1.Guangxi Collaborative Innovation Center of Structure and Property for New Energy and Materials, Guangxi Key Laboratory of Information Materials, School of Material Science and Engineering, Guilin University of Electronic Technology, Guilin 541004, China 2.School of Physics and Technology, Guangxi Normal University, Guilin 541004, China
Fund Project:Project supported by the National Key R&D Program of China (Grant Nos. 2018YFB1502103, 2018YFB1502105), the National Natural Science Foundation of China (Grant Nos. 51801041, 551671062, 5187011196, 51971068), the Natural Science Foundation of Guangxi, China (Grant No. 2017JJB150085), and the Science and Technology Project of Guangxi, China (Grant Nos. 2018AD19052, AA19182014, AD17195073, AA17202030-1)
Received Date:19 August 2020
Accepted Date:10 September 2020
Available Online:25 January 2021
Published Online:05 February 2021
Abstract:The point defect of two-dimensional hexagonal boron nitride (hBN) has recently been discovered to achieve single photon emission at room temperature, and it has become a research hotspot. Despite its important fundamental and applied research significance, the origin of the atomic structure of luminescence defects in hBN is still controversial. In this paper, first-principle calculations based on density functional theory are used to study a defect (CN)3VB in the hexagonal boron nitride monolayer (hBN) where three N atoms near the B vacancy are replaced by C atoms. At the B vacancy of hBN, the three N atoms each carry an in-plane dangling bond and the corresponding unpaired electron, and the unpaired electron can be eliminated by C substitution. We systematically study the geometric structure, electronic structure and optical properties of (CN)3VB defects, analyze the thermodynamic stability of defects through the calculation of the atomic structure, formation energy, and charge state of the defect, and analyze the position in the band gap and its atomic orbital contribution of defect state through energy band structure and wave function. We also analyze its optical properties through dielectric function and absorption coefficient, and predict its luminous photon energy. The results show that the defect can change from a symmetric metastable state to an asymmetric ground state structure with three C atoms connected together through atomic structure relaxation. The formation energy of asymmetric (CN)3VB is 7.94 eV, which is 3.72 eV lower than that of symmetric one. The formation of defects introduces some local defect states contributed by defect dangling σ bonds and reconstructed π bonds in hBN. The defects have valence states between –2 and +2, and the thermodynamic transition energy level of asymmetric (CN)3VB is higher than that of symmetric (CN)3VB. In the transition from the metastable state to the ground state, these defect states can redshift the light absorption boundary of hBN, enhance the absorption intensity of visible light by hBN, and cause internal optical transitions. Among them, there is a visible light transition with an energy threshold around 2.58 eV in the asymmetry (CN)3VB defect. Single boron atom vacancy defect and (CN)3VB have optical transitions near infrared and ultraviolet energy, respectively. The present work will help to further understand the composition and optical properties of point defects in hBN, and provide a theoretical basis for experimentally exploring the origin and properties of the atomic structure of light-emitting point defects. Keywords:hexagonal boron nitride/ carbon doping/ electronic structure/ single photon emission
计算零温零压时, hBN单层沿Brillouin区高对称点方向的能带结构, 选取费米能级EF附近–3—6 eV的能带结构(其中EF = 0), hBN单层价带的顶点在K点, 而导带的底点在$ \varGamma $点, 即价带的最高点与导带最低点不在同一点, 故hBN单层属于间接带隙. 这与Weston等[41]计算的结果一致. hBN单层的带隙宽度为4.366 eV, 大于2.2 eV, 因此可归为宽带隙半导体, 实验中的hBN的带隙约为6 eV[52,53], 与本文计算结果的差异在27%, 显然PBE计算低估了h-BN的带隙, 这是由于DFT计算的是基态近似的结果, 而在真实体系中的能隙属于激发态, 这种情况通常会出现在宽带隙半导体中, 但并不影响对hBN单层电子结构的分析和研究. Huang和Lee[50]通过Heyd-Scuseria-Ernzerhof(HSE)杂化泛函计算出纯净hBN 5.56 eV的带隙, 与实验结果更为接近. 本文主要研究hBN中碳掺杂缺陷的电子结构、光学性质等, 虽然HSE杂化泛函能够得到更接近实验值的带隙, 但是目前并没有研究证明对于缺陷能级的计算, HSE杂化泛函计算比PBE计算更加准确, 所以考虑到HSE杂化泛函的计算量比较大, 本文采用PBE计算. 本征hBN单层如图1所示, 其中图1(a)为本征hBN单层的俯视图和侧视图; 图1(b)为模拟的hBN单层表面扫描隧道显微镜(scanning tunneling microscope, STM)形貌, 由于N原子的电负性比B原子的电负性强, 所以电荷主要集中于N原子周围; 图1(c)是本征hBN单层的能带投影图, 可以看出, 价带的边缘由N原子的${{\rm{p}}_z}$轨道贡献, 而导带的边缘主要由B原子的${{\rm{p}}_z}$轨道贡献. 图 1 本征hBN单层 (a) hBN的俯视图和侧视图; (b) 模拟的hBN单层表面STM形貌, 加载电压为–2 V, 探针与原子表面的距离为0.479564 nm; (c) hBN的能带投影图 Figure1. Intrinsic hBN monolayer: (a) Top view and side view of hBN; (b) the simulated hBN single-layer surface STM morphology, loading voltage is –2 V, the distance between probe and atomic surface is 0.479564 nm; (c) energy band projection view of hBN.
23.2.hBN单层的硼空位缺陷 -->
3.2.hBN单层的硼空位缺陷
VB优化后的局部结构如图2(a)所示. 对称的硼空位(VB)处三个最近的N—N键长度均为2.647 ?, 比优化前增加了0.135 ?. 当打破VB的对称性后, 非对称硼空位(asymmetry VB, as-VB)如图2(d)所示, 自旋向上通道中的一个单一缺陷能级上升到间隙中, 自旋向下通道的简并能级分裂为两个单一能级. as-VB自旋电荷密度图如图2(f)spin所示, 由原来的自旋向下变为自旋向上和自旋向下. 在能量空间上, 图2(d)中标记有1, 2的缺陷能级主要由N原子的${{\rm{p}}_y}$轨道贡献, 标记有3的缺陷能级主要由N原子的${{\rm{p}}_x}$轨道所贡献. 在实空间上, $ \varGamma $点处波函数图中标记有1, 2, 3的缺陷能级均形成σ键类型, 这与能量空间上缺陷能级起源于空位处N原子的${{\rm{p}}_x}$和${{\rm{p}}_y}$轨道一致. 图 2 VB和as-VB的电子结构 (a), (c)分别为VB和as-VB优化后的局部结构俯视图; (b), (d)分别为VB和as-VB的能带投影图; (e), (f)分别为VB和as-VB的缺陷能级在Γ点处波函数的俯视图和自旋电荷密度图 Figure2. Electronic structure of VB and as-VB: (a), (c) The top views of the optimized partial structure of VB and as-VB, respectively; (b), (d) the energy band projection diagrams of VB and as-VB, respectively; (e), (f) the top view of the Γ-point wave functions of the defect levels and spin charge density of VB and as-VB, respectively.
