全文HTML
--> --> -->目前对磷烯纳米带磁性的研究也有一些报道, 例如, Du等[31]研究了裸边ZPNR在4种磁构型下的磁性, 这4种磁构型分别为: 铁磁(FM)αα-αα、反铁磁(AFM-1)αα-ββ、反铁磁(AFM-2)αβ-αβ、反铁磁(AFM-3)αβ-αβ, 研究表明裸边ZPNR的FM 态和AFM-1态无磁性, 而AFM-2态有磁性. Zhu等[32]发现边缘氢饱和的ZPNR没有磁性. Ding等[33]发现OHO 和 OH钝化的ZPNR和APNR具有磁性. Ren等[34]发现边缘用OH/NO2 (或NH2/NO2)与O按2∶1比率的饱和ZPNR可实现自旋极化半金属. Zhou等[30]发现在裸边APNR的边缘掺杂C, O, Si和S原子, APNR有磁性. 然而对磷烯纳米带如何获得半金属, 如何诱发高性能的磁性, 以及诱发磁性的机理等仍需进一步研究.
本文采用基于密度泛函理论(DFT)的第一性原理, 探讨掺杂过渡金属铁(Fe)、钴(Co)和镍(Ni)原子的ZPNR的磁电子学特性, 以及掺杂位置对其影响. 研究表明, 在ZPNR中掺杂不同的杂质表现出不同的特性, 有的有磁性, 有的没有磁性, 提出了在ZPNR中实现半金属的一种新方法, 并且掺杂位置对ZPNR的磁电子学特性有一定的影响. 这些研究结果对于发展基于磷烯的纳米电子器件具有重要意义.
 图 1 掺杂ZPNR的模型结构
图 1 掺杂ZPNR的模型结构Figure1. The geometric structure of doped ZPNRs.
采用基于密度泛函理论(DFT)的第一性原理的ATK(Atomistix ToolKit)软件包对几何结构进行优化和计算电磁性质, 目前这个软件包已被广泛用于纳米结构的研究中[35-38]. 模型结构在非磁状态下进行优化. 结构优化收敛的标准是作用在每个原子上的力小于0.01 eV/?. 为了求解Kohn-Sham 方程, 交换关联势采用广义梯度近似(GGA)中的PBE近似, 赝势采用模守恒赝势, 基函数组选用DZP (double

3.1.结构和稳定性分析
为了便于说明掺杂ZPNR的结构, 在图1(a)中对杂质原子附近的P原子进行了编号, 分别为1—7. 相应的P原子用Pi表示, 其中i = 1, 2, 3, 4, 5, 6, 7. 由图1可见, 取代掺杂后, 杂质原子附近的ZPNR结构发生了一些形变, 最明显的变化就是杂质原子与周边5个P原子(P1—P5)成键, 而原始ZPNR的P原子只与相邻3个P原子成键. 杂质原子与周边5个P原子之间的键长如表1所示, 表中d1—d5分别为杂质原子与其邻近的5个磷原子P1—P5的键长. 作为对比, 表中还列出了未掺杂ZPNR的相关参数. 由表1可见, 未掺杂ZPNR 的d1, d2和d3相等, 都为2.25 ?, 与以前的计算结果一致[11-13]. 掺杂ZPNR 的键长与未掺杂ZPNR的键长相比略有增减, 但相差不大, 其中Ni-ZPNR 的d4和d5增加幅度最大, 大约增加了10%, 而其余键长的增减幅度不足4%. 掺杂后, 杂质原子与P4, P5原子成键, 结果导致杂质原子略微向纳米带对称中心移动.| system | d1/? | d2/? | d3/? | d4/? | d5/? | Eb/eV | ENM–EFM/meV |
| ZPNR | 2.25 | 2.25 | 2.25 | — | — | –5.65 | 0 |
| Fe-ZPNR | 2.27 | 2.27 | 2.17 | 2.32 | 2.32 | –5.69 | 130.11 |
| Co-ZPNR | 2.22 | 2.22 | 2.25 | 2.32 | 2.32 | –5.68 | 0 |
| Ni-ZPNR | 2.26 | 2.26 | 2.25 | 2.50 | 2.48 | –5.65 | 7.23 |
表1掺杂和未掺杂ZPNR的键长、结合能和总能差
Table1.The bond lengths, binding energy, and total energy difference of doped and pristine ZPNRs.
