全文HTML
--> --> -->LiCoO2的结构属于空间群为

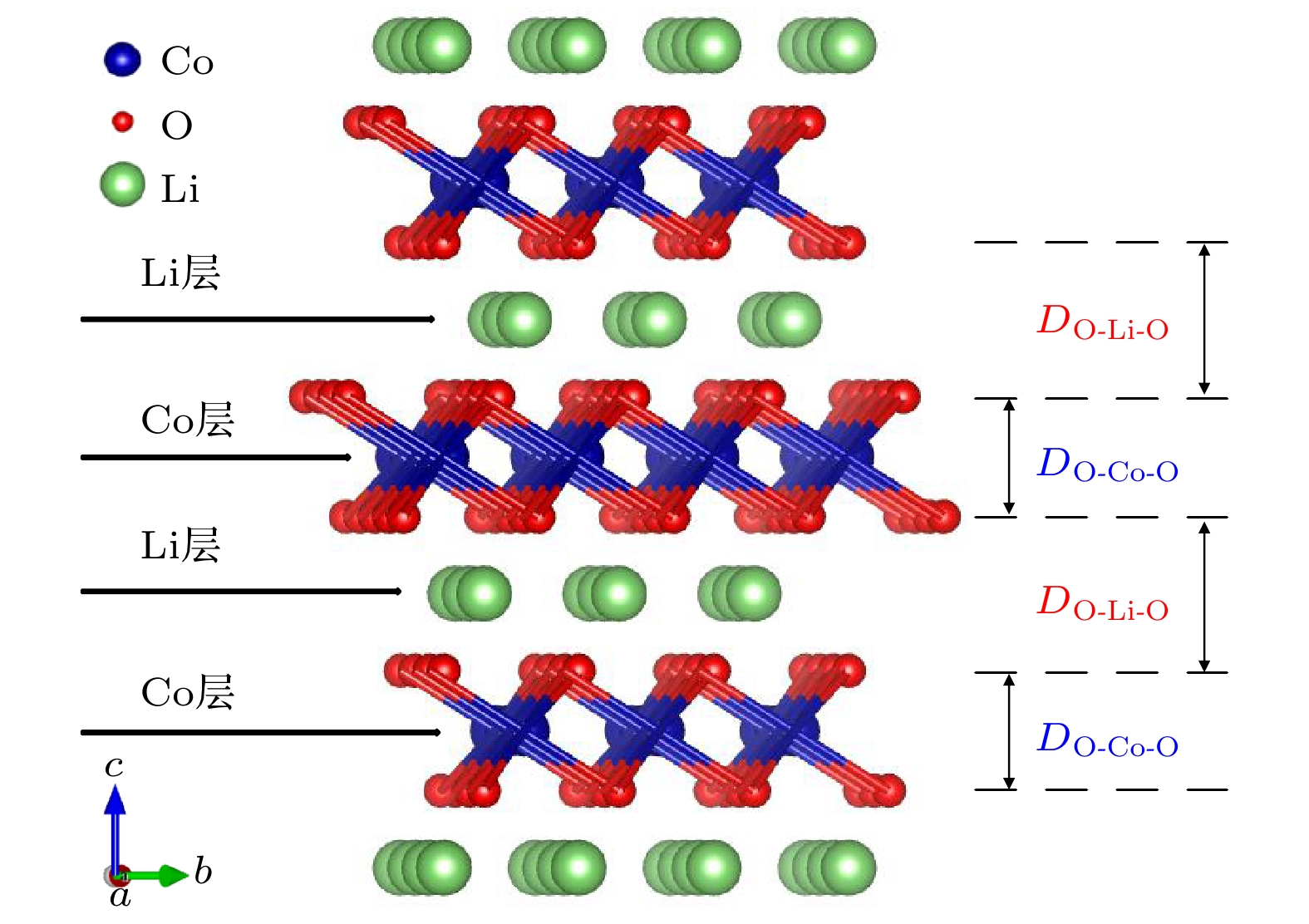 图 1 LiCoO2的晶胞结构, 符号“DO-Co-O”和“DO-Li-O”分别表示过渡金属层(Co层)和锂层(Li层)的厚度
图 1 LiCoO2的晶胞结构, 符号“DO-Co-O”和“DO-Li-O”分别表示过渡金属层(Co层)和锂层(Li层)的厚度Figure1. Unit cell of LiCoO2, the symbols of “DO-Co-O” and “DO-Li-O” are the oxygen distance across the transition metal layer (cobalt layer) and across the Li layer respectively.





3.1.形成能及结构稳定性
在以往的计算中, 人们在分析解释LiCoO2掺杂作用时, 一般都是考虑掺杂元素只替代了Li位或者Co位, 以替代Co位为多数, 但是实际实验中因为制作工艺、掺杂浓度和掺杂温度等因素的影响, 可能存在各种的掺杂组态, 因此需要通过计算各种组态的形成能来加以分析. 绝大多数关于LiCoO2的计算是在2 × 2 × 1的超胞下进行的, 但是, 若是在2 × 2 × 1的超胞下掺入两个以上的Mg原子, 意味着掺入Mg原子浓度过高, 晶胞会发生晶格畸变, 改变所需要的层状结构, 极大程度损害材料的离子导电性. 因此, 我们的计算将LiCoO2扩胞为3 × 3 × 1, 以此降低Mg原子的掺杂浓度, 来减少晶格畸变带来的影响.首先计算了纯的LiCoO2的晶格参数, 计算得到的晶格常数a和c分别为2.827和14.165 ? (c/a = 5.01), 锂层的厚度(DO-Li-O)和过渡金属层的厚度(DO-Co-O)分别为2.654和2.068 ?, 计算结果与其他研究小组的计算结果[29]相符合. 接着, 在(3 × 3 × 1)LiCoO2超胞中, 考虑单个Mg离子的替代情况, 即单离子替代模型(掺杂浓度为3.7%). 计算得到的Mg替代Co位(LiMg0.037Co0.963O2)的形成能为–0.448 eV, 而Mg替代Li位(Li0.963Mg0.037CoO2)的形成能为+2.229 eV. 由此可见, 对于单离子替代, Mg离子在Co位上的替代优于Li位, 掺杂后更容易形成LiCo0.963Mg0.037O2, 而不是形成Li0.963Mg0.037CoO2. 确实, 单离子替代Co位是更有利的, 这与其他研究小组的计算是类似的[20]. 进一步分析Mg掺杂在LiCoO2中Co位对骨架结构稳定性的影响. 结果发现, 单离子替代Co位后晶格常数a和c分别增加至2.830和14.181 ?, 但c/a值与纯的LiCoO2保持不变, 说明掺杂后的骨架结构稳定, 没有发生变形. 但Mg离子掺杂拓宽了掺杂的过渡金属层的厚度DO-Co-O, 由2.068 ?增加至2.096 ?, 这是因为Mg2+离子半径大于Co3+离子的缘故, 而Mg离子掺杂对近邻的锂层的厚度和过渡金属层的厚度影响不大. 但是, 当Mg掺杂浓度提高至7.4%后, 即两个Mg离子的替代情况, 结果发生了有趣的变化. 下面重点分析两个Mg离子的替代情况, 即双离子替代模型, 本文主要考虑两个Mg离子同时替代Co位、分别替代1个Co位和1个Li位、同时替代Li位三种情况.
