全文HTML
--> --> -->目前, 对TBHQ的抗氧化性质及机理的研究较多, 对光谱的研究较少[6]. 逯美红等[7]基于密度泛函理论(density functional theory, DFT)和实验对TBHQ、BHA的拉曼光谱进行对比研究, 并对主要的拉曼特征峰的振动模式进行了指认, 为TBHQ的检测提供了一定的依据, 但该研究仅运用拉曼光谱进行分析且没有对TBHQ的分子间相互作用和激发性质进行研究. Rode等[8]同样基于DFT, 模拟手性对噻吩磺酰胺单体和二聚体的红外、紫外光谱并与实验光谱进行对比研究, 充分研究了其几何结构和理化性质. Snehalatha等[9]对食品添加剂卡莫依辛进行了光谱分析和DFT计算, 通过对比理论和实验红外光谱并进行振动分析, 对了解分子结构具有重要意义, 但是该研究并没有详细解释氢键对红外光谱可能造成的影响. 以上基于量子化学方法, 通过对比实验和理论光谱, 对了解物质的结构、性质及相关检测十分重要. 而目前关于TBHQ的红外、紫外光谱检测和结构性质的研究却很少.
因此, 本文对TBHQ的理论和实验红外、紫外光谱进行比较分析, 一方面基于DFT对红外光谱进行振动分析并考察分子间弱相互作用对红外光谱的影响, 另一方面对紫外光谱的主要激发态做分子轨道和电子空穴分析, 可以充分了解光谱特征、分子结构及激发性质, 对改造、检测相关食品抗氧化剂具有重要意义.
2.1.计算方法
本文利用Gaussian16软件[10]基于密度泛函理论, 对TBHQ分子的红外、紫外光谱进行计算. 首先, 利用B3LYP/6-311G(d, p)级别[11-12]在气相环境下优化分子的结构并进行频率计算. 在几何优化的每一步都精确地计算Hessian矩阵, 优化结果均收敛且频率计算无虚频, 相关结构优化参数如表1所示. 再利用Multiwfn 3.7 [13]软件对频率进行校正并绘制红外光谱. 由于能量的计算对基组的敏感性远高于结构优化和振动分析, 因此, 在结构优化的基础上, 基于含时密度泛函理论(time-dependent density functional theory, TDDFT), 在B3LYP/def2-TZVP级别下[14]计算分子在无水乙醇溶剂中前50个激发态并绘制紫外光谱和电子空穴图[15,16]. 为了充分考虑溶剂效应对结果的影响, 体现溶质与溶剂间的静电相互作用和非静电相互作用, 激发能的计算都是在SMD隐式溶剂模型[17]下进行.| Maximum force/a.u. | RMS force/a.u. | Maximum displacement/a.u. | Root mean square displacement/a.u. |
