-Ag+ | Ag | 0.7803 |
| 4"O | –0.8837 | |
| 5"O | –0.9144 | |
| EGCG-Hg2+ | Hg | 1.7086 |
| 4"O | –0.8860 | |
| 5"O | –0.9045 | |
| EGCG-Cu2+ | Cu | 1.4836 |
| 4"O | –0.8479 | |
| 5"O | –0.8421 | |
| EGCG-Fe2+ | Fe | 1.4342 |
| 4"O | –0.8121 | |
| 5"O | –0.8417 | |
| EGCG-In3+ | In | 1.0761 |
| 4"O | –0.5587 | |
| 5"O | –0.6041 | |
| EGCG-Al3+ | Al | 0.8686 |
| 4"O | –0.5978 | |
| 5"O | –0.7942 | |
| EGCG-Au3+ | Au | 0.8679 |
| 4"O | –0.5229 | |
| 5"O | –0.6244 |
表3EGCG-金属复合物中复合原子的自然布居分析
Table3.Natural population analysis of composite atoms in the EGCG-metal complexes.
2
3.4.EGCG-金属复合物的结合能
金属离子与EGCG之间结合能的大小, 决定了形成的金属复合物是否稳定. 同时也能比较带电量相同的金属复合物结合能, 判断出这些金属离子与EGCG的复合是静电吸引主导, 还是螯合作用主导. 分别计算这七种金属离子与EGCG吸附后的结合能, 结果如图4所示.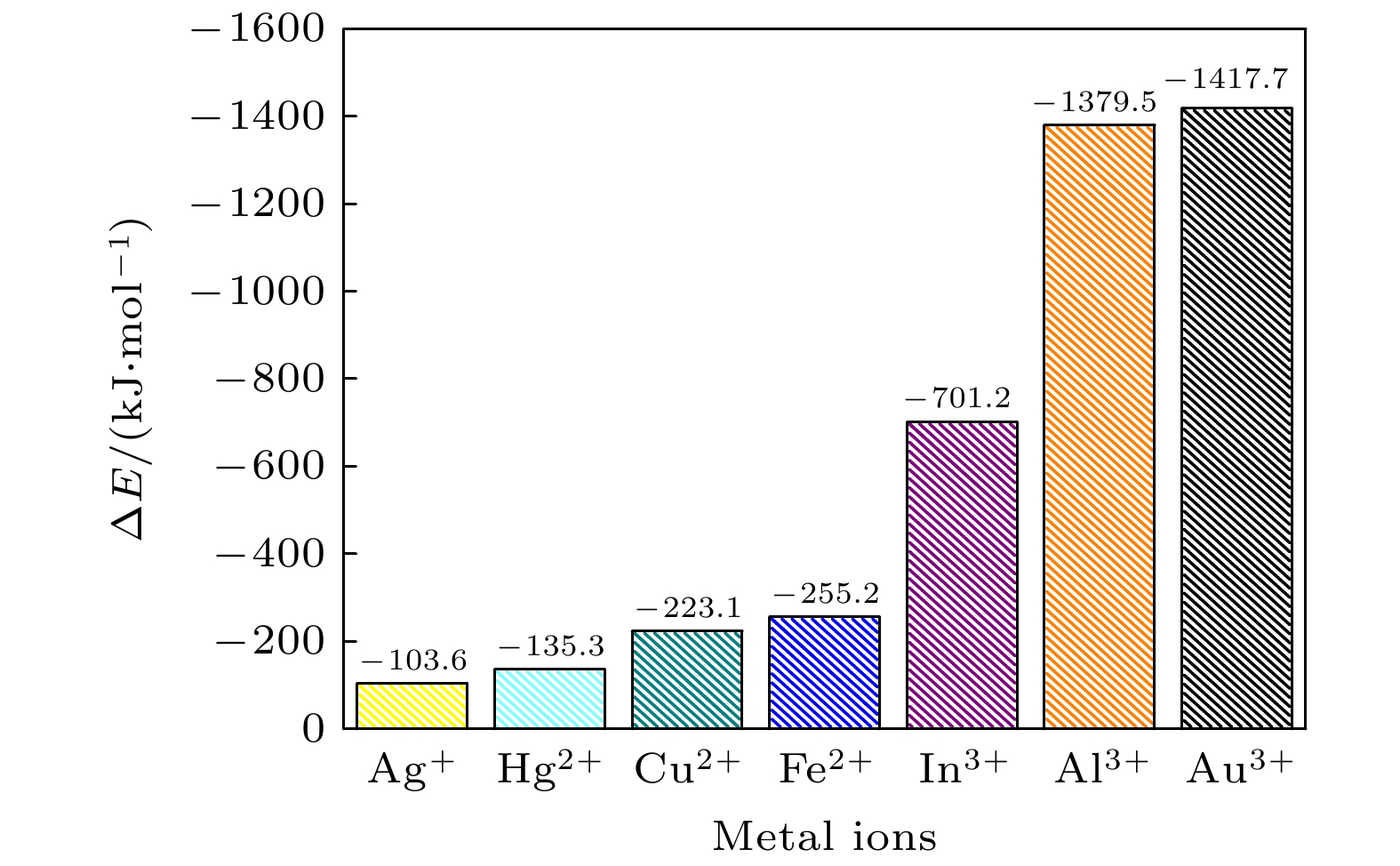 图 4 EGCG-金属复合物的结合能
图 4 EGCG-金属复合物的结合能Figure4. Binding energies of EGCG-metal complexes.
