全文HTML
--> --> -->关于二维材料的研究可以追溯到2004年, 单层自由状态的石墨烯首次在实验上成功剥离出来[1], 推翻了热涨落条件下不允许任何二维介质在有限温度下存在的认知误区. 在此基础上, 研究人员提出了二维材料的概念. 二维材料中载流子迁移、热量扩散均限制在二维平面内, 材料中受限电子、光子等呈现出许多与三维结构中不同的、物理内涵十分丰富的新奇量子现象[2]和效应. 与传统三维材料不同, 二维材料能极大减弱与基底间的耦合, 为电场调控、化学修饰、异质结构建等提供了极大的便利; 二维材料通常是机械易弯曲的, 是发展柔性器件[3]的理想材料; 二维材料由于其超薄的厚度在未来原子层厚度的高集成度光、电、磁性器件等领域具有广泛的应用前景; 另外, 二维材料具有极大的比表面积, 也为催化、超级电容器等研究注入新的活力.
二维材料研究由最初的石墨烯一枝独秀到目前形形色色的二维材料百花齐放. 除了石墨烯, 一系列的二维材料被相继报道[4], 如硅烯[5-11]、锗烯[12,13]、锡烯[14,15]、黑磷[16,17]、金属硫族化合物[18,19]、MXene [20,21]等. 尤其是最近三年, 研究人员通过高通量方法从材料数据库中搜索出数十个家族、总量上千种的新型二维材料, 并构建了二维晶体材料数据库, 是二维材料研究领域新的里程碑. 这些二维材料具有多样的电子结构[22], 覆盖金属、半导体、绝缘体、半金属和超导体等类型, 同时很多材料具有非常优异的力学、磁学和光学性质.
现阶段二维材料的研究主要集中在材料制备、表征、物性研究、修饰改性和应用探索等方面. 得益于理论计算方法和高性能超级计算机的快速发展, 理论计算在目前二维材料研究的快速发展中起到了举足轻重的作用. 理论计算不但能够高效、准确地预测二维材料的结构稳定性以及电子学、力学、光学、磁学等物性, 而且对实验现象也能进行充分的理论解释. 因此, 几乎每一个二维材料领域中的重要成果中均能看到理论计算的身影.
尽管已有一些二维材料的研究综述, 但近年来二维材料的发展日新月异. 在高通量材料计算和二维材料数据库的推动下, 一系列的新型二维材料、二维材料中的新奇物性不断涌现, 关于二维材料研究的报道逐年递增, 如图1所示. 因此, 本综述将从理论计算角度出发梳理二维材料的研究脉络, 回顾近年来发表的一些具有影响力的工作. 在第2节中, 将简要介绍基于密度泛函理论的计算方法. 在第3节中, 将从层状二维材料、非层状二维材料、二维材料异质结三个方面介绍二维材料的研究进展. 在第4节中, 将对理论计算在二维材料研究领域的应用作一个简单的展望.
 图 1 截止到2020年9月以“Two-dimensional materials”为关键词, 从Web of Science网站查询到的近十年的论文发表数
图 1 截止到2020年9月以“Two-dimensional materials”为关键词, 从Web of Science网站查询到的近十年的论文发表数Figure1. Number of publications on two-dimensional materials per year over the last decade. The data is from ISI Web of Science website by September 2020, and the searching keyword is “two-dimensional materials”.