为了研究取代掺杂结构的能量稳定性, 分别取各掺杂ZPNR的超原胞计算结合能Eb, 其定义为[39-41]
为了进一步研究取代掺杂结构的能量稳定性, 分别取各掺杂ZPNR的超原胞计算吉布斯自由能EG, 其定义为[42]
为了检验取代掺杂结构的热稳定性, 我们在几何优化后对其进行了Forcite退火分子动力学模拟[43]. 整个模拟过程使用4个退火循环, 并使用微正则系综. 每个退火循环由初始温度300 K升高至最高温度500 K, 然后冷却至初始温度. 整个模拟过程运行8000步, 总时间为8 ps. 模拟退火后的几何结构如图2所示, 图2(a)—(c)分别为Fe-ZPNR, Co-ZPNR和Ni-ZPNR. 与退火前的ZPNR相比, 杂质原子与周边P原子间的键长略有变化, 但没有产生局部重建. 这表明所有掺杂ZPNR的热稳定性很好.
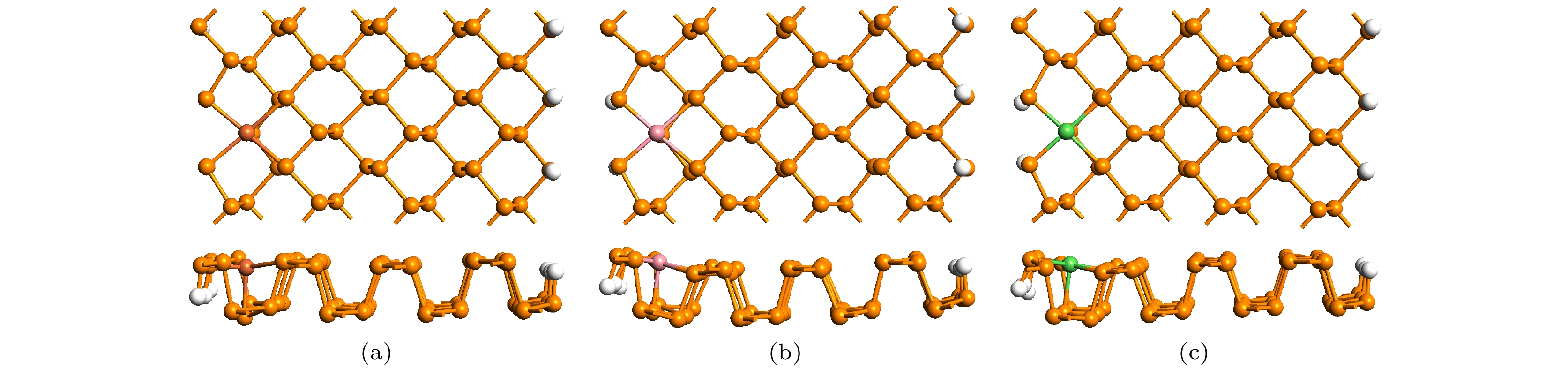 图 2 模拟退火后的模型结构 (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNR
图 2 模拟退火后的模型结构 (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNRFigure2. The geometric structure after anneal simulation: (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNR.