在hBN单层中的每个B原子周围都有3个N原子. 当除去一个硼原子形成VB时, 会留下3个N ${\rm{2 s}}{{\rm{p}}^2}$和3个N ${\rm{2}}{{\rm{p}}_z}$的悬空键. 这些悬空键结合形成局部对称$({{\rm{a}}^{\rm{\sigma }}}, \;{{\rm{a}}^\pi })$和较高价态的反对称$({{\rm{e}}^{\rm{\sigma }}},\;{{\rm{e}}^\pi })$分子轨道. 由前面单个硼原子空位的能带投影图分析得出, 自旋通道中共有3个未占据的缺陷态, 每个缺陷态最多可以接受一个电子, 所以VB可能存在0至–3的电荷态. 图3(a)—(c)分别为${\rm{V}}_{\rm{B}}^{-1}$, ${\rm{V}}_{\rm{B}}^{-2}$, ${\rm{V}}_{\rm{B}}^{-3}$结构优化后的局部俯视图. 随着电荷数的增加, 库仑排斥力增强, N—N键的长度增加, 缺陷结构向外扩张. ${\rm{V}}_{\rm{B}}^{-1}$, ${\rm{V}}_{\rm{B}}^{-2}$, ${\rm{V}}_{\rm{B}}^{-3}$的能带投影图如图3(d)—(f)所示, 当VB处于–3价态时, 由缺陷能级进入到导带中, 所以$ {\rm{V}}_{\rm{B}}^{-3} $不能完全实现, VB的电荷态受近自由电子状态(nearly free electron, NFE)所影响[50]. 图 3 VB在不同价态的电子结构图 (a)?(c)分别为$ {\rm{V}}_{\rm{B}}^{-1} $, $ {\rm{V}}_{\rm{B}}^{-2} $, $ {\rm{V}}_{\rm{B}}^{-3} $优化后的局部结构俯视图; (d)?(f)分别为$ {\rm{V}}_{\rm{B}}^{-1} $, $ {\rm{V}}_{\rm{B}}^{-2} $, ${\rm{V}}_{\rm{B}}^{-3}$的能带投影图 Figure3. Electronic structure diagrams of VB in different valence states: (a)?(c) The top views of the optimized partial structure diagrams of $ {\rm{V}}_{\rm{B}}^{-1} $, $ {\rm{V}}_{\rm{B}}^{-2} $, and $ {\rm{V}}_{\rm{B}}^{-3} $, respectively; (d)?(f) the energy band projection diagrams of $ {\rm{V}}_{\rm{B}}^{-1} $, $ {\rm{V}}_{\rm{B}}^{-2} $, and $ {\rm{V}}_{\rm{B}}^{-3} $, respectively.
通过(1)式和(2)式计算在富氮和贫氮条件下, $ {\rm{V}}_{\rm{B}}^{0} $, $ {\rm{V}}_{\rm{B}}^{-1} $, $ {\rm{V}}_{\rm{B}}^{-2} $, $ {\rm{V}}_{\rm{B}}^{-3}$的形成能, 如图4所示. $ {\rm{V}}_{\rm{B}}^{0} $在富氮和贫氮条件下的形成能分别为7.58和10.11 eV. 此外, 还计算了富氮条件下VB的不同价态之间的热力学转变能级$\varepsilon ({q / {q'}})$, 其中$\varepsilon ({0 / { - 1}})$, $\varepsilon ({{ - 1} / { - 2}})$, $\varepsilon ({{ - 2} / { - 3}})$分别为0.36, 2.55, 3.44 eV, 我们的计算结果均与文献[50]接近. 图 4 在富氮和贫氮条件下, 不同价态的VB形成能为费米能级的函数 Figure4. Formation energies of VB with different valences as a function of Fermi level under the nitrogen-rich and nitrogen-poor conditions.
23.3.hBN单层碳掺杂的缺陷 -->
3.3.hBN单层碳掺杂的缺陷
由前面分析可知, 硼空位缺陷附近的3个氮原子(N 2s22p3)各自带一个在平面内的悬挂键及相应的未配对电子, 而在元素周期表中, 碳原子的价电子数比氮原子的价电子数少1个, 当用碳原子替换氮原子时, 可以消除缺陷中未配对的电子, 因此本文在VB的基础上, 采用碳原子取代硼空位处的3个氮原子, 构成(CN)3VB的缺陷. (CN)3VB优化后的局部结构俯视图如图5(a)所示, 图中C—C的键长均为1.943 ?, 这比未掺杂之前VB最近邻的N—N键长缩短了将近26.6%. 这主要是由于碳原子的电负性比氮原子的电负性弱, C替换N之后C-B的成键能力比B—N的成键能力弱, 所以C—B化学键较长, 从而导致空位附近的碳原子向空位靠拢. 图 5 (CN)3VB和as-(CN)3VB的电子结构 (a), (c)分别为(CN)3VB, as-(CN)3VB优化后局部结构的俯视图; (b), (d)分别为(CN)3VB, as-(CN)3VB的能带投影图; (e) (CN)3VB缺陷能级在Γ点处的波函数图和自旋电荷密度图; (f) as-(CN)3VB缺陷能级在Γ点处的波函数图 Figure5. Electronic structure of (CN)3VB and as-(CN)3VB: (a), (c) The top views of the optimized partial structure of (CN)3VB and as-(CN)3VB; (b), (d) the energy band projection diagrams of (CN)3VB and as-(CN)3VB; (e) the top view of the Γ-point wave functions of the defect levels and spin charge density of (CN)3VB; (f) the top view of the Γ-point wave functions of the defect levels of as-(CN)3VB.
图 8 在富氮和贫氮条件下, 不同价态的(CN)3VB形成能为费米能级的函数 Figure8. Formation energies of (CN)3VB with different valences as a function of Fermi level under the nitrogen-rich and nitrogen-poor conditions.
图 9 在富氮和贫氮条件下, 不同价态的as-(CN)3VB形成能为费米能级的函数 Figure9. Formation energies of as-(CN)3VB with different valences as a function of Fermi level under the nitrogen-rich and nitrogen-poor conditions.
$\varepsilon \left( w \right) = {\varepsilon _1}\left( w \right) + {\rm{i}}{\varepsilon _2}\left( w \right),$
其中$w$表示入射光的频率, ${\varepsilon _1}\left( w \right)$表示复介电函数的实部, ${\rm{i}}{\varepsilon _2}\left( w \right)$表示复介电函数的虚部, ${\rm{i}}$表示虚数单位. 本征hBN, VB, as-VB, (CN)3VB, as-(CN)3VB的复介电函数实部和虚部随入射光能量Ein的变化分别如图(10)(a)—(e)所示. 在对称缺陷中, 复介电函数实部和虚部分别在X和Y方向的张量相同, 表现出各向同性, 但在非对称缺陷中, 实部和虚部分别在X和Y方向的张量不同, 表现出明显的各向异性. 当入射光能量 Ein = 0 时, hBN, VB, as-VB, (CN)3VB, as-(CN)3VB的介电常数值${\varepsilon _0}$(单位: C2·N–1·m–2)分别是2.43, 33.35, 3.50, 32.47, 3.22, 因此对称的VB和(CN)3VB的屏蔽性都比较强. 在低能区域(Ein < 5 eV), 具有缺陷的hBN在复介电函数的实部和虚部均出现了峰值, 并且吸收边界出现红移, 说明硼空位和碳掺杂缺陷提高了hBN对可见光的光学响应程度. 图 10 0 K和0 GPa时hBN, VB, as-VB, (CN)3VB, as-(CN)3VB的复介电函数 Figure10. Complex dielectric functions of hBN, VB, as-VB, (CN)3VB, as-(CN)3VB at 0 K and 0 GPa.