图2给出了两个Mg离子同时替代Co位的各种组态图. 其中, 以某个被Mg替代的Co位标定为“0”位, 而把同层和最近邻层的其他Co的Mg替代位分别编为1—17位(Mg-Mg离子间距d从近到远, 并排除等效位置). 所形成的双Mg离子替代的组态分别记为(0, 1), (0, 2), …, (0, 17). 表1列出了对应的17种组态的形成能. 从表1发现, 双离子替代Co位在同一层情况下, 即组态(0, 1), (0, 2), …, (0, 8), 其中组态(0, 3), (0, 5), (0, 8)的形成能出现了小于0的情况, 而组态(0, 1), (0, 2), (0, 4), (0, 6), (0, 7)的形成能存在大于0的情况, 表示同层的近邻Co位替代在能量上不均匀, 有些可行, 有些不可行. 然而, 双离子替代在异层Co位的情况下, 即组态(0, 9), (0, 10), …, (0, 17)的形成能均在–0.60—–0.90 eV之间, 它们比单个Mg离子替代的形成能–0.448 eV还要低, 这意味着异层Co位的双离子替代是可行的. 进一步分析发现, 在双离子替代异层Co位的情况下, 晶格常数a和c随着掺杂浓度的提升分别增加至2.833和14.194 ?, 但c/a值仍保持不变, 说明掺杂后的骨架结构还是稳定的, 没有发生变形. 此时Mg2+离子同样拓宽了掺杂的过渡金属层的厚度DO-Co-O, 由2.068 ?增加至2.090 ?, 而对近邻的锂层的厚度几乎没有影响.
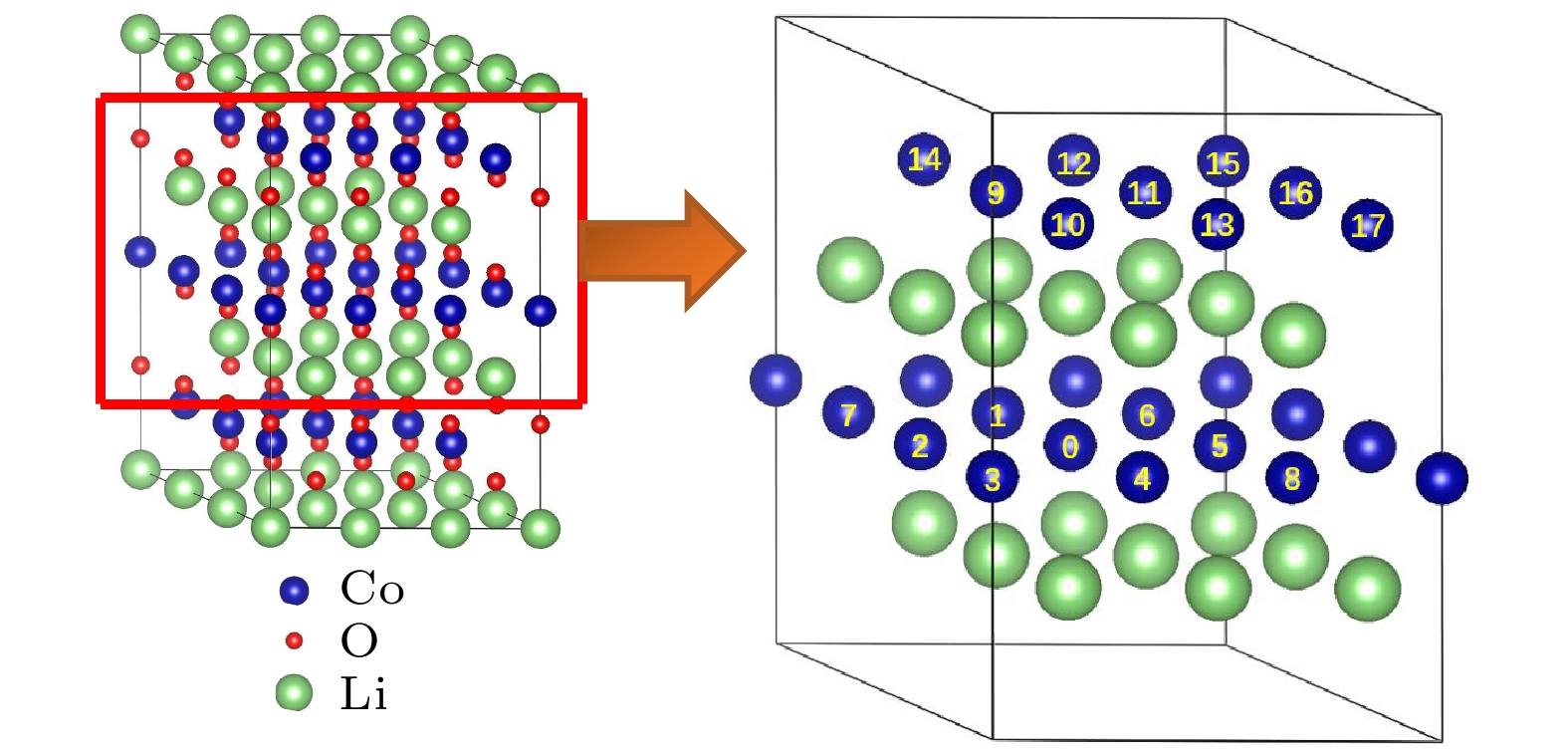 图 2 两个Mg替代两个Co位的各种组态示意图
图 2 两个Mg替代两个Co位的各种组态示意图Figure2. Schematic illustration of various configurations for two Mg replacing two Co sites.