2
3.2.掺杂ZPNR的磁电子学特性
我们首先对掺杂ZPNR进行自旋非极化计算, 其对应的非磁(NM)态的能带结构和态密度(DOS)如图3(a)和图3(b)所示. 图中Fe, Co和Ni分别表示Fe-ZPNR, Co-ZPNR和Ni-ZPNR, 而ZPNR表示未掺杂的ZPNR. 图中虚线表示费米能级, 设为零. 由图3可见, 未掺杂的ZPNR没有能带穿越费米能级, 且在费米能级附近一定能量范围内, ZPNR的DOS为零, 故未掺杂的ZPNR为半导体. 未掺杂的ZPNR的导带底和价带顶都位于Γ点, 故为直接带隙半导体, 其带隙大小为1.51 eV, 这与以前的研究结果是一致的[20-24]. 由图3(a)可见, 掺杂ZPNR都由于掺杂而引入了两条能带, 图中用a, b进行了标示. Fe-ZPNR和Ni-ZPNR各有一条能带(即能带a)穿越费米能级, 而Co-ZPNR没有能带穿越费米能级. 由图2(b)可见, Fe-ZPNR和Ni-ZPNR的DOS在费米能级处都不为零, 而Co-ZPNR的DOS在费米能级处为零. 故当处于NM态时, Fe-ZPNR和Ni-ZPNR为金属, 而Co-ZPNR依然为半导体. Co-ZPNR为间接带隙半导体, 其带隙大小为1.12 eV, 小于未掺杂的ZPNR的带隙. 由此可见, 掺杂Fe或者Ni原子将ZPNR由半导体转变为金属, 而掺杂Co原子则将ZPNR由直接带隙半导体转变成为间接带隙半导体, 且带隙减小.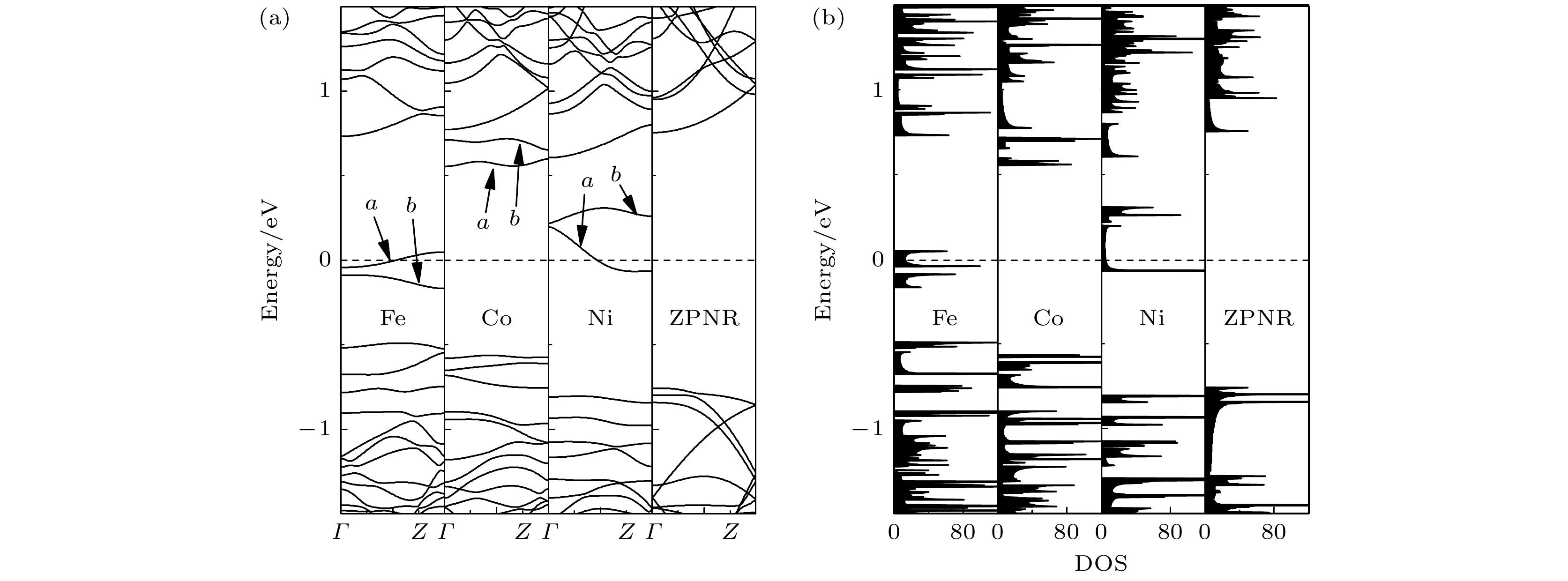 图 3 ZPNR处于NM态的 (a) 能带结构和 (b) 态密度
图 3 ZPNR处于NM态的 (a) 能带结构和 (b) 态密度Figure3. (a) The band structure and (b) density of states of ZPNRs in the nonmagnetic state.