| 0.000006 | 0.000001 | 0.000193 | 0.000036 |
表1TBHQ分子几何结构优化参数
Table1.Optimized geometry structure parameters of TBHQ.
为了研究TBHQ分子间的弱相互作用对红外光谱的影响, 本文利用genmer软件结合molclus 1.9.6软件[18]对该分子的二聚体构型进行搜索, 并得到15个构型. 再调用Gaussian 16软件, 在B3LYP-D3(BJ)/6-311G(d, p)级别对15个二聚体构型进行结构优化, 并采用M062X-D3泛函在6-311+G(2d, p)基组水平上计算它们的单点能[19-23]. 对TBHQ二聚体的弱相互作用的研究, 是利用约化密度梯度(reduced density gradient, RDG)函数方法[24]. RDG函数是用来描述均匀电子分布偏差的实空间函数, 表达式为
2
2.2.实验方法
本文通过实验, 采用KBr压片法对TBHQ红外光谱进行测定. 实验前, 将TBHQ和KBr粉末烘干并按1∶100比例充分研磨, 再压制成均匀透明薄片, 利用Bruker Daltonics公司生产的傅里叶红外变换光谱仪测定. 紫外光谱的测定是采用液相法, 以乙醇为溶剂, 利用MAPADA公司生产的UV-6100S型紫外可见分光光度计测定. 实验采用纯度为99%的食品抗氧化剂TBHQ均为浙江一诺生物科技有限公司生产.3.1.几何结构
食品抗氧化剂TBHQ为白色粉末, 化学分子式为C10H14O2. 利用B3LYP泛函, 在6-311G(d, p)基组水平上将其几何构型优化至局域能量极小值的稳定结构, 如图1所示, 其主要键长、键角和二面角列于表2.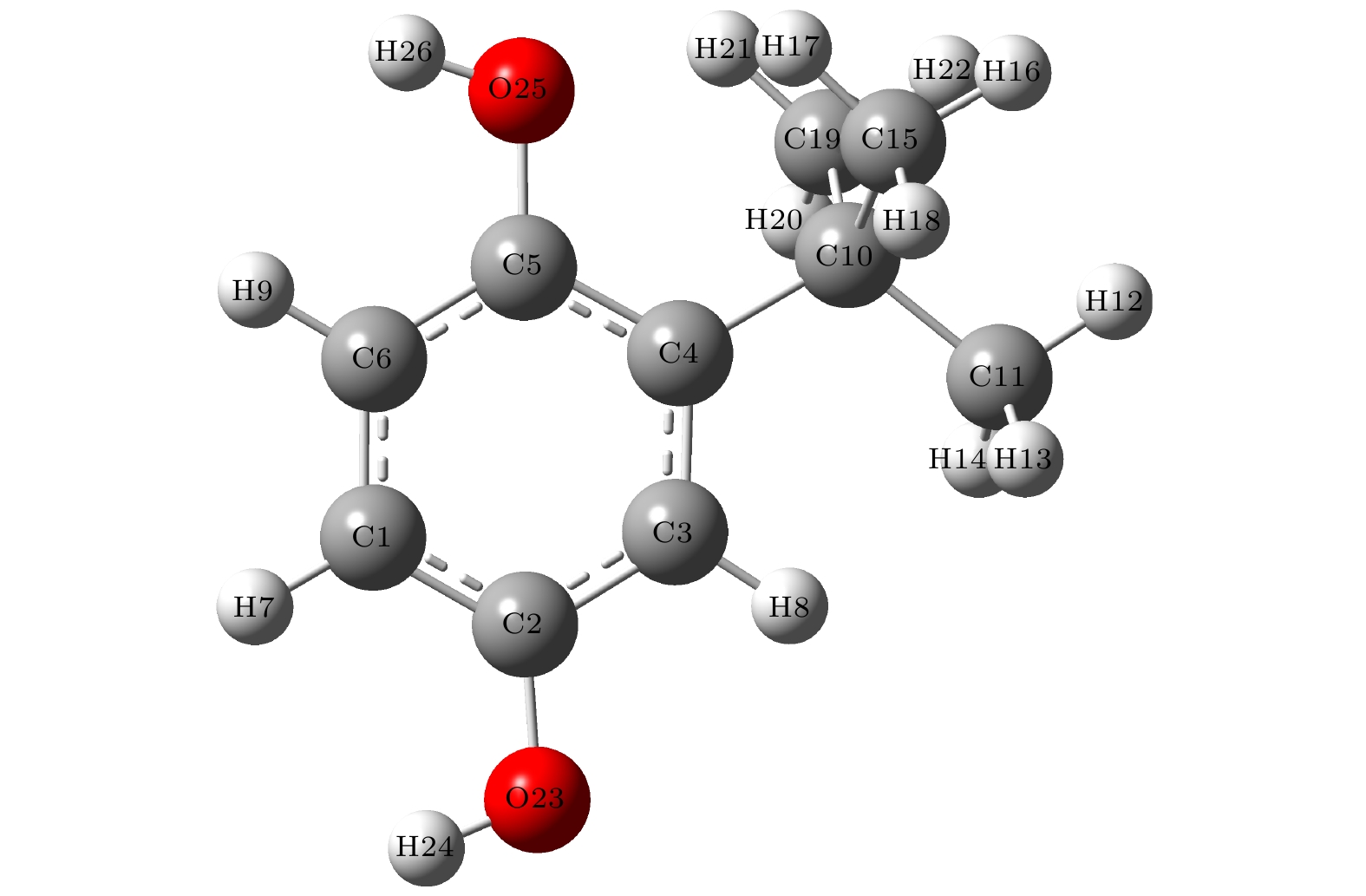 图 1 TBHQ分子几何构型
图 1 TBHQ分子几何构型Figure1. Geometric structure of TBHQ molecule.
| 键长 | R(2, 23) | 0.13725 nm |
| R(4, 10) | 0.15417 nm | |
| R(5, 25) | 0.13791 nm | |
| 键角 | A(2, 23, 24) | 109.0769o |
| A(5, 25, 26) | 108.6475o | |
| A(4, 10, 11) | 111.9551o | |
| 二面角 | D(3, 4, 10, 11) | 0.0003o |
表2TBHQ分子主要键长、键角、二面角
Table2.The main bond length, bond angle, and dihedral angle of TBHQ.