| Mg-Mg间距d/? | 组态 | Eform/eV |
| 2.830 | (0, 1) | 1.651 |
| (0, 2) | 0.196 | |
| (0, 3) | –0.588 | |
| (0, 4) | 0.497 | |
| (0, 5) | –0.604 | |
| (0, 6) | 0.383 | |
| 4.902 | (0, 7) | 0.348 |
| (0, 8) | –1.004 | |
| 4.992 | (0, 9) | –0.887 |
| (0, 10) | –0.886 | |
| (0, 11) | –0.886 | |
| 5.738 | (0, 12) | –0.875 |
| (0, 13) | –0.874 | |
| 6.398 | (0, 14) | –0.872 |
| (0, 15) | –0.870 | |
| (0, 16) | –0.871 | |
| 7.547 | (0, 17) | –0.655 |
表1图2所示的各种掺杂组态的形成能
Table1.Formation energies of the various doping configurations given in Fig. 2.
图3给出了两个Mg离子分别替代1个Co位和1个Li位的各种组态图. 其中, 以某个被Mg替代的Co位标定为“0”位, 把最近邻层的Li位的Mg替代位分别编为1—9, 形成的组态分别记为(0, 1), (0, 2), …, (0, 9). 表2列出了对应的9种组态的形成能. 从表2可以看出, 最近邻三种组态(0, 1), (0, 2)和(0, 3)(Mg-Mg间距d = 2.869 ?)的形成能为负且很低; 尤其是(0, 1)和(0, 3)两种组态的形成能分别为–1.137和–2.228 eV, 这表明两个Mg离子分别替代Co位和Li位的双离子替代在近邻位置容易实现. (0, 5)组态的形成能也存在负值(–0.143 eV), 但比最近邻的三种组态的形成能要高得多. 进一步, 对于替代的两个Mg间距较大的(0, 4), (0, 6), (0, 7), (0, 8)和(0, 9)组态, 其形成能皆大于0, 这是因为两个Mg在距离较远时相互作用弱, Mg替代Li的作用接近于上述Mg单离子替代Li的情况, 故形成能都大于零, 不容易实现. 进一步分析双离子替代在近邻Co-Li位对骨架结构稳定性的影响. 结果发现, Mg掺杂后晶格常数a和c都增大, 分别为2.833和14.194 ?, 但c/a值与纯的LiCoO2保持不变, 说明Mg离子替代近邻Co-Li位后的骨架结构稳定, 没有发生变形. 但掺杂的锂层的厚度DO-Li-O减小, 由2.654 ?减小至2.639 ?, 这主要归因于Mg2+离子与O2–离子之间有着更强的库仑相互作用, 而掺杂的过渡金属层的厚度DO-Co-O增加明显, 由2.068 ?增加至2.112 ?. 而与之近邻的锂层和过渡金属层的厚度变化不大.
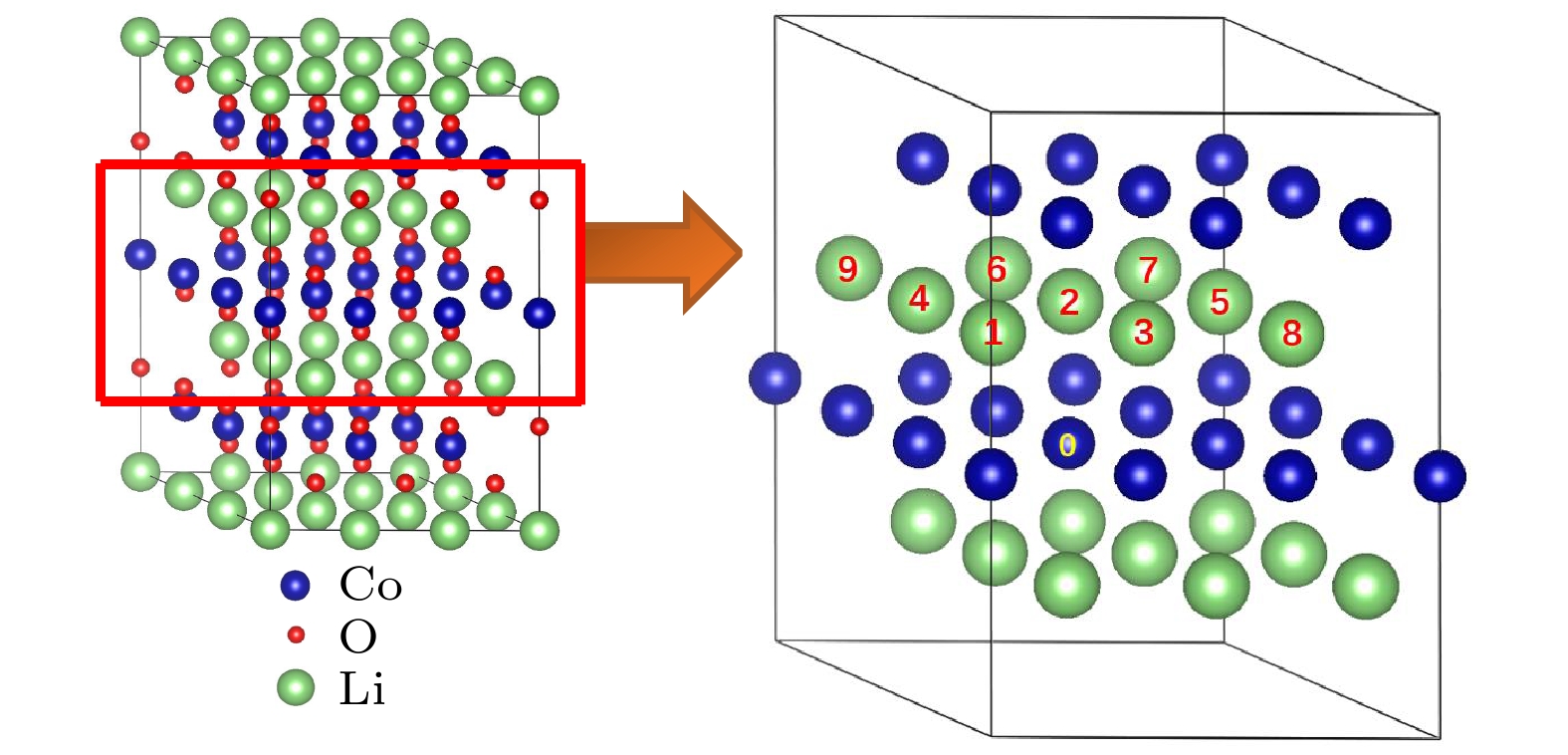 图 3 两个Mg分别替代1个Co位和1个Li位的各种组态示意图
图 3 两个Mg分别替代1个Co位和1个Li位的各种组态示意图Figure3. Schematic illustration of various configurations for two Mg replacing one Co site and oneLi site respectively.