2.1.密度泛函理论
20世纪60年代发展起来的密度泛函理论, 是一种将多电子体系近似转化为单电子问题的方法. 基于此理论的第一性原理计算, 已经在多门学科的研究中取得了大量辉煌的成果. 密度泛函理论的奠基人之一沃尔特·科恩(Walter Kohn)也因此于1998年获得了诺贝尔化学奖. 下面简要介绍本文涉及的密度泛函理论的基本内容.3
2.1.1.绝热近似
晶体属于复杂的多粒子体系, 其定态薛定谔方程可以写作:





















3
2.1.2.Hohenberg-Kohn定理
与量子力学中将波函数Ψ作为基本变量不同的是, 密度泛函理论是将电子密度函数作为核心变量去描述多电子体系基态的各种性质. 1964年, Hohenberg和Kohn提出两个基本定理.定理一 作用于多电子体系的外势






定理二 在粒子数不变的情况下, 能量泛函


根据上述定理, 基态能量可写成泛函形式:



3
2.1.3.Kohn-Sham方程[23]




则


Kohn-Sham方程的重要之处在于: 在形式上能够将多电子体系的基态问题转变成单电子在有效势场中运动的问题, 其中电子之间复杂的相互作用都包含在泛函项


3
2.1.4.交换关联泛函
局域密度近似(LDA)[24]是一种广泛使用的近似, 在此近似下交换关联泛函可以表示为






广义梯度近似(GGA)[25,26] 则引入电子密度和梯度函数再加入自旋, 交换关联泛函可以表示为


3
2.1.5.赝势方法
因为原子核区域内的势场发散并十分复杂, 价电子的波函数需要许多平面波基矢展开. 而在离原子核较远的区域, 其势场比较平滑, 价电子的波函数类似平面波函数. 赝势[30]方法就是利用价电子的波函数没有急剧震荡的特性, 有效地减小了波函数展开需要的平面波基矢, 极大地提高了计算效率.2
2.2.材料稳定性分析
对于理论预言的新材料, 需要分析其结构是否稳定、有限温度下结构是否发生相变或者被破坏、实验合成潜力等.3
2.2.1.动力学稳定性计算
声子谱是表示组成材料原子的集体振动模式. 对于一个包含n个原子的原胞, 声子谱总共有3n支声子, 其中有3条声学支, 3n – 3条光学支. 声学支表示原胞的整体振动, 光学支表示原胞内原子间的相对振动. 材料的振动频率可表示为
3
2.2.2.热稳定性计算
通过声子谱判断材料稳定之后, 便可通过分析分子动力学进一步判断材料在一定温度下的稳定性[31]. 分子动力学方法: 首先构建超胞, 然后施加一定温度, 运行一段时间之后观察原胞结构是否遭到破坏来判断该材料能否在该温度下稳定存在. 通过吉布斯自由能计算, 可以比较不同构型在不同温度下的稳定性, 判断材料是否会在一定温度下发生相变.3
2.2.3.热力学稳定性计算
形成能是指由相应单质合成化合物所释放的能量. 以二元化合物AxBy为例, 其形成能



如果需要判断材料处于稳态还是亚稳态, 可以分析该材料的凸包图(convex hull能量). 如图2所示, 红色虚线是计算稳态AxBy的形成能得到的凸包图, 其纵轴是形成能, 横轴是元素B在AxBy中的占比. 若化合物在红色虚线上方例如插图中的D, 那么它可能分解为A和B, 或者是声子谱没有虚频的亚稳态; 若化合物在红色虚线下方例如插图中的C, 那么它是稳定态.