吸收系数 I 和复介电函数的虚部${\rm{i}}{\varepsilon _2}\left( w \right)$密切相关, 因此可以通过介电函数的虚部得到光学吸收谱, 如图11所示, 为了清楚显示吸收光谱在低能区间的吸收峰, 图11(a)—(e)中的左图纵坐标较右图放大约6倍, 本征hBN在可见光波段范围几乎不吸收光子, 直到Ein = 4.4 eV时, 开始吸收光能量, 这与前面计算出的本征hBN的带隙为4.37 eV相符合. 在硼原子空位与碳原子掺杂之后, 吸收边界出现较大的红移, 并且在0.2 eV均出现了吸收峰, 这可能是由于VB和(CN)3VB的价带穿越费米能级EF导致的. 当打破对称结构时, as-VB和as-(CN)3VB的吸收峰发生了蓝移, 且由吸收橙红色光转变为吸收青蓝色光为主. 在较高能量的波段中, 本征hBN与有缺陷的hBN的吸收光谱大致相同. 图 11 0 K和0 GPa时hBN, VB, as-VB, (CN)3VB, as-(CN)3VB的吸收系数 Figure11. Absorption coefficients of hBN, VB, as-VB, (CN)3VB, as-(CN)3VB at 0 K and 0 GPa.
损失函数L描述的是光电子在均匀介质中穿过时的能量损失情况, 本征hBN和缺陷hBN的吸收函数如图12所示, 本征hBN和缺陷hBN在Ein = 7 eV和Ein = 18 eV处具有明显的能量损失峰. 但是, 有缺陷的hBN在低能区域(Ein < 5 eV)引入了许多损失峰, 这与图11吸收光谱在低能区时具有吸收峰相对应, 并且经过碳掺杂后, hBN的损失峰主要集中在可见光波段. 说明具有空位和碳掺杂缺陷的hBN在低能段时, 能级间相互作用比较强. 图 12 0 K和0 GPa时hBN, VB, as-VB, (CN)3VB, as-(CN)3VB的损失函数 Figure12. Loss function of hBN, VB, as-VB, (CN)3VB, as-(CN)3VB at 0 K and 0 GPa.
为了探究hBN发光缺陷的原子结构起源, 根据前面计算的hBN缺陷的电子结构和光学性质, 对缺陷带隙中可能存在的内部能级跃迁(基态到激发态)进行分析. VB的电子结构模型如图13(b)所示, 在价带以上0.41 eV的潜在基态到价带以上2.07 eV的潜在激发态之间有单一的跃迁, 导致约1.7 eV的跃迁, 这与图13(d)中VB的介电函数虚部在Ein为1.7 eV处的峰值相对应, 在吸收光谱上对应波长为730 nm的红色光. as-VB的电子结构模型如图13(e)所示, 自旋向上和自旋向下通道中均存在价带以上0.14 eV的潜在基态到价带以上0.89 eV的潜在激发态之间的内部跃迁, 而且两个自旋通道的内部跃迁几乎为简并能级, 导致约0.75 eV的跃迁, 这与图13(f)中as-VB的介电函数虚部在Ein为0.75 eV处的峰值相一致, 为深红外光. 图 13 hBN单层硼原子空位的模型图 (a), (d)分别为VB和as-VB优化后的局部结构图; (b), (e)分别为VB和as-VB的电子结构模拟图, 黑色箭头和灰色箭头分别代表占据态和未占据态; (c), (f)分别为VB和as-VB的介电虚部在X和Y方向张量 Figure13. Model diagram of boron vacancies in hBN monolayer: (a), (d) The optimized local structure diagrams of VB and as-VB; (b), (e) simulated electronic structures diagrams of VB and as-VB, where black and grey arrows indicate occupied and unoccupied states; (c), (f) the tensors of the dielectric imaginary part of VB and as-VB in the X and Y directions.
(CN)3VB的电子结构模型如图14(b)所示: 在自旋向上通道中, 存在一个价带以上0.89 eV的潜在基态到价带以上4.14 eV的潜在激发态之间的单一跃迁, 导致约3.25 eV的光学跃迁; 在自旋向下通道中, 存在一个价带以上0.46 eV的潜在基态到价带以上4.51 eV的潜在激发态之间的单一跃迁, 导致4.05 eV的光学跃迁; 分别与图14(c)中(CN)3VB的介电函数虚部在Ein约为3.25 和4.05 eV处的峰值相对应, 为波长307和382 nm的紫外光. 在非对称的碳掺杂缺陷中, as-(CN)3VB的电子结构模型如图14(e)所示, 自旋向上和自旋向下通道中均存在价带以上1.30 eV的潜在基态到价带以上3.88 eV的潜在激发态之间的内部跃迁, 两个自旋通道的内部跃迁能级为双简并能级, 导致约2.58 eV的跃迁, 与图14(f)中as-(CN)3VB的介电函数虚部在Ein为2.58 eV处的峰值相一致, 且其吸收光为波长570 nm的青色光, 这是与图11中非对称碳掺杂缺陷的吸收光谱相符合, 说明非对称的碳掺杂缺陷在可见光波段为单光子发射提供了一个潜在的跃迁途径. 图 14 hBN单层碳掺杂的模型图 (a), (d) (CN)3VB和as-(CN)3VB优化后的局部结构图; (b), (e) (CN)3VB和as-(CN)3VB的电子结构模拟图, 黑色箭头和灰色箭头分别代表占据态和未占据态; (c), (f) (CN)3VB和as-(CN)3VB的介电虚部在X和Y方向张量 Figure14. Model diagram of carbon doping in hBN monolayer: (a), (d) The optimized local structure diagrams of (CN)3VB and as-(CN)3VB; (b), (e) simulated electronic structures diagrams of (CN)3VB and as-(CN)3VB, where black and grey arrows indicate occupied and unoccupied states; (c), (f) the tensors of the dielectric imaginary part of (CN)3VB and as-(CN)3VB in the X and Y directions.



