| Mg-Mg间距d/? | 组态 | Eform/eV |
| 2.869 | (0, 1) | –1.137 |
| (0, 2) | –0.497 | |
| (0, 3) | –2.228 | |
| 4.030 | (0, 4) | 0.978 |
| (0, 5) | –0.143 | |
| 4.924 | (0, 6) | 0.319 |
| (0, 7) | 0.346 | |
| (0, 8) | 0.344 | |
| 6.346 | (0, 9) | 0.756 |
表2图3所示的各种掺杂组态的形成能
Table2.Formation energies of the various doping configurations given in Fig.3.
图4给出了两个Mg离子同时替代Li位的各种组态图. 其中, 以某个被Mg替代的Li位标定为“0”位, 而把同层和最近邻层的其他Li位的Mg替代位分别编为1—17, 形成的组态分别记为(0, 1), (0, 2), …, (0, 17). 表3列出了对应的17种组态的形成能. 从表3可以看到, 最近邻6种组态(0, 1), (0, 2), …, (0, 6)(Mg-Mg间距d = 2.830 ?)的形成能均为负且比较低, 在–0.40— –1.00 eV之间; 同层内的(0, 7)和(0, 8)组态(d = 4.902 ?)的形成能也是负的, 但相对高一些. 而异层的(0, 9), (0, 10), …, (0, 17)组态的形成能大多数都大于零, 表明不容易形成, 这也是因为两个Mg在距离较远时相互作用弱, Mg替代Li的作用接近于上述Mg单离子替代Li的情况. 同样地, Mg掺杂在LiCoO2中Li位也会对骨架结构产生影响, 分析两个Mg离子替代同层Li位的情况, 结果发现, Mg掺杂后晶格常数a和c都有增加, 但c/a值下降至4.99, 说明掺杂后的骨架结构略有变形. Mg离子掺杂明显减小了掺杂的锂层的厚度DO-Li-O, 由2.654 ?减小至2.613 ?, 这主要归因于Mg2+离子与O2–离子之间较强的库仑相互作用. 由于掺杂的锂层“塌陷”, 致使近邻的过渡金属层的厚度DO-Co-O明显增大, 由2.068 ?增加至2.101 ?, 而近邻的锂层的厚度变化不大.
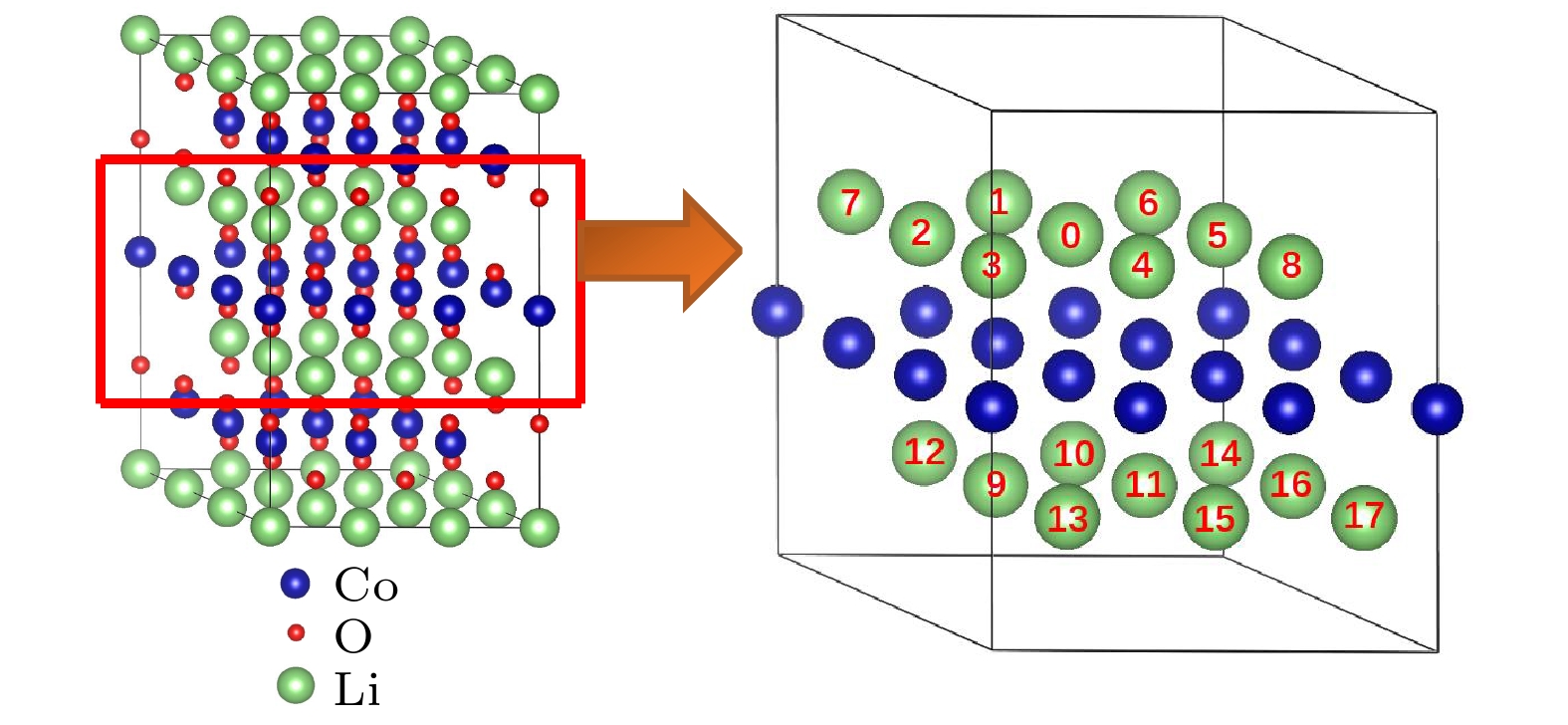 图 4 两个Mg替代两个Li位的各种组态示意图
图 4 两个Mg替代两个Li位的各种组态示意图Figure4. Schematic illustration of various configurations for two Mg replacing two Li sites.