2
3.2.红外光谱
在结构优化的相同级别下, 对TBHQ分子的频率进行计算, 无虚频. 再利用Multiwfn 3.7软件对所有频率进行校正, 校正因子为0.967, 并绘制气相下4000—400 cm–1的红外光谱. 实验上, 利用KBr压片法测定红外光谱. 其中, 计算光谱为图中的红色谱线, 对应于左边Y轴的摩尔吸收系数, 实验光谱为图中的蓝色谱线, 对应于右边Y轴的透过率. TBHQ的红外光谱如图2所示, 其特征吸收峰的振动分析如表3所示. 图 2 TBHQ红外光谱
图 2 TBHQ红外光谱Figure2. IR spectra of TBHQ.
| 序号 | 计算光谱/cm–1 | 实验光谱/cm–1 | 振动分析 |
| 1 | 3709.24 | 3288.54 | 酚羟基的O—H伸缩振动 |
| 2 | 3034.34 | 3001.16 | 苯环上C—H伸缩振动 |
| 3 | 2989.00 | 2962.58 | 丁基上C—H伸缩振动 |
| 4 | 1581.86 | 1589.30 | 苯环的环伸缩振动 |
| 5 | 1483.16 | 1485.15 | 苯环的环伸缩振动及C—H弯曲振动 |
| 6 | 1440.48 | 1442.72 | C—H弯曲振动 |
| 7 | 1284.43 | 1309.63 | C—O伸缩振动及C—H弯曲振动 |
| 8 | 1169.72 | 1184.26 | O—H面内弯曲振动及C—H面内弯曲振动 |
| 9 | 917.64 | 933.52 | C—H弯曲振动 |
| 10 | 757.59 | 771.51 | 苯环上C—H面外弯曲振动 |
表3TBHQ分子红外光谱振动分析
Table3.Vibration analysis of IR spectra of TBHQ.
通过对TBHQ理论光谱和实验光谱的对比分析可知特征吸收峰整体吻合较好. 位于3001.16 cm–1处苯环上的C—H伸缩振动, 1589.30, 1485.15 cm–1处苯环的环伸缩振动和771.51 cm–1处苯环上C—H面外弯曲振动的吸收峰为芳烃的特征吸收峰; 2962.58 cm–1处C—H伸缩振动和1442.72 cm–1处C—H弯曲振动为烷烃的特征吸收峰; 3288.54 cm–1处O—H伸缩振动和1309.63 cm–1处C—O伸缩振动为酚的特征吸收峰. 2358.88 cm–1处的小吸收峰可能是由于样品中含有碳氮三键的杂质引起. 而理论光谱和实验光谱在3000 cm–1以上的区域存在较大差异, 可能是由于分子间氢键的作用造成.
为了探究TBHQ分子之间是否形成氢键[25], 首先利用genmer软件结合molclus1.9.6软件对该分子的二聚体构型进行搜索, 并得到15个构型, 再调用Gaussian16软件, 利用B3LYP-D3(BJ)泛函, 结合6-311G(d, p)基组对15个二聚体构型进行结构优化, 并在M062X-D3/6-311+G(2d, p)下计算它们的单点能[23]. 15个二聚体能量如图3所示. 再通过Multiwfn 3.7软件结合VMD软件[26]绘制出TBHQ二聚体的RDG函数等值面图[24,27]. 通过分析可知, 二聚体3, 8, 10, 13和15都存在分子间氢键的作用, 它们的RDG函数等值面图如图4所示. 图中, 等值面为蓝色区域表示氢键、强卤键等强吸引作用, 绿色区域表示范德瓦耳斯作用, 红色区域表示在环、笼中出现较强的位阻效应等强互斥作用[23].
 图 3 TBHQ二聚体的单点能
图 3 TBHQ二聚体的单点能Figure3. Conformational energy of the dimer.
 图 4 TBHQ二聚体RDG函数等值面图 (a) 二聚体3; (b) 二聚体8; (c) 二聚体10; (d) 二聚体13; (e) 二聚体15
图 4 TBHQ二聚体RDG函数等值面图 (a) 二聚体3; (b) 二聚体8; (c) 二聚体10; (d) 二聚体13; (e) 二聚体15Figure4. RDG function isosurface map of dimer: (a) Dimer 3; (b) dimer 8; (c) dimer 10; (d) dimer 13; (e) dimer 15.
二聚体8和10的能量较低, 为–1079.7784744和–1079.7784757 Hartree, 且二聚体8和10的总均方根位移(root mean square displacement/deviation)为0.00004117 nm, 它们的结构十分相似, 极可能为TBHQ二聚体的稳定结构, 因此, 做出二聚体10的红外光谱如图5所示. 而二聚体3和13的结构也较为相似. 通过对图4进行分析可知, TBHQ二聚体RDG函数等值面在两个羟基之间形成蓝色等值面, 即形成了分子间氢键. 通过对比单分子的红外光谱可知, 二聚体的红外光谱在3563.85 cm–1处出现一个吸收峰, 由振动分析可知, 其主要是由于形成分子间氢键的两个酚羟基的O—H伸缩振动形成, 而3709.24 cm–1处的吸收峰为未形成分子间氢键的酚羟基的O—H伸缩振动形成. 所以, 分子间氢键的形成削弱了O—H键的强度, 导致吸收频率降低. 不同的分子间氢键使O—H键的削弱程度不同, 在叠加效果下使得O—H伸缩振动在红外光谱3670—3070 cm–1处出现一个宽峰. 而理论光谱是模拟气相环境下单分子的红外光谱, 并不存在分子间氢键的作用, 导致理论光谱和实验光谱在3000 cm–1以上的区域存在较大差异.