 图 1 本征hBN单层 (a) hBN的俯视图和侧视图; (b) 模拟的hBN单层表面STM形貌, 加载电压为–2 V, 探针与原子表面的距离为0.479564 nm; (c) hBN的能带投影图
图 1 本征hBN单层 (a) hBN的俯视图和侧视图; (b) 模拟的hBN单层表面STM形貌, 加载电压为–2 V, 探针与原子表面的距离为0.479564 nm; (c) hBN的能带投影图




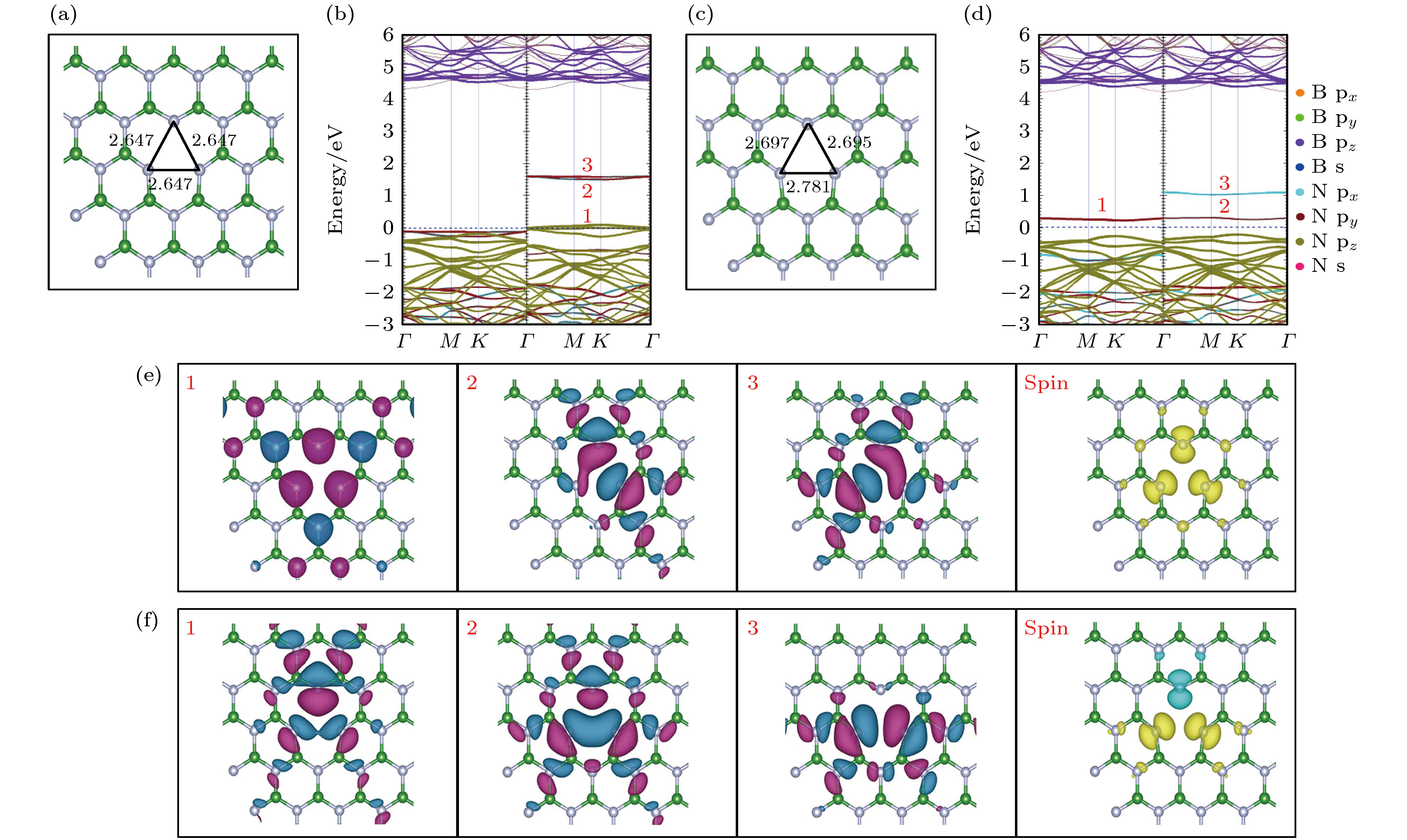 图 2 VB和as-VB的电子结构 (a), (c)分别为VB和as-VB优化后的局部结构俯视图; (b), (d)分别为VB和as-VB的能带投影图; (e), (f)分别为VB和as-VB的缺陷能级在Γ点处波函数的俯视图和自旋电荷密度图
图 2 VB和as-VB的电子结构 (a), (c)分别为VB和as-VB优化后的局部结构俯视图; (b), (d)分别为VB和as-VB的能带投影图; (e), (f)分别为VB和as-VB的缺陷能级在Γ点处波函数的俯视图和自旋电荷密度图