| Mg-Mg间距d/? | 组态 | Eform/eV |
| 2.830 | (0, 1) | –0.965 |
| (0, 2) | –0.442 | |
| (0, 3) | –0.635 | |
| (0, 4) | –0.491 | |
| (0, 5) | –0.723 | |
| (0, 6) | –0.965 | |
| 4.902 | (0, 7) | –0.126 |
| (0, 8) | –0.226 | |
| 4.992 | (0, 9) | 1.883 |
| (0, 10) | –0.019 | |
| (0, 11) | 0.208 | |
| 5.738 | (0, 12) | 0.459 |
| (0, 13) | 0.729 | |
| (0, 14) | 0.223 | |
| 6.398 | (0, 15) | 2.305 |
| (0, 16) | 0.993 | |
| 8.060 | (0, 17) | –0.182 |
表3图4所示的各种掺杂组态的形成能
Table3.Formation energies of the various doping configurations given in Fig.4.
综上所述, 基于上述的形成能计算容易发现, 对于单个Mg离子替代, Mg更倾向于替代Co位. 然而, 在双离子替代情况下, 两个Mg离子容易异层同时替代Co位; 两个Mg离子容易同层同时替代Li位; 两个Mg离子更容易最近邻分别替代Co位和Li位, 且这种情况的形成能是最低的. 也就是Mg离子倾向分散性地替代Co位, 同时也有可能聚集性地替代Li位或Co-Li位. Mg掺杂在LiCoO2中Co位、Li位和Co-Li位会影响骨架结构的稳定性, Mg掺杂LiCoO2后, 晶格常数a和c都增加了, 但不同的掺杂方式对LiCoO2的锂层的厚度和过渡金属层的厚度的影响不同, 具体表现为: Mg掺杂在LiCoO2中Co位时, 掺杂的过渡金属层的厚度增大, 而与之近邻的锂层和过渡金属层的厚度变化不大; Mg掺杂在LiCoO2中Li位, 会明显减小掺杂锂层的厚度, 由于掺杂的锂层“塌陷”, 会引起近邻的过渡金属层厚度变大; Mg掺杂在LiCoO2中Co-Li位时, 掺杂锂层的厚度减小, 而掺杂的过渡金属层的厚度增大, 与之近邻的锂层和过渡金属层的厚度变化不大.
2
3.2.态密度与电子态
下面重点研究形成能小于零的掺杂组态的电子态. 图5给出了纯的LiCoO2和单个Mg离子替代Co位LiCo0.963Mg0.037O2的态密度(DOS)图. 从图5(a)可以看出, 计算得到的纯LiCoO2带隙为1.56 eV, 接近实验测量值1.7—2.7 eV [30]. 而从图5(b)可以看出, Mg单替代Co位, 所获得的LiCo0.963Mg0.037O2的费米能级位置由间隙处转移到价带边缘, 体现金属性的特点, 从而提高了电子电导. 同时还研究了Mg掺杂后的磁性, 发现掺杂后LiCo0.963Mg0.037O2材料表现出一定的磁性, 但是磁矩较小, 这个磁性是由歧化反应(2Co3+→Mg2+ + Co4+)导致的, 它来源于Mg掺入引起的Co4+. 