 图 2 凸包图, 用于判断材料稳态和亚稳态[32]
图 2 凸包图, 用于判断材料稳态和亚稳态[32]Figure2. Convex hull diagram is used to estimate the ground state and metastable state of a material[32].
3.1.层状二维原子晶体材料
在2017年以前, 人们对二维材料的研究主要集中在石墨烯、过渡金属硫族化合物、黑磷等少部分具有层状块体结构的材料上. 尽管它们中的很多材料在某些方面具有优异性能, 但一些三维体系中存在的物性还未在二维材料中发现, 一个非常重要的原因是已经发现和研究的二维材料种类太少. 如何广泛研究并快速扩充二维材料种类是整个二维材料研究领域亟待解决的问题. 基于密度泛函理论的高通量计算是一种非常高效的材料预测手段, 例如高通量计算方法已成功应用于寻找锂空气电池和锂离子电池材料[33,34]、储氢材料[35]、闪烁体材料[36]、电催化剂[37]和光伏材料[38]等. 通过从ICSD (inorganic crystal structure database), COD (crystallography open database)等体相材料数据库中筛选出的层状材料进行第一性原理高通量计算就可以很好地去理解和预测一些潜在的二维材料及其性质.2017年, Choudhary等[39]基于高通量计算提出了一种从体相材料中识别二维材料的方法. 由于PBE计算晶格常数通常数值会偏高, 尤其是在仅有范德华(van der Waals, vdW)相互作用的方向上. 文章于是利用这一特点作为筛选二维材料的有效判据. 具体来说就是将一种材料在ICSD中实验上测得的晶格常数和在Materials-Project中基于密度泛函理论(DFT)计算的a, b, c三个方向上的晶格常数作比较, 如果两个晶格常数之间相对误差




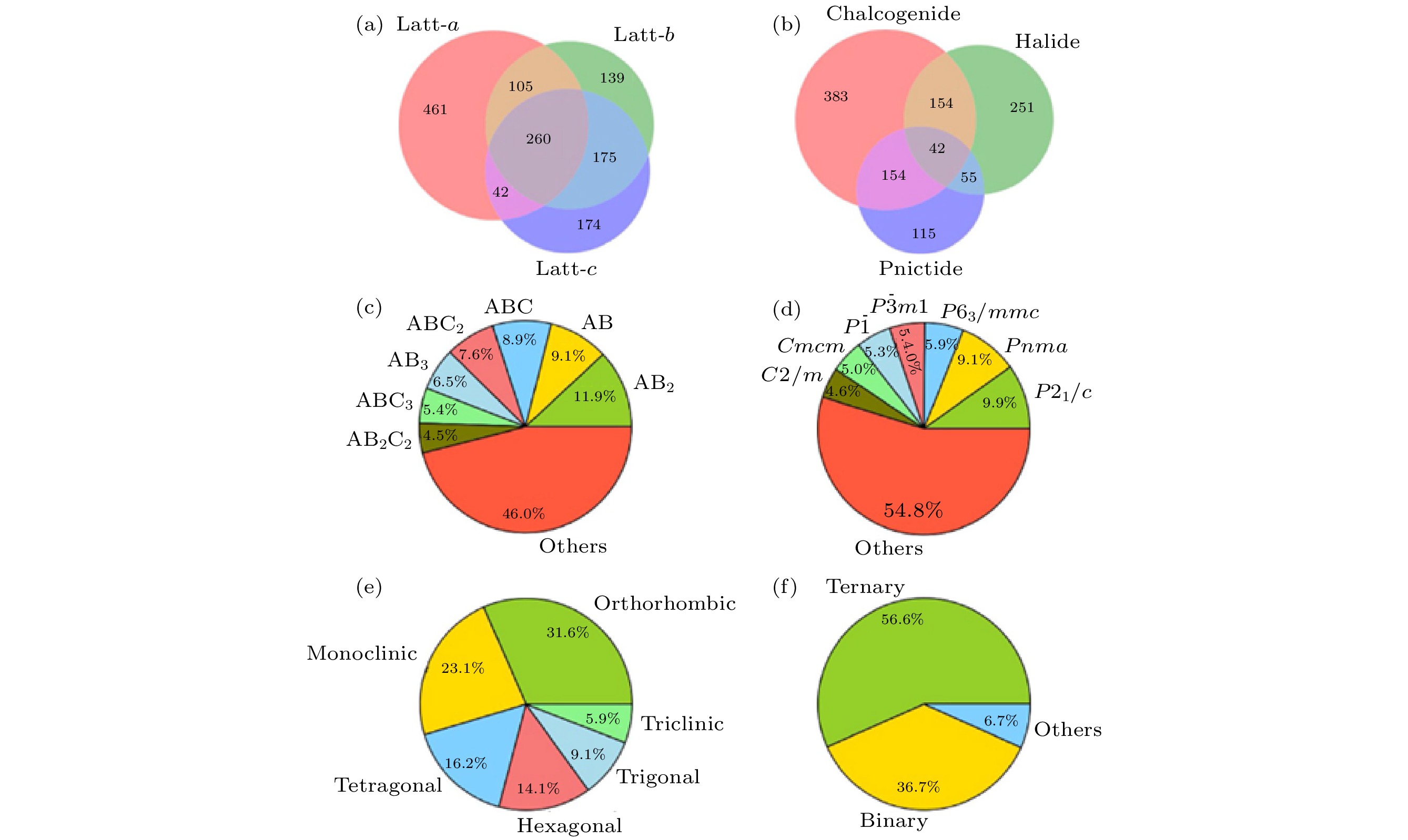 图 3 预测的层状材料结构、成分等信息统计 (a) 晶格常数的相对误差; (b) 化学元素组成; (c) 晶体类型; (d) 晶体空间群; (e) 晶系; (f) 元素种类[39]
图 3 预测的层状材料结构、成分等信息统计 (a) 晶格常数的相对误差; (b) 化学元素组成; (c) 晶体类型; (d) 晶体空间群; (e) 晶系; (f) 元素种类[39]Figure3. Classification of predicted layered materials in term of (a) relative error in lattice constants; (b) chemical compositions; (c) crystal prototypes; (d) crystal space groups; (e) crystal systems; (f) number of distinct chemical constituents[39].