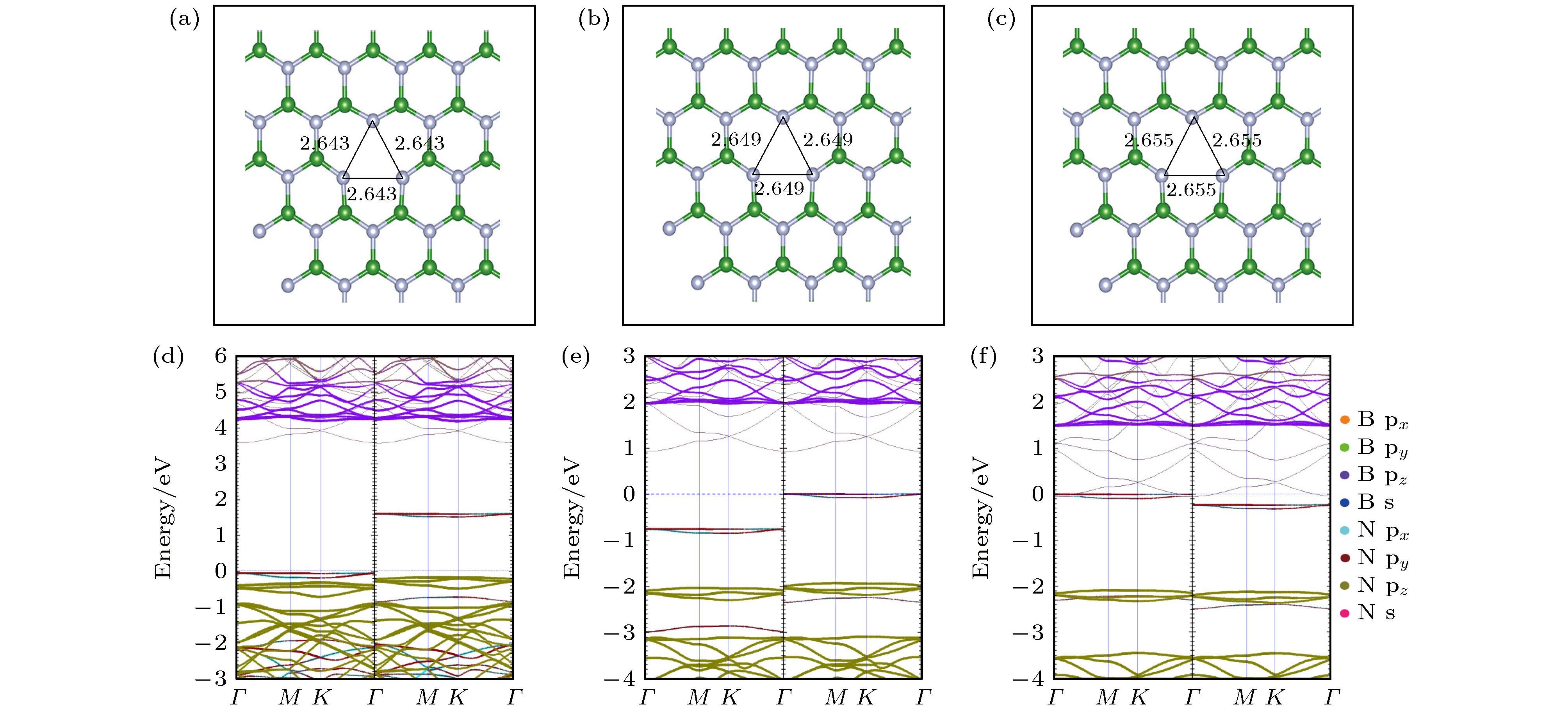 图 3 VB在不同价态的电子结构图 (a)?(c)分别为
图 3 VB在不同价态的电子结构图 (a)?(c)分别为




















 图 4 在富氮和贫氮条件下, 不同价态的VB形成能为费米能级的函数
图 4 在富氮和贫氮条件下, 不同价态的VB形成能为费米能级的函数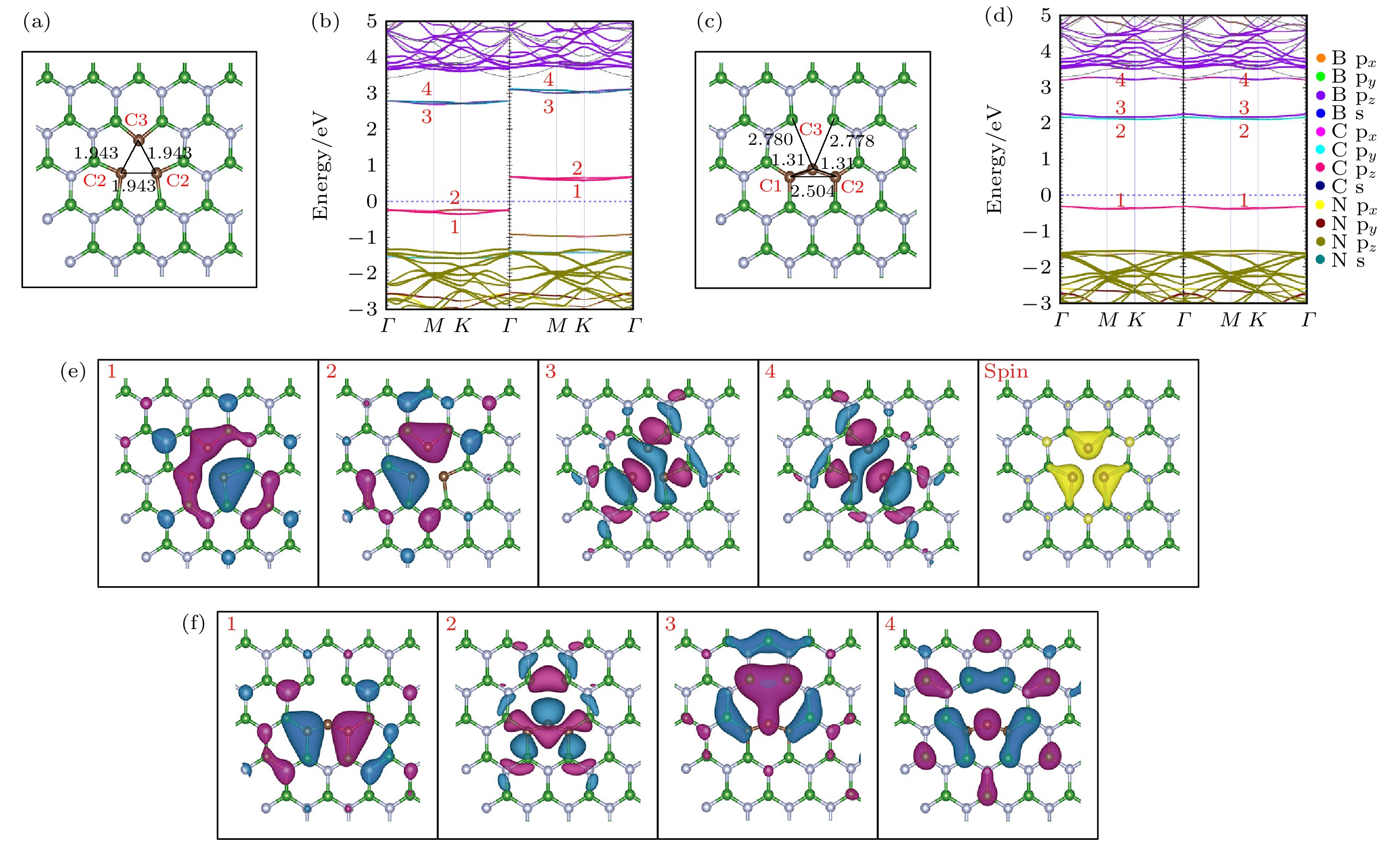 图 5 (CN)3VB和as-(CN)3VB的电子结构 (a), (c)分别为(CN)3VB, as-(CN)3VB优化后局部结构的俯视图; (b), (d)分别为(CN)3VB, as-(CN)3VB的能带投影图; (e) (CN)3VB缺陷能级在Γ点处的波函数图和自旋电荷密度图; (f) as-(CN)3VB缺陷能级在Γ点处的波函数图
图 5 (CN)3VB和as-(CN)3VB的电子结构 (a), (c)分别为(CN)3VB, as-(CN)3VB优化后局部结构的俯视图; (b), (d)分别为(CN)3VB, as-(CN)3VB的能带投影图; (e) (CN)3VB缺陷能级在Γ点处的波函数图和自旋电荷密度图; (f) as-(CN)3VB缺陷能级在Γ点处的波函数图




