图6(a)和图6(b)分别给出了纯的LiCoO2和LiCo0.963Mg0.037O2的分波态密度(PDOS). 从图6(a)可以看出, Co-3d电子是价带顶和导带底电子态的最主要贡献, 费米能级附近的电子态主要来自Co-3d和O-2p轨道间的杂化作用. 而掺入Mg后, LiCo0.963Mg0.037O2中Mg原子的态密度在总态密度中的占比很低, 对总态密度几乎没有贡献, 这与Hoang[31]的计算结果是一致的.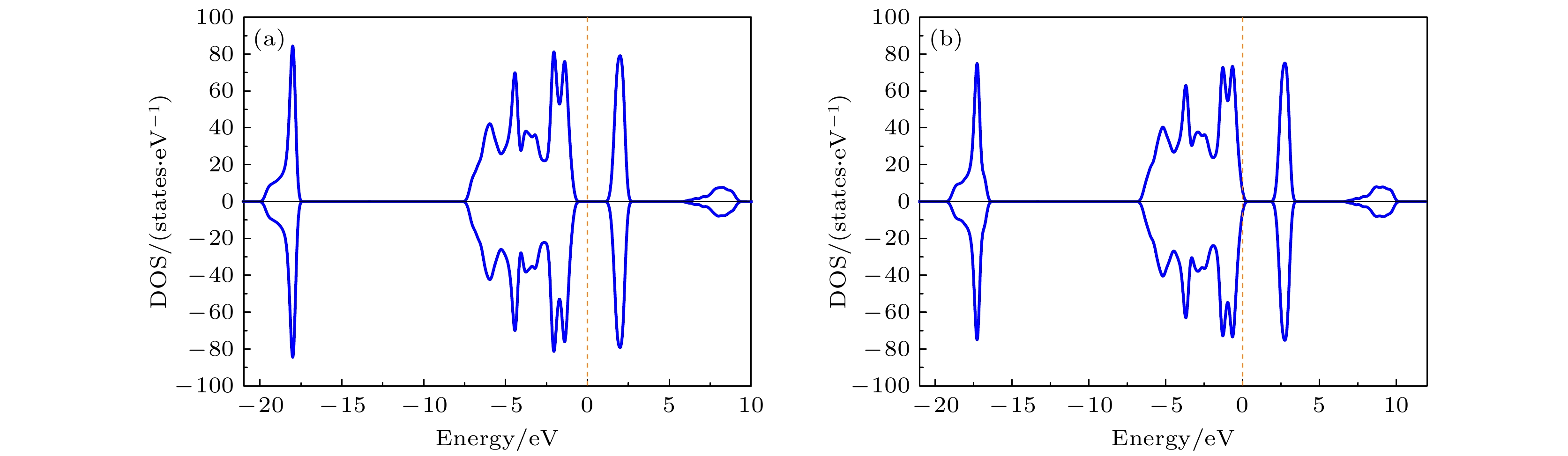 图 5 态密度(DOS)图 (a) 纯的LiCoO2; (b) LiCo0.963Mg0.037O2. 费米能级设为零
图 5 态密度(DOS)图 (a) 纯的LiCoO2; (b) LiCo0.963Mg0.037O2. 费米能级设为零Figure5. Density of states (DOS): (a) Pure LiCoO2; (b) LiCo0.963Mg0.037O2. The Fermi level is set to be zero.
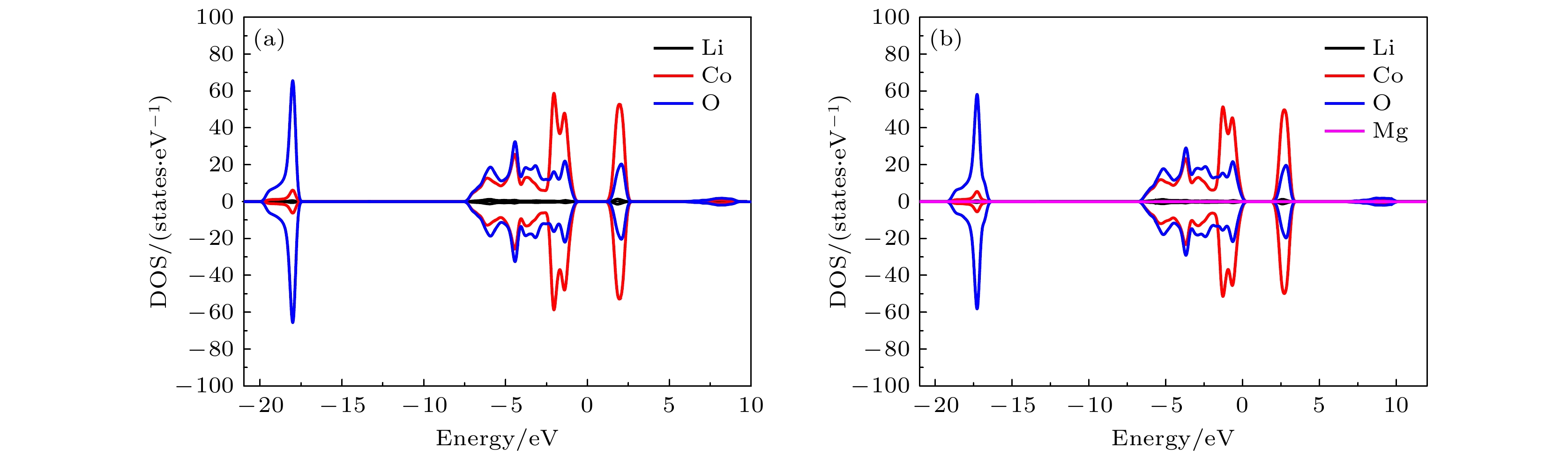 图 6 分波态密度(PDOS)图 (a) 纯的LiCoO2; (b) LiCo0.963Mg0.037O2
图 6 分波态密度(PDOS)图 (a) 纯的LiCoO2; (b) LiCo0.963Mg0.037O2Figure6. Partial density of states (PDOS): (a) Pure LiCoO2; (b) LiCo0.963Mg0.037O2.
图7给出了上述两个Mg离子同时替代Co位、分别替代1个Co位和1个Li位两种模型中形成能为负的各种组态的态密度(DOS)图. 作为比较, 图中同时给出了纯的LiCoO2的态密度, 如图7(a)所示. 图7(b)—图7(d)分别表示与图2对应的两个Mg替代同层的两个Co位时组态(0, 3), (0, 5)和(0, 8)的态密度; 图7(e)—图7(h)分别表示与图2对应的两个Mg替代异层的两个Co位时组态(0, 9), (0, 12), (0, 14)和(0, 17)的态密度. 可以看到, 与纯的LiCoO2不同, 在两个Mg替代Co位后, 材料全部呈现为金属性, 电子导电性显著提升. 同时还可以看到, 异层两个Co位处于较远距离下, 掺杂后的态密度与单个Mg替代Co位时的态密度(图5(b))几乎一样. 因此, 若Mg掺杂全部替代Co位, 则异层的Mg之间影响甚微, 可视为独立的掺杂位置. 然而, 当另一个Mg替代在近邻的Li位时, 产生了一个很有趣的现象. 图7(i)—图7(l)分别表示与图3对应的两个Mg分别替代1个Co位和1个Li位时组态(0, 1), (0, 2), (0, 3)和(0, 5)的态密度. 可以看到, 与两个Mg同时替代Co位的情况不同, 在两个Mg分别替代近邻的Co位和Li位后, 材料仍然保持半导体态, 并且组态(0, 1), (0, 2)和(0, 5)在带隙中间出现了电子局域态, 使电子电导增强. 而(0, 3)组态没有出现局域态, 其DOS态与纯的LiCoO2接近, 只是带隙略微降低.