2018年, Mounet等[42]通过不同的原子几何结构算法识别潜在的二维层状材料, 并通过高通量计算搜索了其中机械易剥离的材料. 他们通过对比ICSD和COD结构数据库中108423种实验已知的结构的原子间距和原子共价键长, 识别出5619种具有层状结构的材料, 再使用考虑范德瓦耳斯修正的密度泛函理论计算研究了这些层状材料的层间结合能, 发现一共有1825种材料可能剥离出二维的单层结构, 其中易剥离的材料有1036种, 潜在可剥离的材料有789种, 如表1所列. 文章还描述了易剥离的材料中比较有研究潜力的、具有较小原胞的258种化合物的声子谱、电子结构、磁性和拓扑性质, 其中表2列出了这258种化合物中有56种化合物具有磁性, 包括37种铁磁体和19种反铁磁体.
| Unique to the ICSD | Unique to the COD | Common to both | Total sum | |
| Experimental data | ||||
| CIF inputs | 99212 | 87070 | 186282 | |
| Unique 3D structures (set A) | 34548 | 60354 | 13521 | 108423 |
| Layered 3D structures (set B) | 3257 | 1180 | 1182 | 5619 |
| DFT calculations | ||||
| Layered 3D, relaxed (set C) | 2165 | 175 | 870 | 3210 |
| Binding energies (set D) | 1795 | 126 | 741 | 2662 |
| 2D easily exfoliable (EE) | 663 | 79 | 294 | 1036 |
| 2D potentially exfoliable (PE) | 524 | 34 | 231 | 789 |
| Total | 1187 | 113 | 525 | 1825 |
表1数据库统计[42]
Table1.Database statistics[42].
| Ferromagnetic | Antiferromagnetic | |
| Metals | Co(OH)2, CoO2, ErHCl, ErSeI, EuOBr, EuOI, FeBr2, FeI2, FeTe, LaCl, NdOBr, PrOBr, ScCl, SmOBr, SmSI, TbBr, TmI2, TmOI, VS2, VSe2, VTe2, YCl, YbOBr, YbOCl | CoI2, CrSe2, FeO2, FeOCl, FeSe, PrOI, VOBr |
| Semiconductors | CdOCl, CoBr2, CoCl2, CrOBr, CrOCl, CrSBr, CuCl2, ErSCl, HoSI, LaBr2, NiBr2, NiCl2, NiI2 | CrBr2, CrI2, LaBr, Mn(OH)2, MnBr2, MnCl2, MnI2, VBr2, VCl2, VI2, VOBr2, VOCl2 |
表2易剥落的磁性化合物[42]
Table2.Easily exfoliable magnetic compounds[42].
C2DB[43]是一个基于高通量计算的开放式二维材料数据库, 该数据库包含30多种不同晶体结构家族的共1500余种二维材料的电子结构、热力学、力学、磁学和光学性质. 数据库中常见二维材料结构类型如图4所示, 这些丰富多彩的结构类型也表明二维材料家族还有巨大的潜力值得挖掘. 特别强调的是计算电子能带使用了杂化密度泛函(HSE06)和多体微扰理论(G0W0). C2DB数据库的筛选流程是首先通过结构识别算法从体相材料中识别出层状材料及其单层结构和成分信息. 通过元素替换方式构成可能的其他初始理论晶格结构模型, 再按照图5所示的流程计算新二维材料的结构和性质. 此外, 目前已经创建的二维材料数据库还有materials web[44]和2D materials encyclo-pedia[45]等.
 图 4 C2DB数据库中典型的二维材料[43]
图 4 C2DB数据库中典型的二维材料[43]Figure4. Example of two dimensional materials prototypes in the C2DB[43].
 图 5 C2DB数据库的工作流程图[43]
图 5 C2DB数据库的工作流程图[43]Figure5. The workflow of producing data of C2DB[43].