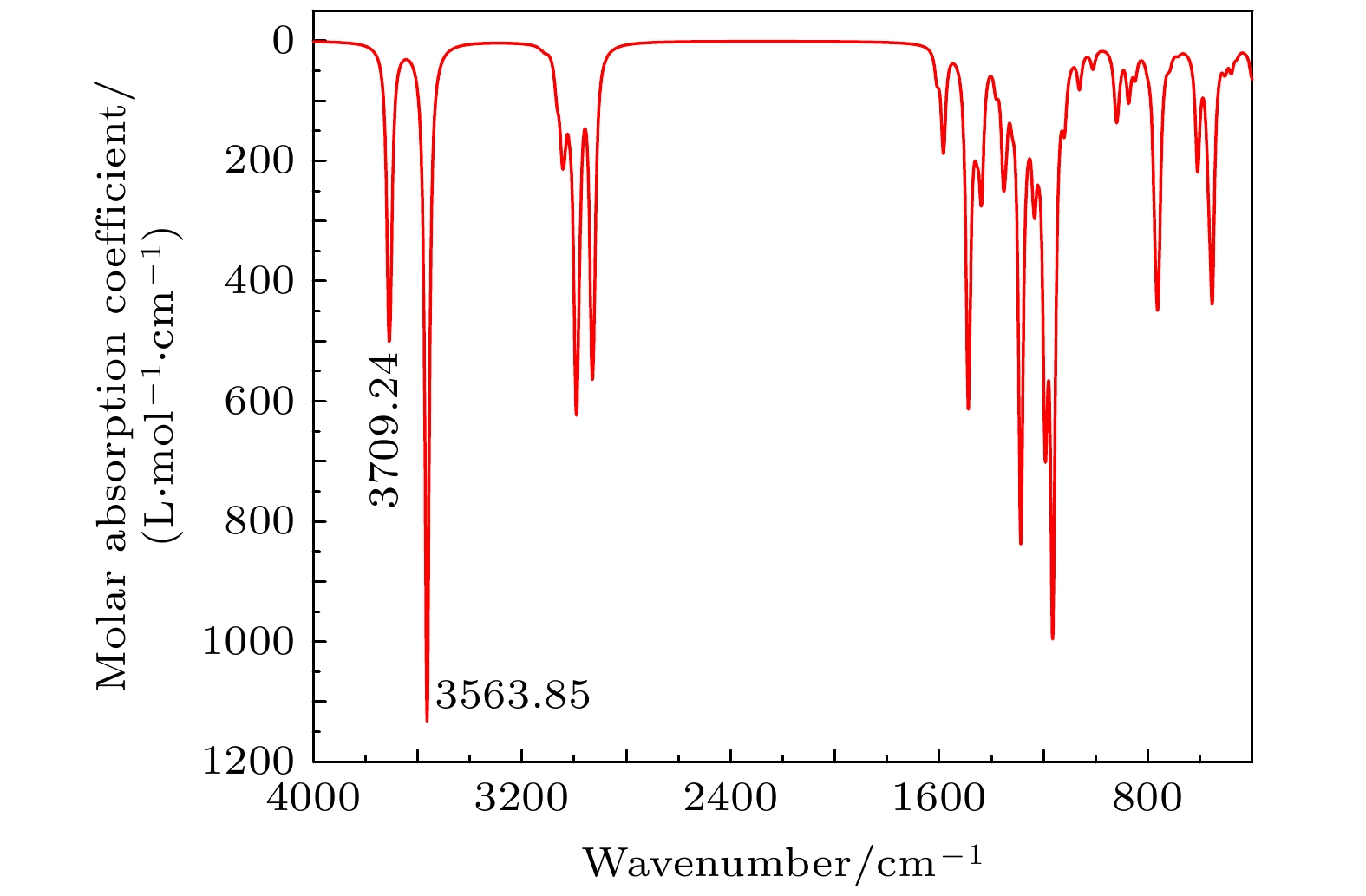 图 5 二聚体10的红外光谱
图 5 二聚体10的红外光谱Figure5. IR spectrum of dimer 10.