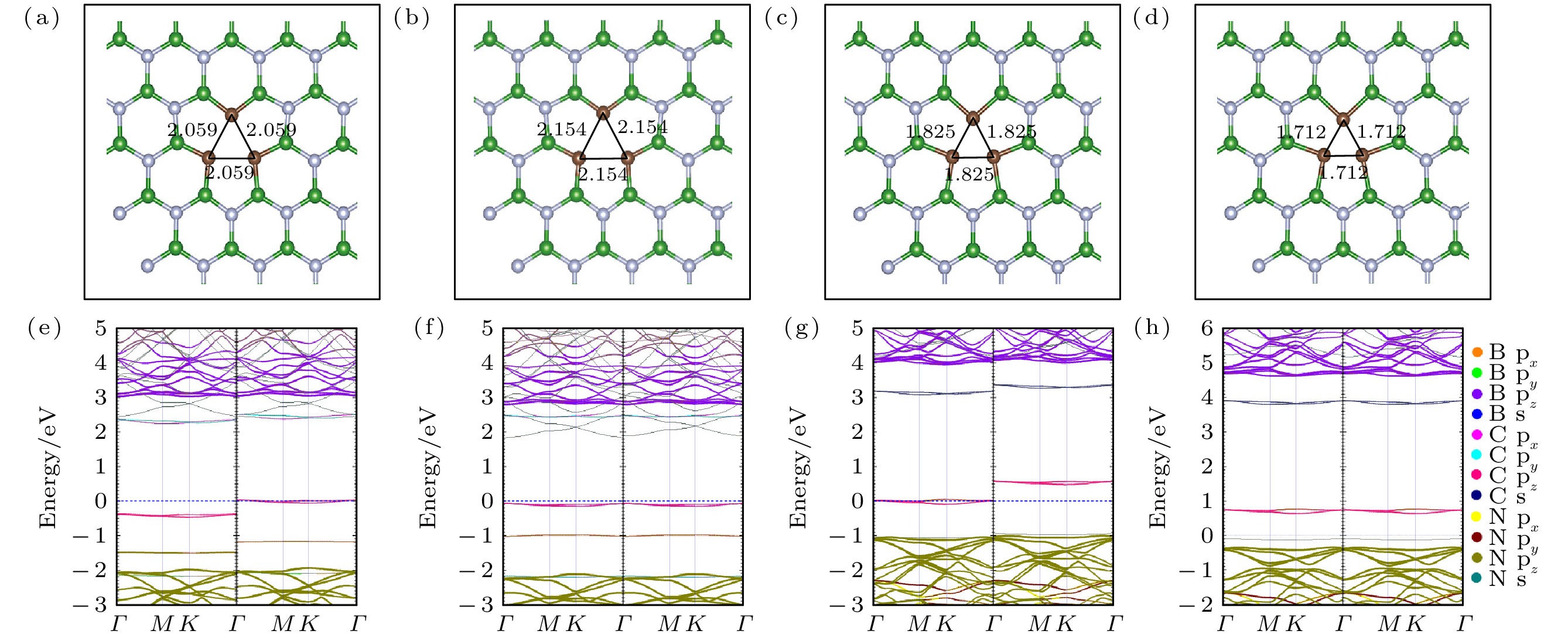 图 6 (CN)3VB不同价态优化后的局部结构和能带投影图 (a)?(d)分别为(CN)3
图 6 (CN)3VB不同价态优化后的局部结构和能带投影图 (a)?(d)分别为(CN)3

































 图 7 as-(CN)3VB不同价态优化后的局部结构和能带投影图 (a)?(d)分别为as-(CN)3
图 7 as-(CN)3VB不同价态优化后的局部结构和能带投影图 (a)?(d)分别为as-(CN)3















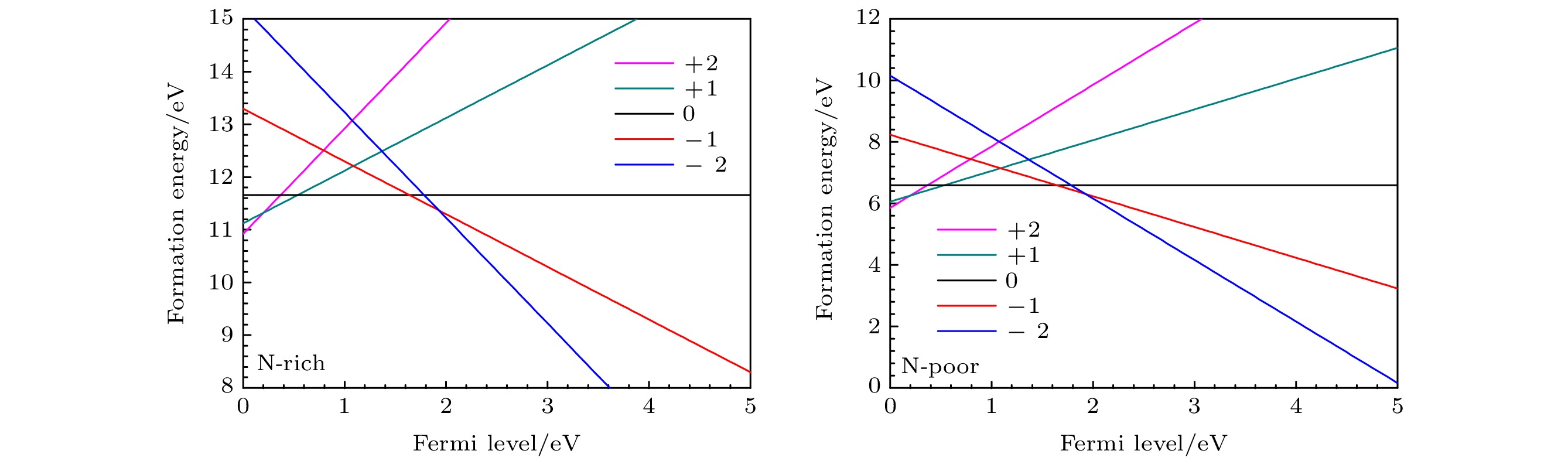 图 8 在富氮和贫氮条件下, 不同价态的(CN)3VB形成能为费米能级的函数
图 8 在富氮和贫氮条件下, 不同价态的(CN)3VB形成能为费米能级的函数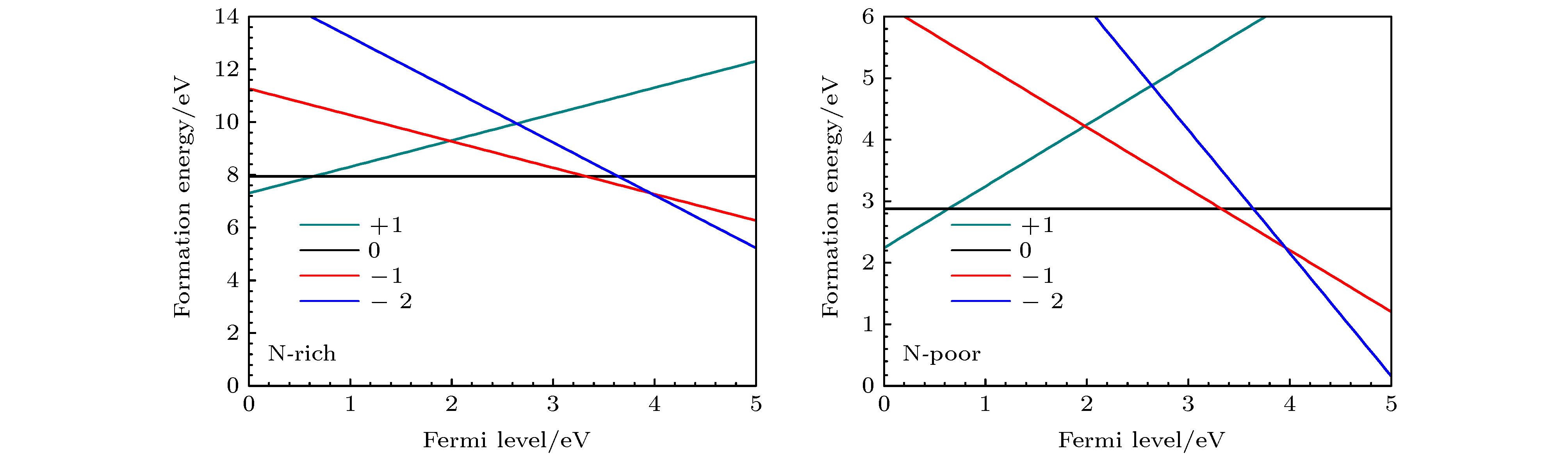 图 9 在富氮和贫氮条件下, 不同价态的as-(CN)3VB形成能为费米能级的函数