2
3.2.非层状二维原子晶体材料
对于体相为层状结构的二维材料, 机械剥离是得到高质量二维单层结构的有效方法. 随着分子束外延等自下而上的制备手段的发展, 一些体相为非层状结构的二维单层结构也被实验合成出来, 并展现出许多新奇的物性. 硅烯和锗烯是这一类二维材料中最早研究的材料. 在剥离出石墨烯之前, 就有理论预测[5]: 单层Si和Ge与石墨烯的平面蜂窝状晶格结构不同, 是翘曲的蜂窝状结构, 并能导致发生量子自旋霍尔效应[46,47]. 受到硅烯、锗烯研究工作的启发, 其他二维单层非层状材料也取得了一系列进展.3
3.2.1.As和Sb
2015年以前, 大多数报道的二维半导体都具有较小的带隙, 极大地限制了它们在蓝光、紫外光光电器件中的应用. 2015年, Zhang等[48]基于第一性原理计算预测了具有宽带隙和高稳定性的新型二维单元素的半导体砷烯和锑烯. 其中, 体相和单层的As和Sb结构具有与硅烯和锗烯类似的翘曲蜂窝状晶格结构, 体相砷和单层砷烯的结构如图6所示, 体相砷层间距离、键长和键角分别是2.04 ?, 2.49 ?和97.28°. 相比之下, 在砷烯中由于晶格畸变, 键长和键角变化为2.45 ?和92.548°. 体相锑的层间距离、键长和键角分别是1.84 ?, 2.84 ?和93.28°, 锑烯中键长和键角分别为2.45 ?和92.548°. 为了判断这两种新型二维晶体的稳定性, 文章计算了砷烯和锑烯的声子谱如图7所示, 结果显示两种材料的声子谱中均没有虚频, 表明砷烯和锑烯动力学稳定. 2017年Wu等[49]在PdTe2衬底上生长了锑烯, 2018年Shao等[50]也在Ag(111)衬底上成功合成了高质量的锑烯. 体相As和Sb通常是半金属, 而它们的单层结构是带隙为2.49 eV和2.28 eV的间接带隙半导体. 这种电子结构有望在未来具有高开关比的晶体管、蓝光或紫外光光电器件以及机械传感器等方面具有潜在的应用. 另外, 砷烯和锑烯在双轴应变下都能够转变成直接带隙半导体, 在纳米电子和光电器件等领域同样具有潜在的应用. 图 6 体相砷的 (a) 俯视图和(b) 顶视图; 翘曲单层砷烯的(c) 俯视图和(d) 侧视图[48]
图 6 体相砷的 (a) 俯视图和(b) 顶视图; 翘曲单层砷烯的(c) 俯视图和(d) 侧视图[48]Figure6. (a) Side view and (b) top view of the structure of arsenic; (c) top view and (d) side view of a buckled As monolayer (arsenene)[48].
 图 7 层砷烯和层锑烯的声子谱图[48]