为了更清楚地分析杂质原子对能带结构的影响, 图4绘出了掺杂ZPNR能带结构中能带a的电荷密度图, 图4(a)—(c)分别为Fe-ZPNR, Co-ZPNR和Ni-ZPNR, 等值面取0.3 |e|/?3. 由图4可见, 掺杂ZPNR的能带a由杂质原子起主要作用, 而起次要作用的是杂质原子附近的几个P原子. 由此可见, 能带a正是由于掺杂而引入的杂质能带, 并导致Fe-ZPNR和Ni-ZPNR由半导体转变为金属, 而Co-ZPNR的带隙小于未掺杂的ZPNR的带隙.
 图 4 部分能带的电荷密度 (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNR
图 4 部分能带的电荷密度 (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNRFigure4. The charge density of partial band: (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNR.
为了更进一步分析杂质原子中哪个轨道的电子起主要作用, 图5绘出了掺杂ZPNR的投影态密度(PDOS)图, 图5(a)—(c)分别为Fe-ZPNR, Co-ZPNR和Ni-ZPNR. 图中的Fe, Co和Ni分别表示投影到Fe, Co和Ni原子的PDOS, 而s, p和d则分别表示投影到相应杂质原子的s, p和d轨道的PDOS. 可见, 杂质原子的PDOS主要由其d轨道的电子贡献, 即掺杂ZPNR的杂质原子对电子特性起主要作用的是d轨道的电子.
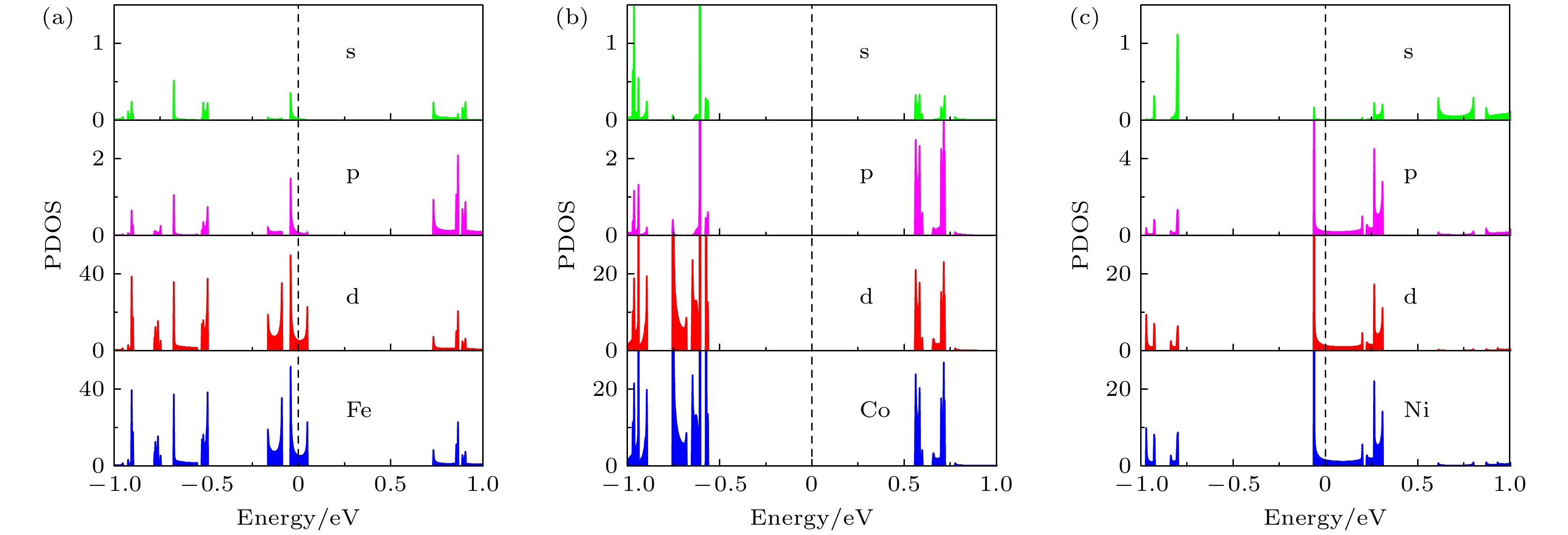 图 5 掺杂ZPNRs投影态密度 (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNR
图 5 掺杂ZPNRs投影态密度 (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNRFigure5. The partial density of states of ZPNRs: (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNR.