2
3.3.紫外光谱
基于TDDFT, 在B3LYP/def2-TZVP级别下, 利用SMD溶剂模型, 计算分子在无水乙醇溶剂中前50个激发态并绘制紫外光谱, 再利用Multiwfn 3.7软件分析主要激发态的分子轨道特征和电子-空穴图[16]. 实验上, 利用液相法将样品溶于乙醇溶液并测定紫外光谱. 为了更好的将理论光谱和实验光谱进行对比, 本文仅关注200—340 nm波段振子强度大于0.05的激发态, TBHQ分子的紫外光谱如图6和7所示, 激发性质如表4所示.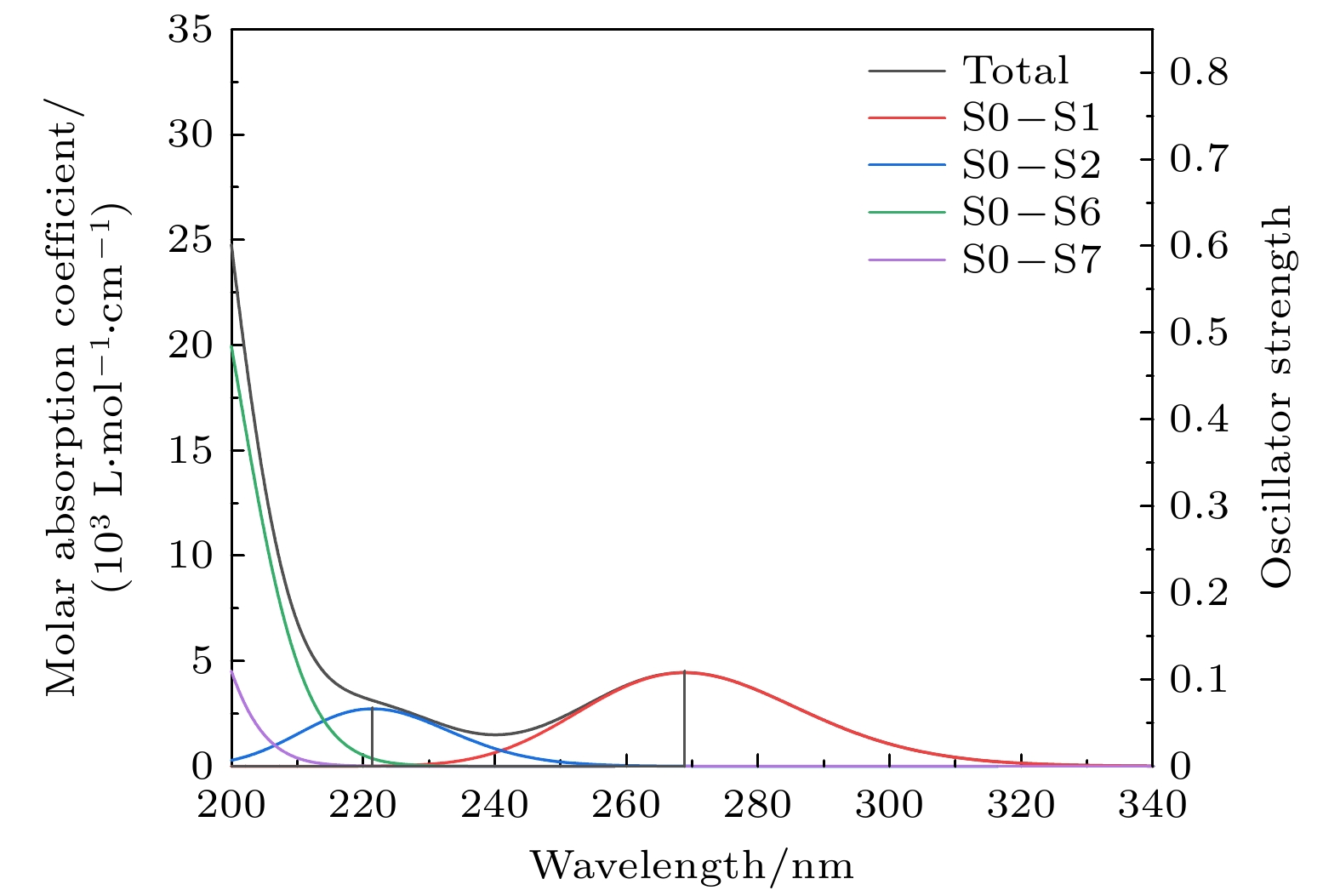 图 6 TBHQ分子理论紫外光谱
图 6 TBHQ分子理论紫外光谱Figure6. Theoretical UV spectrum of TBHQ.