图 7 层砷烯和层锑烯的声子谱图[48]Figure7. Phonon dispersions of arsenene and antimonene[48].
3
3.2.2.单层CuSe
2018年, Gao等[51]报道了通过直接硒化Cu(111)表面的方法可以成功制备出具有一维摩尔图案的单层CuSe. 高分辨率扫描隧道显微镜(STM)和低能电子衍射谱(LEED)的测量结果表明CuSe具有六角蜂窝状结构. 通过第一性原理计算进一步确定样品是Cu(111)衬底上生长的具有翘曲的蜂窝状晶格单层CuSe. 图8(a)是Cu(111)衬底上单层CuSe的高分辨率STM图像, 可以清楚地观察到一维摩尔图案. 图8(b)是Cu(111)衬底上单层CuSe的低能电子衍射(LEED)图案, 红色和黄色圆圈分别表示单层CuSe和Cu(111)晶格的衍射斑点, 可以看出单层CuSe与Cu(111)表面之间有30°的旋转. 为了研究Cu(111)表面上单层CuSe的原子构型, 文章构建了 (
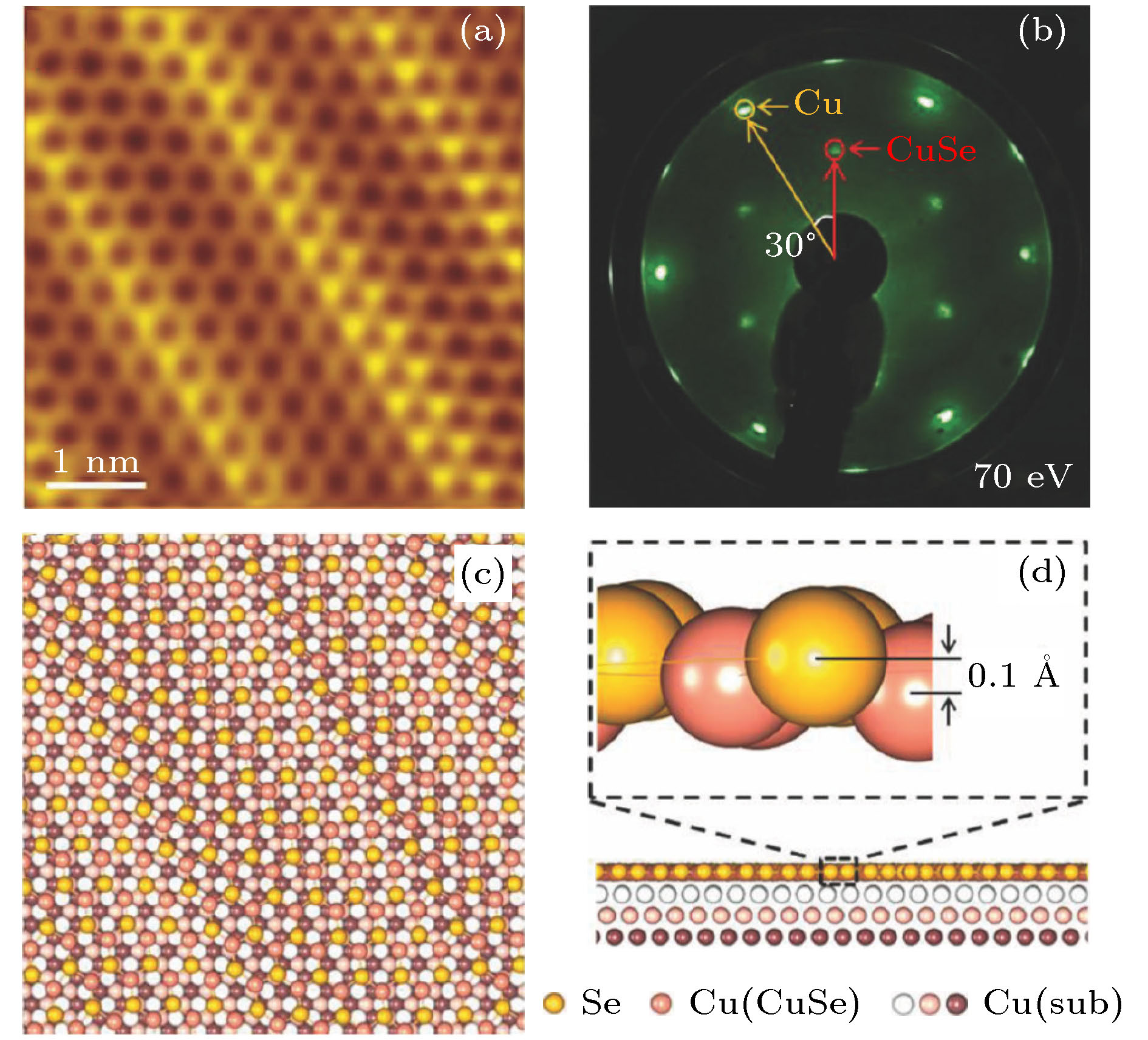 图 8 Cu(111)上单层CuSe的(a) 高分辨率STM图像, (b) LEED图; Cu(111)上单层CuSe优化原子结构模型的(c) 俯视图, (d) 侧视图[51]
图 8 Cu(111)上单层CuSe的(a) 高分辨率STM图像, (b) LEED图; Cu(111)上单层CuSe优化原子结构模型的(c) 俯视图, (d) 侧视图[51]Figure8. Monolayer CuSe on Cu(111): (a) High-resolution STM image, (b) LEED pattern; optimized atomic structure of monolayer CuSe on Cu(111): (c) top view, (d) side view[51].
通过考虑自旋-轨道耦合(SOC)效应的第一性原理计算对CuSe/Cu(111)电子结构进行理论模拟, 如图9(a)所示: 在能量低于–2.0 eV区域有非常大的电子态密度, 这主要是由Cu(111)基底贡献的; 而在–2.0 eV以上能带主要由CuSe单层贡献, 并有两个孔状带. 实验上ARPES对样品测量结果如图9(b)所示, 与理论计算结果十分吻合, 进一步表明单层CuSe结构稳定并能在实验上成功制备. 另外, 第一性原理计算还研究了单层自由状态下CuSe的电子结构, 发现单层CuSe具有狄拉克节点线费米子(DNLF), 其受到镜面对称性的保护. 这种DNLF存在于自旋-轨道耦合带隙内的拓扑非平庸边缘态上. 如果在弱耦合衬底(例如石墨烯)上合成CuSe, 则可以使用ARPES直接观察到有趣的DNLF特性.