现在我们对掺杂ZPNR进行自旋极化计算以研究掺杂诱发的磁性. 定义NM态与铁磁(FM)态之间的总能差为

为了分析掺杂ZPNR的磁电子学特性, 图6展示了掺杂ZPNR处于FM态的能带结构和态密度, 图6(a)—(d)分别为未掺杂ZPNR, Fe-ZPNR, Co-ZPNR和Ni-ZPNR. 图6中蓝色实线和红色实线分别表示α和β自旋(下同). 由图6(a)和图6(c)可见, 未掺杂ZPNR和Co-ZPNR的能带是简并的, 没有能带穿越费米能级, 在费米能级附近一定能量范围内, ZPNR的DOS为零, 故未掺杂ZPNR和Co-ZPNR为半导体, 其带隙大小分别为1.51和1.12 eV. 能带简并这一特性结合总能差ΔE = 0, 进一步表明未掺杂ZPNR和Co-ZPNR没有磁性. 未掺杂ZPNR没有磁性, 这一结果与之前的研究结果[32]是一致的. 由图6(b)和图6(d)可见, Fe-ZPNR和Ni-ZPNR在费米能级附近的能带都发生了分裂, 表明Fe-ZPNR和Ni-ZPNR具有磁性. Fe-ZPNR的α和β自旋都没有能带穿越费米能级, 且在费米能级处的态密度都为零, 故Fe-ZPNR为自旋半导体, 且为半-半导体. Fe-ZPNR的α和β自旋的导带底和价带顶都位于Γ点, 故都为直接带隙, 其带隙大小分别为0.87和0.13 eV. Ni-ZPNR的β自旋没有能带穿越费米能级, 但α自旋有能带穿越费米能级, 且在费米能级处β自旋的态密度不为零, 而α自旋的态密度为零, 故Ni-ZPNR为半金属. Ni-ZPNR的β自旋为间接带隙, 其带隙大小为0.90 eV. 可见, 掺杂Fe原子可使ZPNR由无磁性半导体转变为磁性半导体, 而掺杂Ni原子可使ZPNR由无磁性半导体转变为磁性半金属.