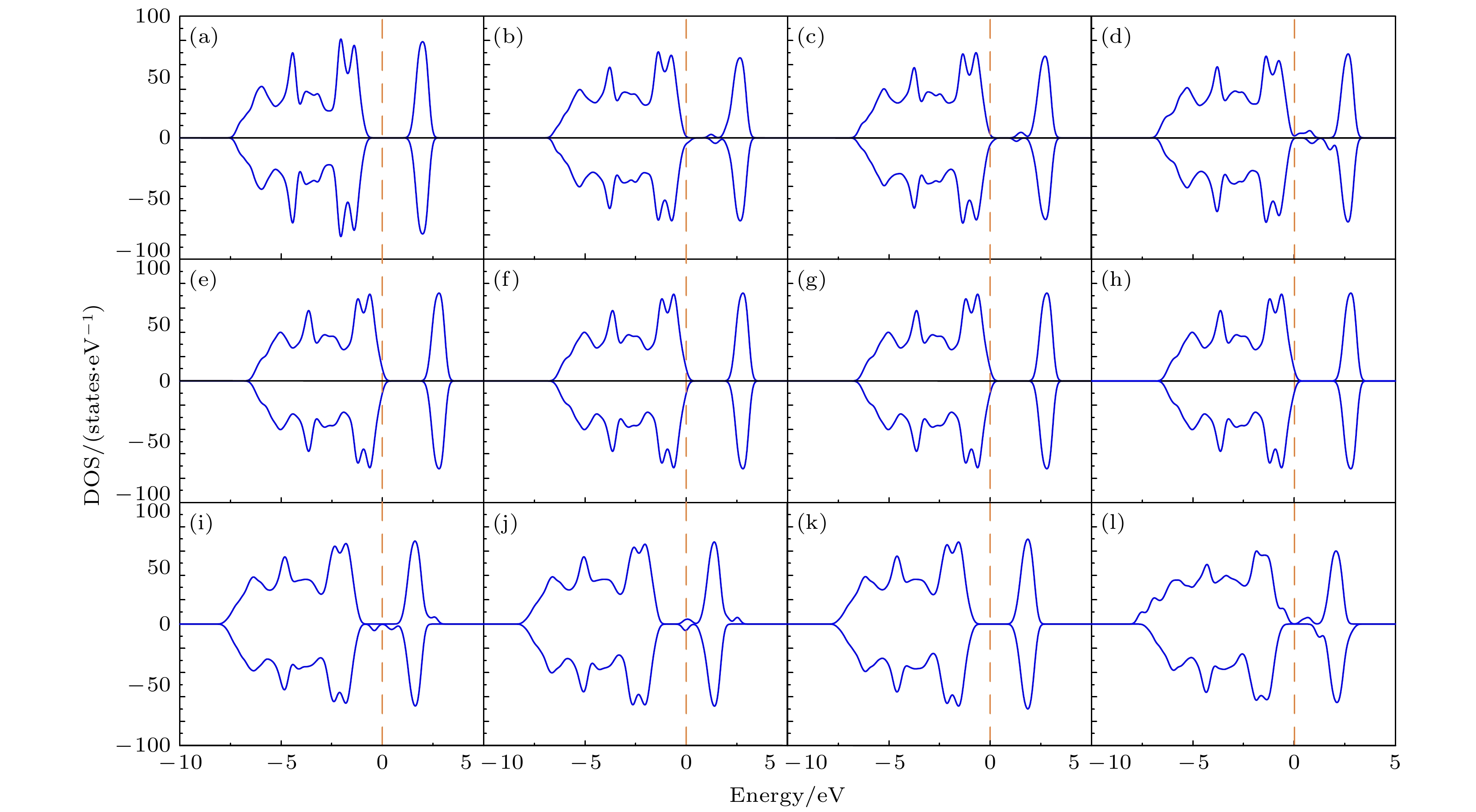 图 7 态密度(DOS)图 (a) 纯的LiCoO2; 图2所示的两个Mg替代同层的两个Co位时对应组态 (b) (0, 3), (c) (0, 5), (d) (0, 8); 图2所示的两个Mg替代异层的两个Co位时对应组态 (e) (0, 9), (f) (0, 12), (g) (0, 14), (h) (0, 17); 图3所示的两个Mg分别替代1个Co位和1个Li位时对应组态 (i) (0, 1), (j) (0, 2), (k) (0, 3), (l) (0, 5). 费米能级设为零
图 7 态密度(DOS)图 (a) 纯的LiCoO2; 图2所示的两个Mg替代同层的两个Co位时对应组态 (b) (0, 3), (c) (0, 5), (d) (0, 8); 图2所示的两个Mg替代异层的两个Co位时对应组态 (e) (0, 9), (f) (0, 12), (g) (0, 14), (h) (0, 17); 图3所示的两个Mg分别替代1个Co位和1个Li位时对应组态 (i) (0, 1), (j) (0, 2), (k) (0, 3), (l) (0, 5). 费米能级设为零Figure7. Density of states (DOS): (a) Pure LiCoO2; (b) (0, 3), (c) (0, 5), (d) (0, 8)configurations for two Mg atoms replacing two Co sites in the same layer given in Fig. 2; (e) (0, 9), (f) (0, 12), (g) (0, 14), (h) (0, 17) configurations for two Mg atoms replacing two Co sites in different layers given in Fig. 2; (i) (0, 1), (j) (0, 2), (k) (0, 3), (l) (0, 5) configurations for two Mg atoms replacing one Co site and one Li site respectively given in Fig. 3. The Fermi level is set to be zero.