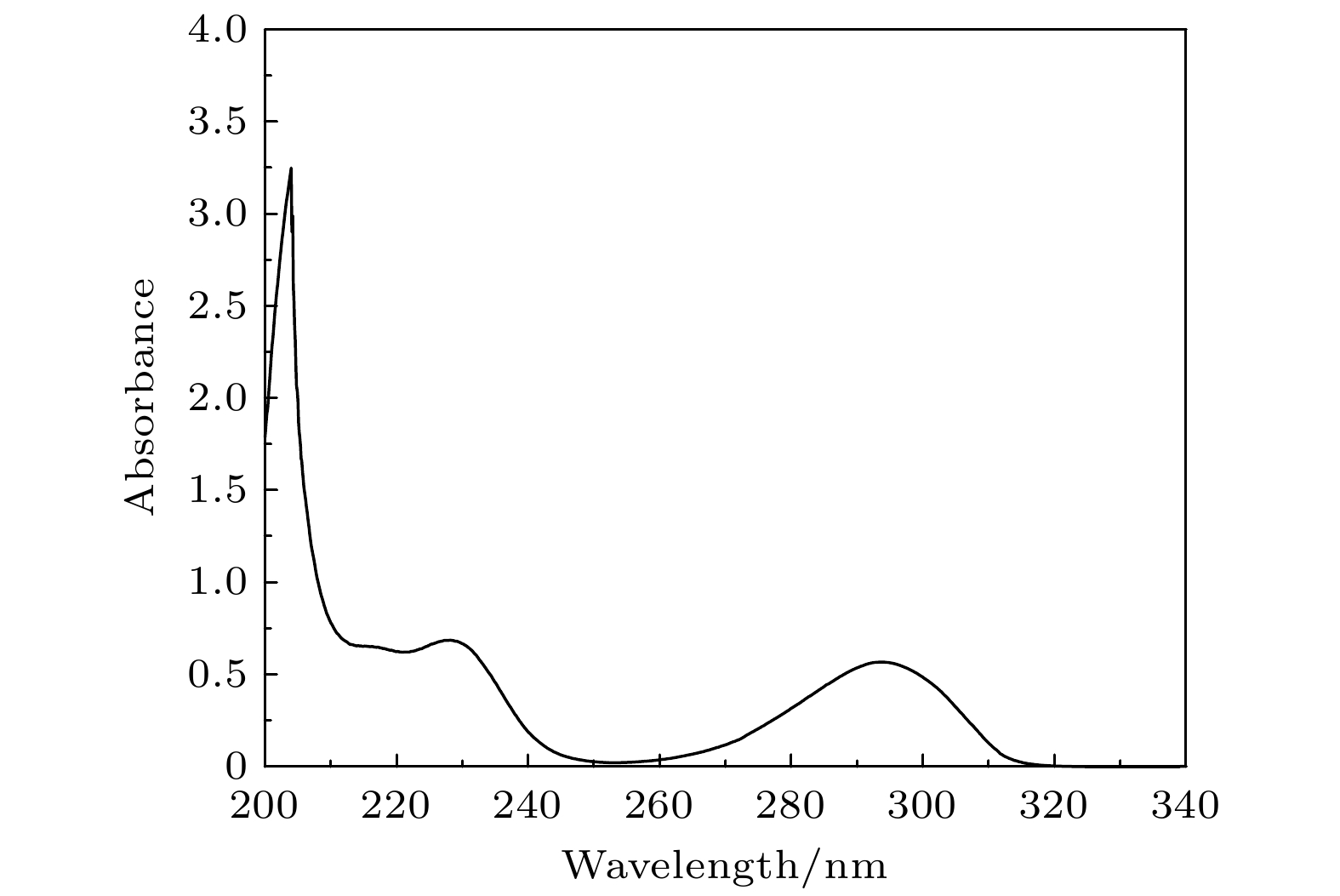 图 7 TBHQ分子实验紫外光谱
图 7 TBHQ分子实验紫外光谱Figure7. Experimental UV spectrum of TBHQ.
| 理论吸收峰值 | 实验吸收峰值 | 激发态 | 激发能/eV | 波长/nm | 振子强度 | 轨道跃迁 | 贡献权重/% |
| 221.4 nm 268.8 nm | 229.1 nm 293.7 nm | 1 | 4.6118 | 268.84 | 0.1100 | 45→46 44→47 | 91.91 7.86 |
| 2 | 5.6003 | 221.39 | 0.0676 | 45→47 44→46 | 78.65 20.57 | ||
| 6 | 6.4329 | 192.73 | 0.6902 | 44→46 45→47 | 77.09 20.07 | ||
| 7 | 6.7109 | 184.75 | 0.5669 | 44→47 45→46 | 89.30 7.24 |
表4TBHQ分子的激发性质
Table4.Excited state properties of TBHQ.