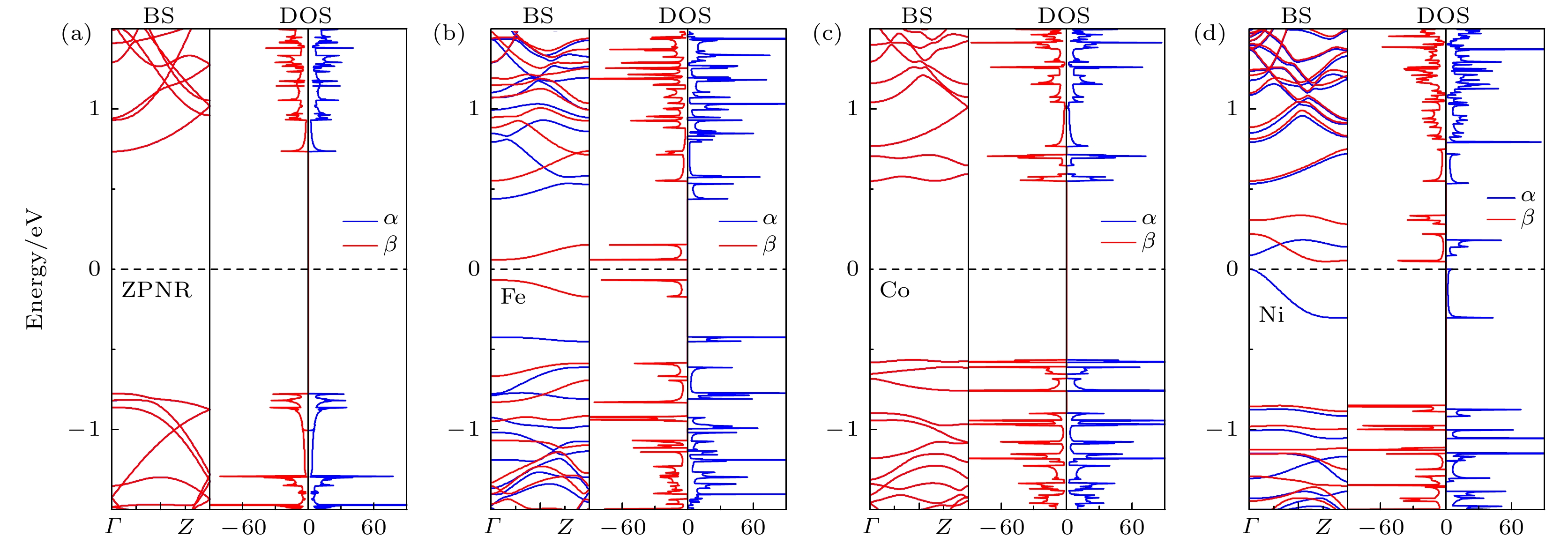 图 6 ZPNR处于FM态的能带结构和态密度 (a) ZPNR; (b) Fe-ZPNR; (c) Co-ZPNR; (d) Ni-ZPNR
图 6 ZPNR处于FM态的能带结构和态密度 (a) ZPNR; (b) Fe-ZPNR; (c) Co-ZPNR; (d) Ni-ZPNRFigure6. The band structure and density of states of ZPNRs in the ferromagnetic state: (a) ZPNR; (b) Fe-ZPNR; (c) Co-ZPNR; (d) Ni-ZPNR.
为了分析磁性的来源, 图7给出了掺杂ZPNR在FM态的自旋极化电荷密度等值面图, 图7(a)—(c)分别为Fe-ZPNR, Co-ZPNR和Ni-ZPNR. 图7中红色和蓝色分别代表α和β自旋, 等值面取0.02 |e|/?3. 自旋极化电荷密度



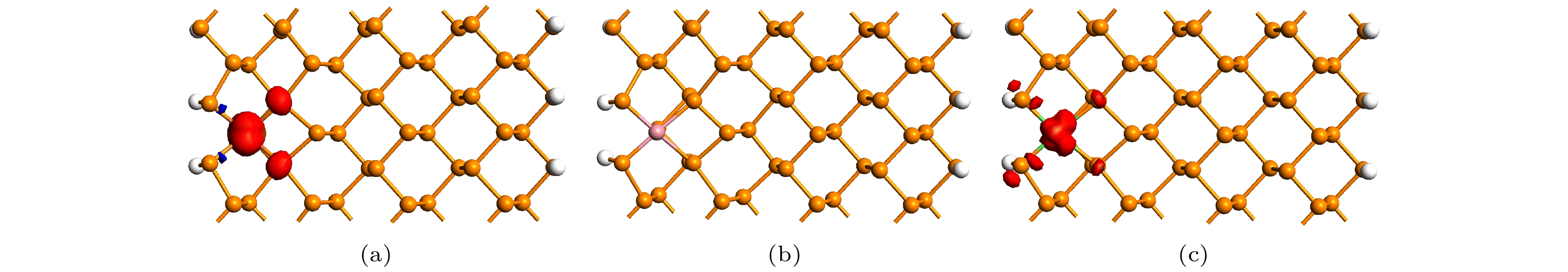 图 7 自旋极化电荷密度等值面图 (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNR
图 7 自旋极化电荷密度等值面图 (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNRFigure7. The isosurface plots of spin polarization charge density in the ferromagnetic state: (a) Fe-ZPNR; (b) Co-ZPNR; (c) Ni-ZPNR