图 9 在富氮和贫氮条件下, 不同价态的as-(CN)3VB形成能为费米能级的函数




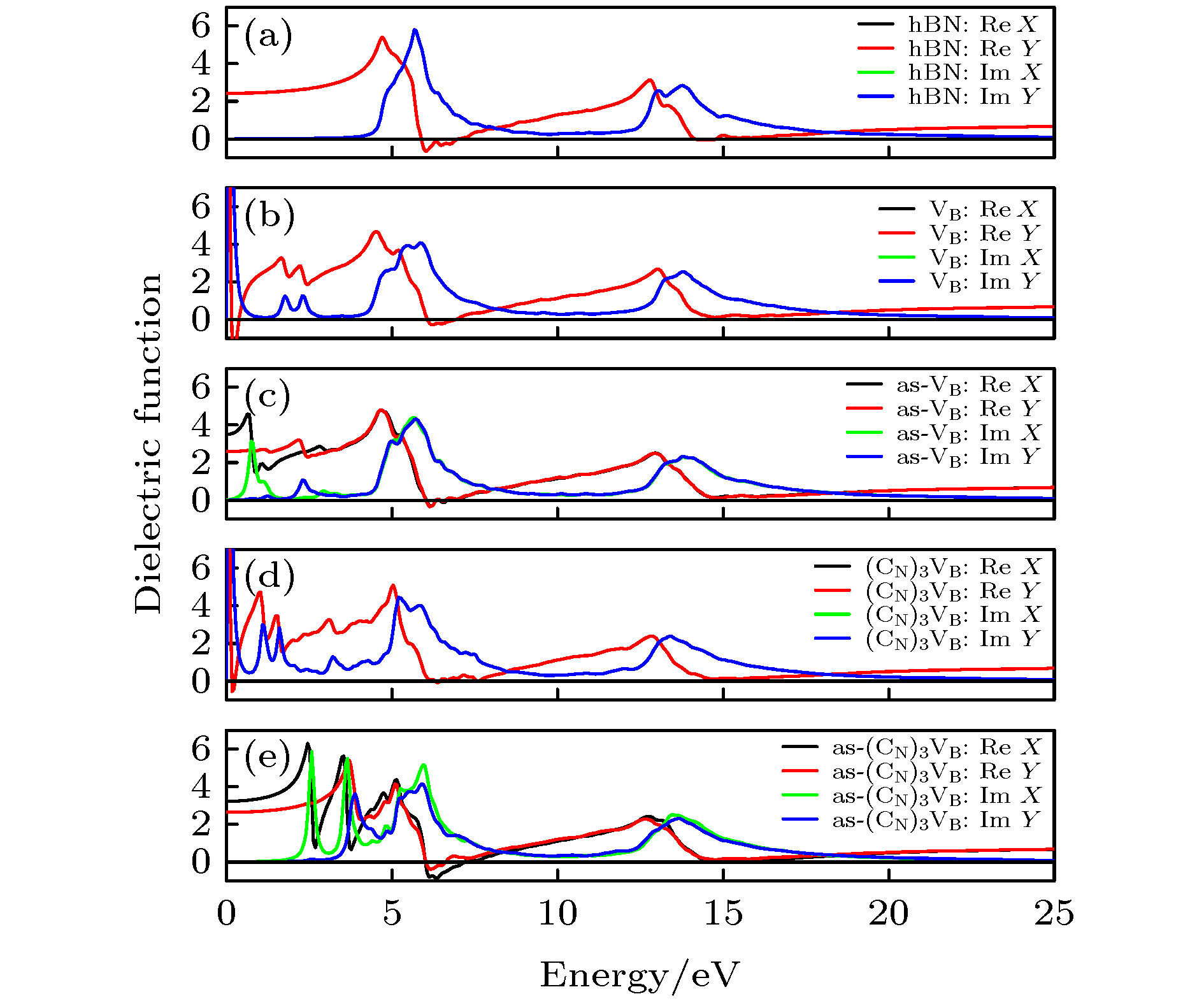 图 10 0 K和0 GPa时hBN, VB, as-VB, (CN)3VB, as-(CN)3VB的复介电函数
图 10 0 K和0 GPa时hBN, VB, as-VB, (CN)3VB, as-(CN)3VB的复介电函数
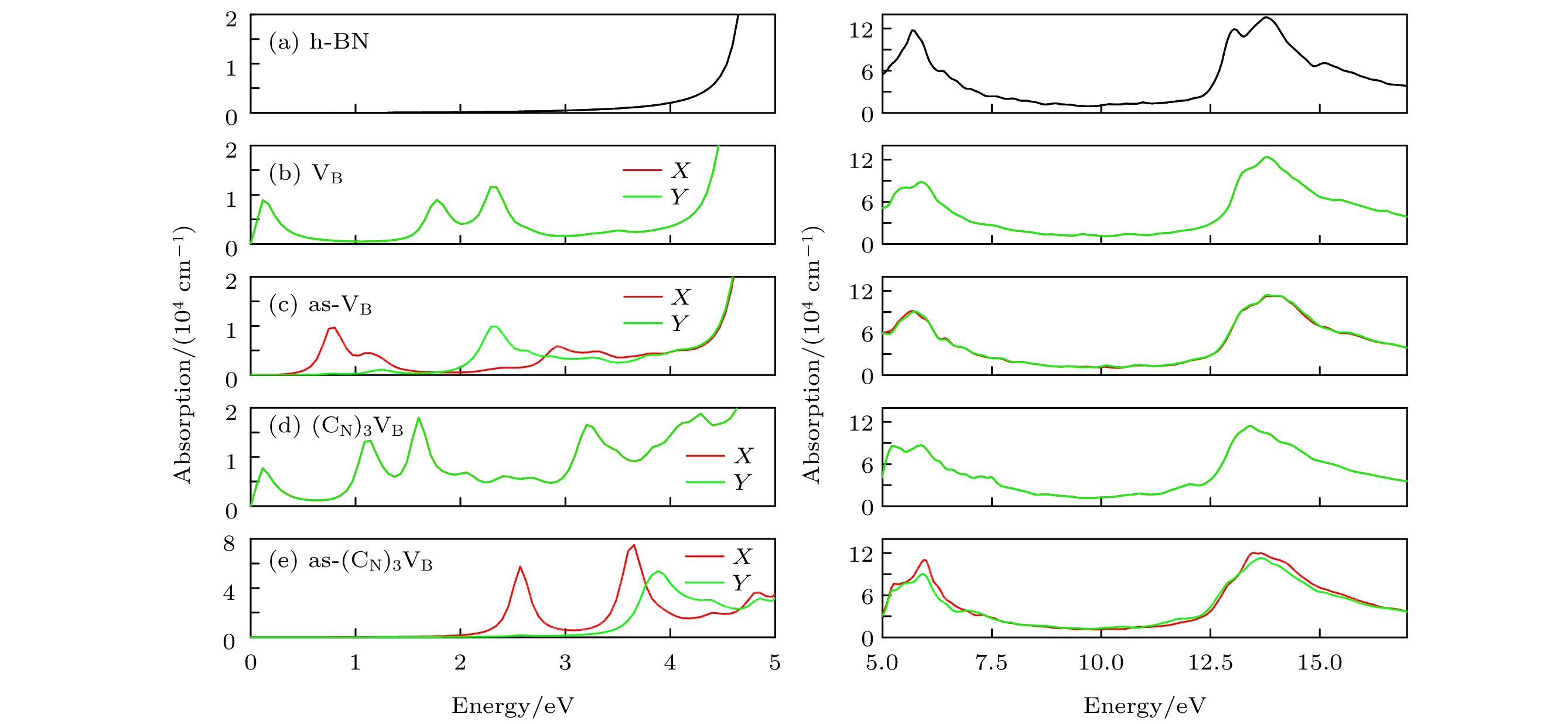 图 11 0 K和0 GPa时hBN, VB, as-VB, (CN)3VB, as-(CN)3VB的吸收系数
图 11 0 K和0 GPa时hBN, VB, as-VB, (CN)3VB, as-(CN)3VB的吸收系数 图 12 0 K和0 GPa时hBN, VB, as-VB, (CN)3VB, as-(CN)3VB的损失函数
图 12 0 K和0 GPa时hBN, VB, as-VB, (CN)3VB, as-(CN)3VB的损失函数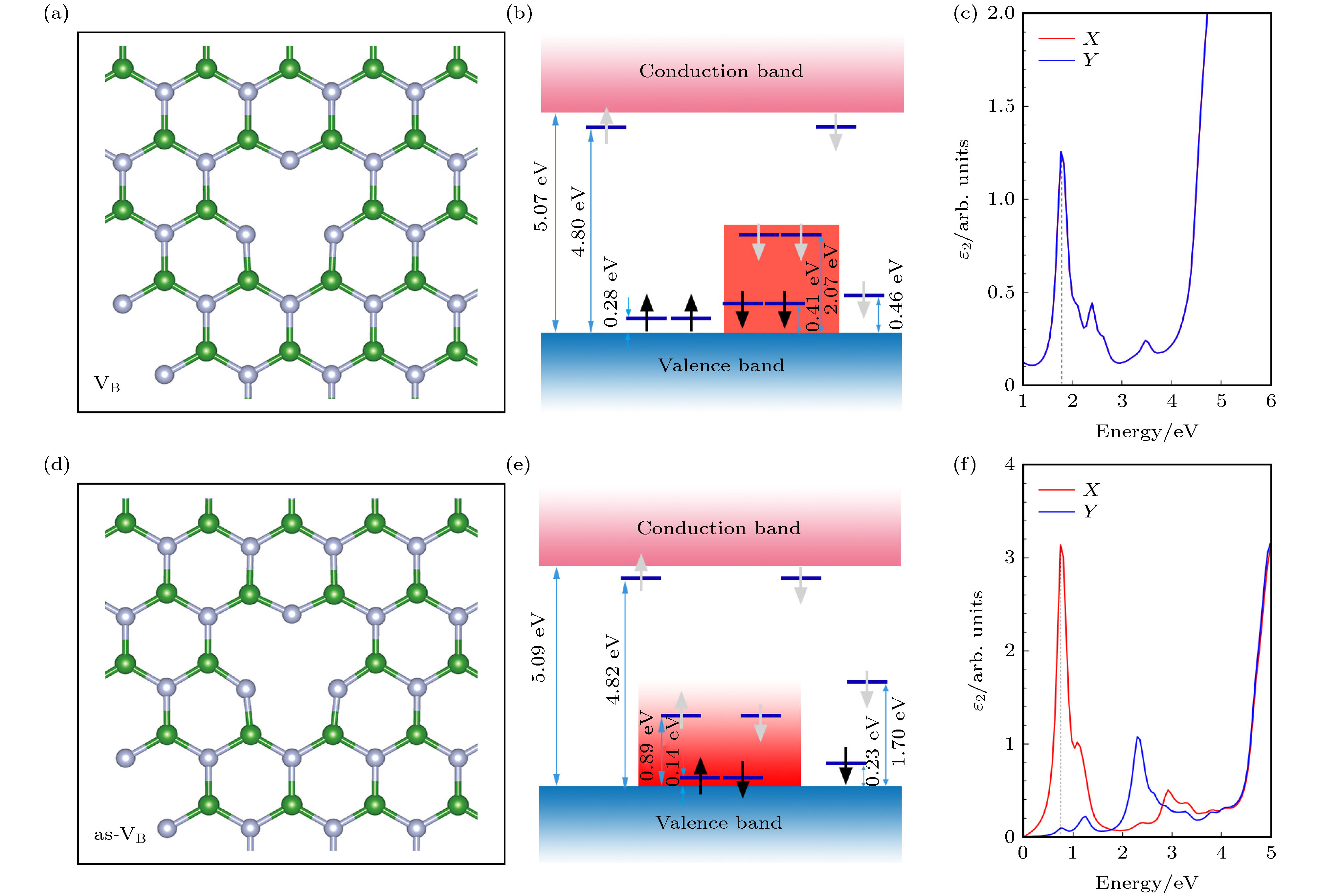 图 13 hBN单层硼原子空位的模型图 (a), (d)分别为VB和as-VB优化后的局部结构图; (b), (e)分别为VB和as-VB的电子结构模拟图, 黑色箭头和灰色箭头分别代表占据态和未占据态; (c), (f)分别为VB和as-VB的介电虚部在X和Y方向张量
图 13 hBN单层硼原子空位的模型图 (a), (d)分别为VB和as-VB优化后的局部结构图; (b), (e)分别为VB和as-VB的电子结构模拟图, 黑色箭头和灰色箭头分别代表占据态和未占据态; (c), (f)分别为VB和as-VB的介电虚部在X和Y方向张量 图 14 hBN单层碳掺杂的模型图 (a), (d) (CN)3VB和as-(CN)3VB优化后的局部结构图; (b), (e) (CN)3VB和as-(CN)3VB的电子结构模拟图, 黑色箭头和灰色箭头分别代表占据态和未占据态; (c), (f) (CN)3VB和as-(CN)3VB的介电虚部在X和Y方向张量
图 14 hBN单层碳掺杂的模型图 (a), (d) (CN)3VB和as-(CN)3VB优化后的局部结构图; (b), (e) (CN)3VB和as-(CN)3VB的电子结构模拟图, 黑色箭头和灰色箭头分别代表占据态和未占据态; (c), (f) (CN)3VB和as-(CN)3VB的介电虚部在X和Y方向张量