通过对TBHQ紫外光谱的对比分析可知, 理论和实验光谱整体吻合较好. 理论光谱最大吸收峰位于200 nm以下的真空紫外区, 200 nm以上有两个小的吸收峰, 分别位于221.4 nm和268.8 nm, 主要由基态跃迁至1, 2, 6, 7激发态形成. 其中, 电子由第45轨道(最高占据轨道)激发到第46轨道(最低空轨道)对S0—S1激发的贡献较大; 电子由第45轨道激发到第47轨道对S0—S2激发的贡献较大; 电子由第44轨道激发到第46轨道对S0—S6激发的贡献较大; 电子由第44轨道激发到第47轨道对S0—S7激发的贡献较大. 通过绘制以上4个轨道的波函数等值面图, 对激发特征进行分析, 如图8所示. 其中, 轨道波函数正相位用红色等值面表示, 负相位用蓝色等值面表示, 等值面大小未标注时皆为0.05. 通过分析可知, 第44轨道的苯基上具有π轨道特征, 丁基上的C—C键具有σ轨道特征; 第45轨道具有π和n轨道特征, 当等值面大小为0.18时n轨道特征更明显; 第46轨道表现出π*轨道特征; 第47轨道表现出n和π*轨道特征, 当等值面大小为0.14时π*轨道特征更明显. 因此, 268.8 nm处的吸收峰为n→π*和π→π*跃迁形成; 221.4 nm处的吸收峰为n→π*和π→π*跃迁形成; 200 nm以下的吸收峰为π→π*和σ→π*跃迁形成. 由实验光谱可知, 其最大吸收峰同样位于200 nm以下, 在229.1 nm和293.7 nm处有吸收峰, 而靠近200 nm附近出现了尖峰, 主要是由于该波段接近真空紫外区, 受制于仪器自身测量范围和精度的影响所造成的误差. 通过对上述光谱的分析可知, 实验光谱相对理论光谱出现轻微红移, 一方面可能是由于乙醇溶剂的极性造成了π→π*跃迁谱带向长波方向位移, 另一方面, 可能是由于理论光谱是模拟单分子在溶剂下的光谱情况, 而实验光谱是较多分子在溶剂下形成的光谱.
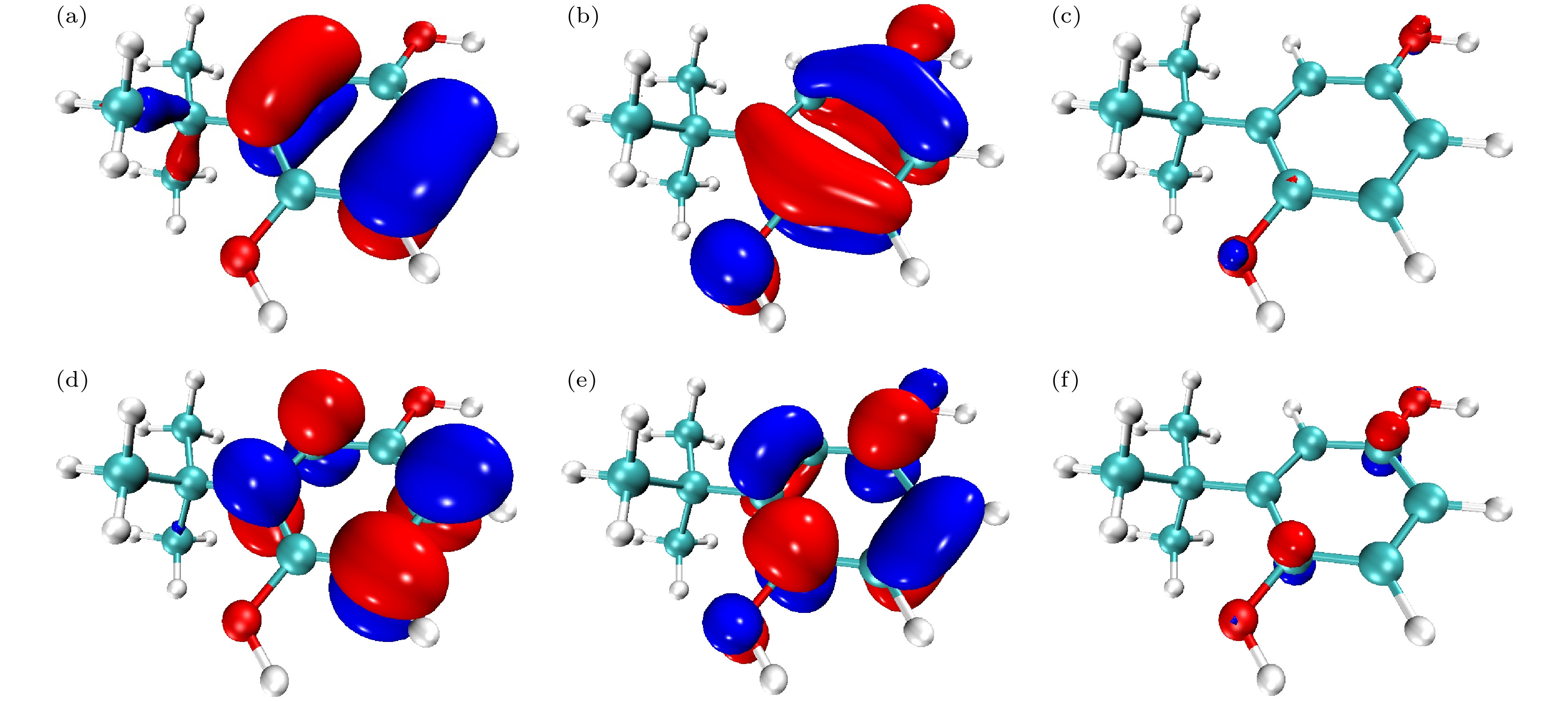 图 8 分子轨道波函数等值面图 (a) 第44轨道; (b) 第45轨道; (c) 第45轨道(等值面为0.18); (d) 第46轨道; (e) 第47轨道; (f) 第47轨道(等值面为0.14)
图 8 分子轨道波函数等值面图 (a) 第44轨道; (b) 第45轨道; (c) 第45轨道(等值面为0.18); (d) 第46轨道; (e) 第47轨道; (f) 第47轨道(等值面为0.14)Figure8. Isosurface of molecular orbital wavefunction: (a) The 44th molecular orbital; (b) the 45th molecular orbital; (c) the 45th molecular orbital (isosurface = 0.18); (d) the 46th molecular orbital; (e) the 47th molecular orbital; (f) the 47 th molecular orbital (isosurface = 0.14).