对ZPNR磁矩计算结果表明, 未掺杂ZPNR和Co-ZPNR超原胞的总磁矩为0μB, Fe-ZPNR和Ni-ZPNR超原胞的总磁矩分别为1.00μB和0.89μB, μB为玻尔磁子. 磁矩计算结果同样表明, 对Fe-ZPNR和Ni-ZPNR的磁矩贡献最大的是杂质原子. Fe和Ni原子的磁矩分别为0.96μB和0.43μB, 分别对其超原胞的总磁矩贡献了96.0%和48.3%. 由此可进一步说明掺杂Fe-ZPNR和Ni-ZPNR的磁性主要由杂质原子贡献. 而未掺杂ZPNR和Co-ZPNR超原胞的总磁矩为零, 也进一步表明未掺杂ZPNR和Co-ZPNR没有磁性.
掺杂诱发ZPNR的磁性, 可做如下解释. 由于P原子最外层具有5个价电子, 每个P原子与周边3个P原子成键, 即有3个电子与周边原子的电子配对, 剩余2个电子配对. 按照能量最小原理, 原子内的电子先填充4s层, 再填充3d层. 对于Fe, Co和Ni原子来说, 它们的4s层都填充2个电子, 而3d层填充的电子数量分别为6, 7和8个. 由前面的分析知道, 在掺杂ZPNR中, 杂质原子与周边5个P原子成键, 且杂质原子对电子特性起主要作用的是d轨道的电子. Fe原子的3d层有6个电子, Fe原子与周边5个P原子成键, 剩下1个未配对电子. Co原子的3d层有7个电子, Co原子与周边5个P原子成键, 剩下的两个电子配对. 而Ni原子的3d层有8个电子, Ni原子与周边5个P原子成键, 剩余3个电子, 其中2个电子配对, 剩下1个未配对电子. 正是由于在ZPNR中掺杂Fe和Ni原子出现了未配对电子, 才导致Fe-ZPNR和Ni-ZPNR具有磁性. 而在ZPNR中掺杂Co原子没有出现未配对电子, 因此Co-ZPNR没有磁性.