图8给出了与图4对应的两个Mg同时替代Li位时形成能为负的各种组态的态密度(DOS)图. 可以看出, 与纯的LiCoO2态密度不同, 在两个Mg替代Li位后, 各组态的费米面均向导带底偏移, 体现N型半导体的特点; 同时带隙明显减小, 且出现了电子局域态, 使电子电导增强. 尤其是(0, 17)组态, 尽管两个Mg替代Li位的距离比较远, 但仍然有–0.182 eV的形成能, 形成的DOS态接近金属态, 如图8(f)所示, 其机制还有待进一步研究. 图9给出了对应图8的6个组态的分波态密度(PDOS), 可以看出, 态密度图中出现的那些电子占据态都是由Co-3d和O-2p的电子轨道间耦合形成的.
 图 8 态密度(DOS)图. 图4中所示的两个Mg替代同层的两个Li位时对应组态 (a) (0, 1), (b) (0, 2), (c) (0, 3), (d)(0, 8); 图4中所示的两个Mg替代异层的两个Li位时对应组态 (e) (0, 10), (f) (0, 17). 费米能级设为零
图 8 态密度(DOS)图. 图4中所示的两个Mg替代同层的两个Li位时对应组态 (a) (0, 1), (b) (0, 2), (c) (0, 3), (d)(0, 8); 图4中所示的两个Mg替代异层的两个Li位时对应组态 (e) (0, 10), (f) (0, 17). 费米能级设为零Figure8. Density of states (DOS): (a) (0, 1), (b) (0, 2), (c) (0, 3), (d) (0, 8) configurations for two Mg atoms replacing two Li sites in the same layer given in Fig. 4; (e) (0, 10), (f) (0, 17) configurations for two Mg atoms replacing two Li sites in different layers given in Fig. 4. The Fermi level is set to be zero.
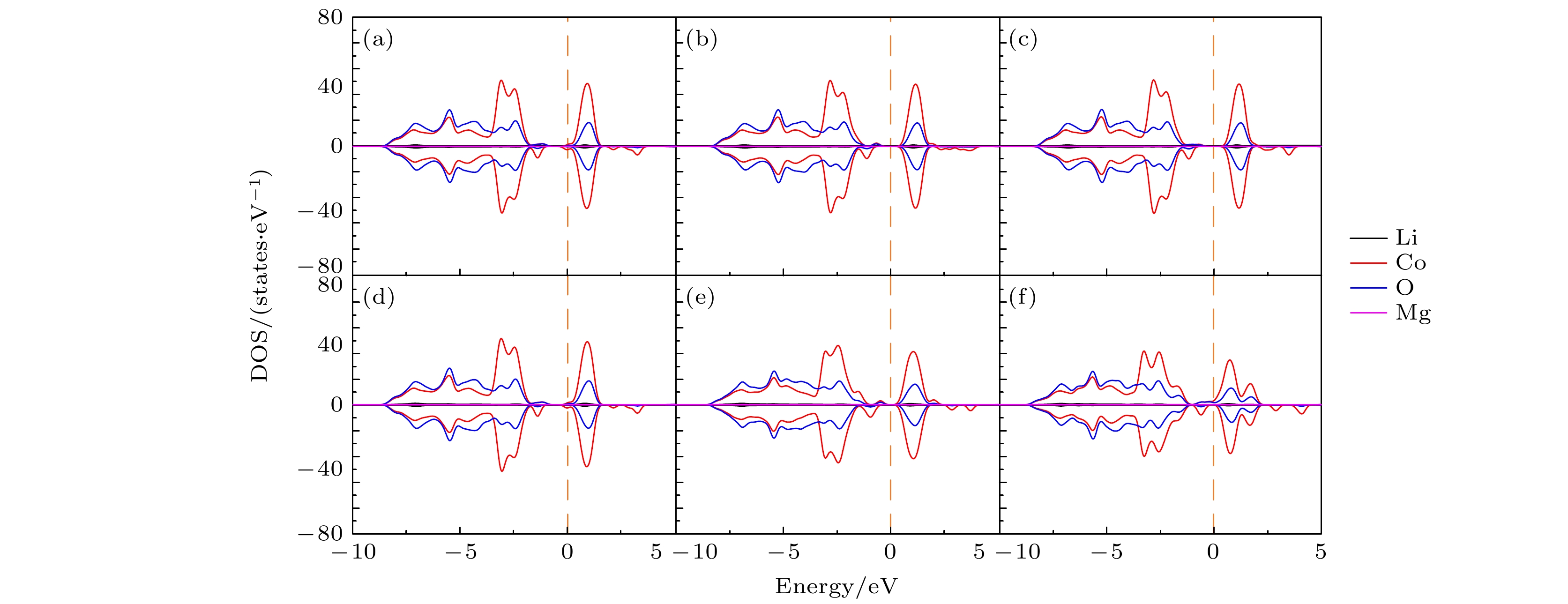 图 9 对应图8中6个组态的分波态密度(PDOS) (a) (0, 1); (b) (0, 2); (c) (0, 3); (d) (0, 8); (e) (0, 10); (f) (0, 17)
图 9 对应图8中6个组态的分波态密度(PDOS) (a) (0, 1); (b) (0, 2); (c) (0, 3); (d) (0, 8); (e) (0, 10); (f) (0, 17)Figure9. Partial density of states (PDOS) of the 6 configurations corresponding to Fig. 8: (a) (0, 1); (b) (0, 2); (c) (0, 3); (d) (0, 8); (e) (0, 10); (f) (0, 17).
综上所述可以看到, 在形成能小于零情况下, Mg单替代和双替代Co位, 其DOS态的结构类似, 都是形成费米面穿过价带顶的金属态, 明显提高LiCoO2材料的电子电导率; Mg单替代Li位在能量上是不支持的, 但双替代近邻的Li位在能量上是允许的, 其DOS态的结构类似, 形成的费米面向导带底偏移, 同时出现有明显的电子局域态, 较大地提高了LiCoO2材料的电子电导率; 两个Mg分别替代近邻的Co位和Li位的组态在能量上也是允许的, 但其DOS态与纯的LiCoO2相比变化不大, 仍然保持半导体态.
本工作的计算在国家超级计算天津中心天河一号执行, 在此表示感谢.