 图 9 Cu(111)上单层CuSe (a) 沿K–Γ–K方向测量的ARPES图; (b) 理论计算的能带图[51]
图 9 Cu(111)上单层CuSe (a) 沿K–Γ–K方向测量的ARPES图; (b) 理论计算的能带图[51]Figure9. Monolayer CuSe on Cu(111): (a) ARPES intensity plots measured along the K–Γ–K direction; (b) calculated band structure [51]
2
3.3.二维材料异质结
范德瓦耳斯异质结构是在一种或两种以上的二维材料上堆垛另一种二维材料, 层与层之间通过范德瓦耳斯力相结合[52]. 强大的层内共价键让二维材料具有很高的面内稳定性, 而相对较弱的范德瓦耳斯力也足以让堆垛在一起的二维材料紧密结合. 随着二维晶体家族的范围逐渐扩展, 在原子精度上可以创建大量的范德瓦耳斯异质结构, 但是受限于实验合成效率问题仅研究了一部分的二维材料异质结. 通过理论计算可以高效模拟大批量异质结物性. 最近, 理论报道了许多不同类型的范德瓦耳斯异质结构中的电、光和磁等性能.根据能带排列方式, 半导体二维材料组成的异质结一般分为三种类型[53], 分别为I型、II型和III型. I型异质结是指异质结中能隙较小的材料同时具有导带的最小值和价带的最大值, 如图10(a)和图10(b)所示. 能隙较大的材料中激发的电子和空穴可以按照箭头指示方向转移到能隙较小的材料中, 而能隙较小的材料中激发的载流子不能转移到能隙较大的材料中. I型异质结在光学器件中有着非常广泛的应用, 例如发光二极管(LED)[54]和激光器. II型异质结是指导带的最小值和价带的最大值分别位于不同的材料中, 如图10(c)和图10(d)所示. 能隙较大的材料激发的电子可以转移到能隙较窄的材料中, 空穴不可以转移; 而能隙较窄的材料激发的空穴可以转移到能隙较宽的材料中, 电子不可以转移. III型异质结是指一种材料价带的最大值仍然小于另一种材料导带的最小值, 这种异质结构并不常见. 总结来说, 当材料A和材料B构成异质结, 若CBMA > CBMB > VBMB > VBMA, 则为I型异质结; 若CBMB > CBMA > VBMB > VBMA, 则为II型异质结; 若 CBMB > VBMB > CBMA> VBMA, 则为III型异质结, 其中CBM代表导带底, VBM代表价带顶.
 图 10 电荷转移示意图 (a), (b) I型异型结; (c), (d) II型异型结[53]
图 10 电荷转移示意图 (a), (b) I型异型结; (c), (d) II型异型结[53]Figure10. Schematic of allowed charge transfer: (a), (b) Type-I heterostructures; (c), (d) type-II heterostructures[53].