2
3.3.掺杂位置对磁电子学特性的影响
为了研究掺杂位置对掺杂ZPNR磁电子学特性的影响, 在此以掺杂Ni原子为例进行分析, 其能带结构如图8所示, 图8(a)和图8(b)分别为NM态和FM态, 图8(b)中的插图为费米能级附近能带结构的局部放大图. 为了说明方便, 将Ni原子取代图1(a)中标注x, 6和7的P原子的ZPNR分别用Nix-ZPNR, Ni6-ZPNR和Ni7-ZPNR表示. 标注x, 6和7的P原子分别位于第1, 第2和第3条P原子链靠纳米带对称中心一侧的中间位置. 图8中的6和7分别代表Ni6-ZPNR和Ni7-ZPNR. 结合图3(a)和图8(a)可见, 当处于NM态时, 在ZPNR的3个不同位置掺杂Ni原子, 都在费米能级附近引入了两条杂质能带, 并且有一条穿越费米能级, 故ZPNR属性不变, 都为金属. 结合图6(d)和图8(b)可见, 当处于FM态时, 在ZPNR的3个不同位置掺杂Ni原子, 费米能级附近的能带都发生了分裂, 但呈现出不同的特性. Ni6-ZPNR的磁电子学特性与Nix-ZPNR相同, α自旋有能带穿越费米能级, 但β自旋没有能带穿越费米能级, 即呈现为半金属. Ni6-ZPNR的β自旋为直接带隙, 其带隙大小为0.74 eV. 但Ni7-ZPNR的磁电子学特性与Nix-ZPNR不同, 此时α和β自旋都没有能带穿越费米能级, 即呈现为磁性半导体. Ni7-ZPNR的α和β自旋都为间接带隙, 其带隙大小分别为0.16 eV和 0.74 eV. 由此可见, 当处于FM态时, 在靠近纳米带边缘位置掺杂Ni原子, ZPNR表现为磁性半金属, 而在靠近纳米带对称中心位置掺杂Ni原子, ZPNR则表现为磁性半导体.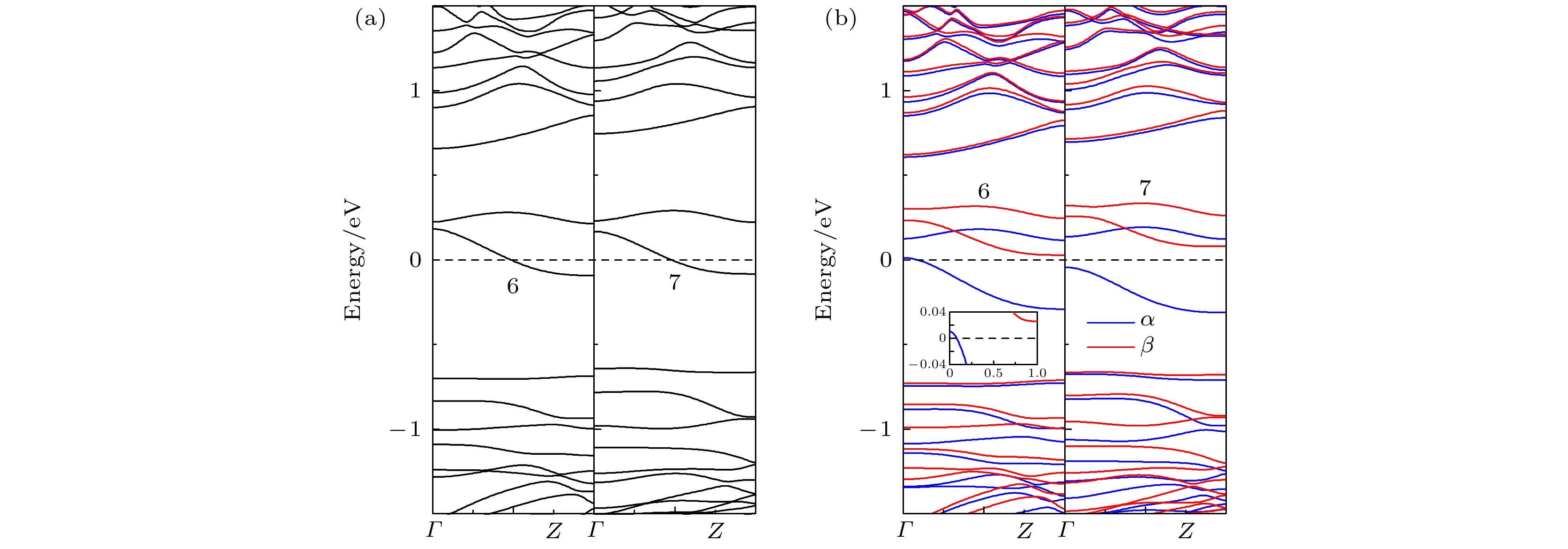 图 8 改变掺杂位置时ZPNR的能带结构 (a) NM; (b) FM
图 8 改变掺杂位置时ZPNR的能带结构 (a) NM; (b) FMFigure8. The band structure of ZPNRs with different doping position: (a) NM; (b) FM.