对比不同金属复合物的结合能, 我们发现复合物的结合能与金属离子所带电荷量有所联系. 一般来说, 金属离子所带的电荷量越大, 越容易进行电荷转移, 其金属复合物的结合能越低; 而对于带电量相同的金属离子, EGCG与其反应的结合能越低, 形成的复合物也就越稳定.
由图4可以看出, Ag+离子与EGCG的结合能只有–103.6 kJ/mol, 是七种复合物中结合能最高的. 对比带电量均为正二价的金属离子, Hg2+离子与EGCG的结合能是–135.3 kJ/mol, 高于成键的键能, 这也证实EGCG-Hg2+复合物中没有形成螯合键. EGCG-Fe2+复合物(


随后观察正三价的金属离子与EGCG复合后的结合能, 发现同为“腔状结构”的EGCG-In3+与EGCG-Al3+, EGCG-Au3+复合物结合能差异较大. EGCG-In3+复合物的结合能为–701.2 kJ/mol, 而EGCG-Al3+和EGCG-Au3+结合能分别是–1379.5 和–1417.7 kJ/mol. 根据前面几方面的考察, 归结原因是EGCG-In3+复合物中, In3+离子与EGCG距离较远导致静电吸引作用较弱, 虽然也存在芳环堆积作用, 但整体的吸附能力远小于EGCG-Au3+复合物. 而In3+, Al3+和Au3+离子与EGCG之间的吸附应该都是静电作用与芳环堆积作用共同主导.
2
3.5.EGCG-金属复合物的弱相互作用
Johnson等[24]提出了一种可视化研究弱相互作用的方法, 称为RDG或NCI方法. 这种方法不仅可以指出哪里存在弱相互作用, 还能可视化地了解弱相互作用的强度与类型. 其中将原子间弱相互作用的强弱用色彩加以区分, 即被设为蓝 > 绿 > 红. 蓝色区域代表相互作用强度较强, 最常见的就是氢键和卤键等作用. 绿色区域说明相互作用强度很弱, 范德瓦耳斯作用区域符合这个特征. 红色区域则为位阻效应区域, 对应的是环中周围原子间的互斥效应. 为了考察金属离子与EGCG的相互作用, 采用Multiwfn[31]+VMD[32]软件绘制了这几种金属复合物的RDG函数等值面图(图5), 用来判断是否存在弱相互作用. 图 5 EGCG-金属复合物的RDG函数等值面图 (a) EGCG-Ag+; (b) EGCG-Hg2+; (c) EGCG-Cu2+; (d) EGCG-Fe2+; (e) EGCG-In3+; (f) EGCG-Al3+; (g) EGCG-Au3+
图 5 EGCG-金属复合物的RDG函数等值面图 (a) EGCG-Ag+; (b) EGCG-Hg2+; (c) EGCG-Cu2+; (d) EGCG-Fe2+; (e) EGCG-In3+; (f) EGCG-Al3+; (g) EGCG-Au3+Figure5. RDG function isosurface diagrams of EGCG-metal complexes: (a) EGCG-Ag+; (b) EGCG-Hg2+; (c) EGCG-Cu2+; (d) EGCG-Fe2+; (e) EGCG-In3+; (f) EGCG-Al3+; (g) EGCG-Au3+.
通过观察发现, 在图5(a)和图5(b)中, 复合后的结构没有发生明显的变化且金属离子与D环4"O, 5"O之间都形成了明显的蓝色等值面, 这表明了EGCG与金属离子两者之间存在较强的弱作用(即存在静电吸引作用). 同时图5(c)中的Cu2+离子与EGCG的螯合键之间也形成了蓝色的等值面, 这是因为EGCG-Cu2+复合物中螯合作用强度一般, 导致两者间仍存在静电吸引; 并且B环与D环之间还呈现了小量的黄绿色等值面, 证实还存在小量芳环堆积作用. 而图5(d)中Fe2+离子与EGCG中D环4"O, 5"O形成的螯合键之间并没有任何颜色的等值面(即没有弱作用存在), 这也反映了EGCG-Fe2+复合物主要以螯合键的形式复合在一起. 此外, 图5(e)—(g)中不仅金属离子与D环4"O, 5"O之间存在蓝色等值面, B环与D环之间还呈现了大面积的黄绿色等值面(即存在芳环堆积作用). 当然如果仔细对比图5(e)与图5(g)可以看出, In3+离子与D环4"O, 5"O之间的等势面呈现的是淡绿色, 而EGCG-Au3+复合物中的等势面则是深蓝色, 这也从侧面展示了Au3+离子与EGCG之间的静电吸引作用更强.