通过对TBHQ紫外光谱的分析可知, 在200—340 nm波段, 其主要由基态跃迁至第1, 2, 6, 7激发态形成, 因此对这4个激发做电子空穴分析. 通过Multiwfn3.7结合VMD软件, 分别做出它们的电子空穴图, 如图9所示, 其等值面值均为0.002. 其中, 绿色区域为电子等值面, 蓝色区域为空穴等值面. 表5为相关激发的电子空穴相关参数. 其中, Sr衡量电子和空穴的重叠程度, 最大值1说明完全重叠, 最小值0说明完全不重叠; D指数衡量电子与空穴质心之间的距离; H指数是衡量电子与空穴的总体平均分布广度; t指数是衡量电子与空穴的分离程度, t > 0说明电子与空穴分离较为充分, t < 0 说明电子与空穴没有充分分离; 空穴离域指数(hole delocalization index, HDI)和电子离域指数(electron delocalization index, EDI)分别衡量空穴和电子的离域情况, 它们的数值越小, 离域程度越高, 分布的均匀程度越大[23]. 通过结合图形和相关参数分析可知, 这4个激发的电子和空穴主要分布于苯环和酚羟基上, Sr指数都较大, 尤其是S0—S2和S0—S6激发的电子和空穴几乎完全重叠; 而D指数较小, 说明电子与空穴质心很接近; t指数都为负值, 说明电子和空穴没有充分分离; H指数都较为接近, 说明电子和空穴的总体分布广度都差不多, 基本都覆盖于体系中的苯环和酚羟基上; 这4个激发的HDI和EDI值并不是很大且都较为接近, 说明空穴和电子的离域特征明显, 离域于整个苯环上. 通过以上分析可知, 这4个激发为电子局域激发.
 图 9 TBHQ的电子空穴图 (a) S0—S1; (b) S0—S2; (c) S0—S6; (d) S0—S7
图 9 TBHQ的电子空穴图 (a) S0—S1; (b) S0—S2; (c) S0—S6; (d) S0—S7Figure9. Electron-hole distribution diagram of TBHQ: (a) S0–S1; (b) S0–S2; (c) S0–S6; (d) S0–S7.
| Sr/a.u | D/nm | H/nm | t/nm | HDI | EDI | |
| S0—S1 | 0.789 | 0.0075 | 0.2115 | –0.1259 | 9.63 | 8.81 |
| S0—S2 | 0.917 | 0.0034 | 0.2130 | –0.1292 | 8.81 | 8.72 |
| S0—S6 | 0.913 | 0.0271 | 0.2139 | –0.1134 | 8.00 | 7.88 |
| S0—S7 | 0.762 | 0.0223 | 0.2127 | –0.1179 | 8.63 | 9.39 |
表5电子空穴相关参数
Table5.Electron-hole distribution related parameters of TBHQ.
通过对TBHQ光谱的对比研究, 可以充分了解红外、紫外光谱的形成机理, 并充分了解TBHQ的分子结构和激发性质. 这对了解、改造以及检测食品抗氧化剂TBHQ具有重要意义.