2016年, ?z?elik等[55]研究了常见二维半导体材料的能带结构, 其中包括IV和III-V族化合物的单层材料、V族元素的单层材料、过渡金属二卤化物(TMD)和过渡金属三卤化物(TMT). 利用基于密度泛函理论的第一性原理计算, 作者研究了903种不同组合的异质结类型, 建立了异质结周期表, 如图11所示, 其中绿色、红色和蓝色框分别代表I型、II型和III型异质结, 对角线分开的左下区域和右上区域分别是PBE和HSE06的计算结果. 这种预测为未来的实验与理论研究提供了有价值的参考.
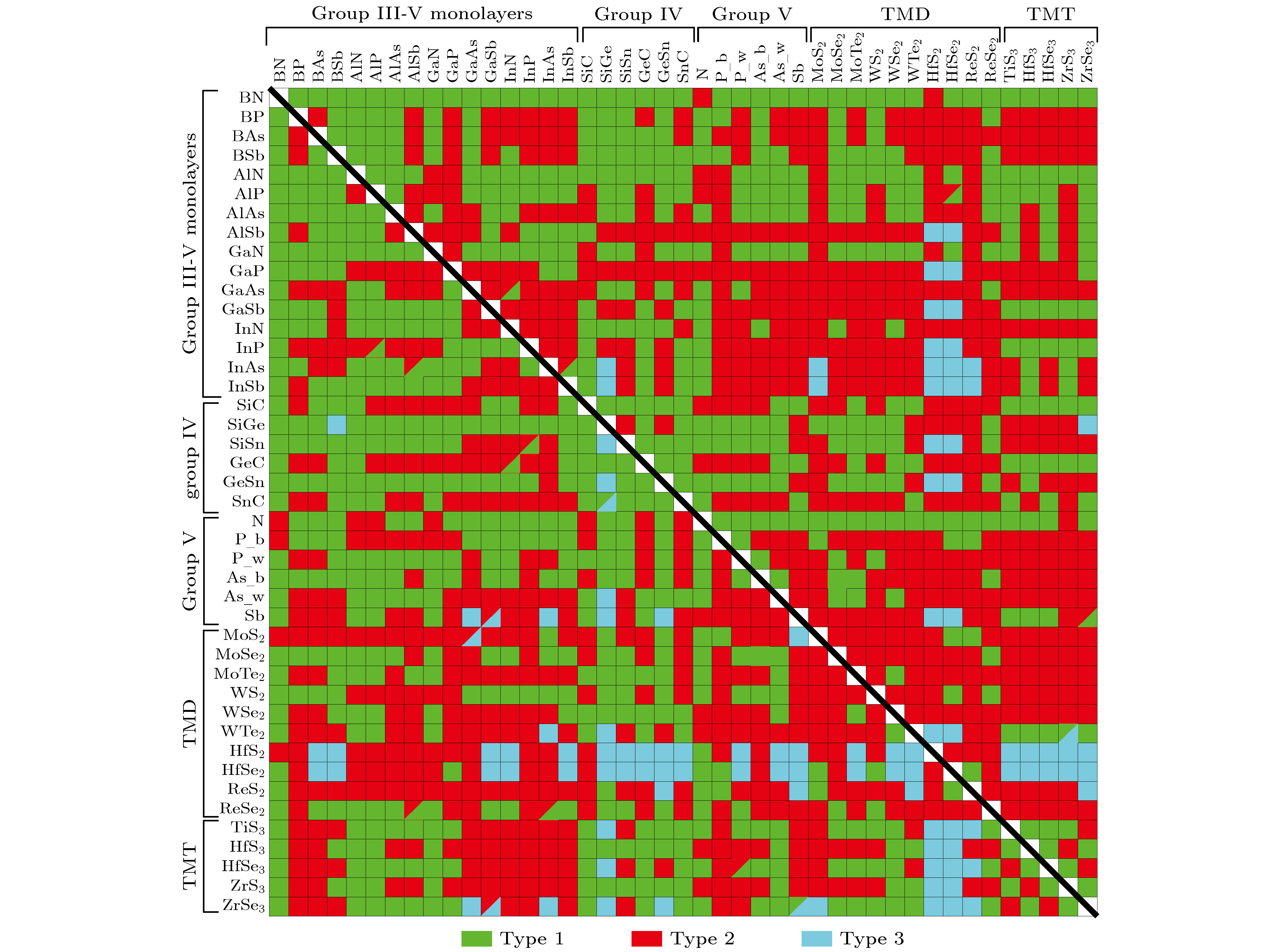 图 11 异质结周期表[55]
图 11 异质结周期表[55]Figure11. Periodic table of heterostructures[55].
单层的过渡金属二卤化物(TMDs)具有良好的光学性能, 若两个TMD单层组成范德瓦耳斯异质结, 则层内激子和层间激子将会同时存在. 层内激子是指激发的电子和空穴在同一层中, 层间激子[56-59]是指电子和空穴在不同层中. 这样的TMD双层异质结在超快电荷转移、超快层间热激子形成等领域有着潜在的应用. 由较典型的MoS2, MoSe2, WS2和WSe2这4种材料组成的MoSe2/MoSe2[60], MoS2/WS2[61-65], MoS2/WSe2[66-72], MoSe2/WSe2[73], MoSe2/WSe2[74]和WS2/WSe2[75]这6种范德瓦耳斯异质结均为II型异质结, 并且它们具有相似的电子和光学性质.
2018年, Torun等[76]对MoS2/WS2异质结进行第一性原理计算. MoS2/WS2异质结的激子效应光谱如图12(a)所示, 其中蓝色、红色、绿色分别是MoS2, WS2和MoS2/WS2的吸收光谱. 在MoS2/WS2中, X1激子是层间激子, 其能量比MoS2层中层内激子X2和WS2层中层内激子X3低. 投影能带结构如图12(b)所示, 激子在K点处从价带的最大值跃迁到导带的最小值. 具有激子效应异质结的能带排列如图12(c)所示, MoS2/WS2的层间激子X1的结合能为0.43 eV, 比MoS2层和WS2层的层内激子X2, X3的结合能数值小70 meV. 这是因为电荷载流子在空间上的分离降低了层间激子结合能, 层间较弱的库仑作用使层间激子的结合能远小于层内激子的结合能, 导致层间激子是最低能级, 任一层中的层内激子都是激发态. 突出的激子效应在超薄材料的载流子动力学领域有着重要的应用.
 图 12 (a) MoS2, WS2和MoS2/WS2的吸收光谱图; (b) 布里渊区K点附近的电子能带; (c) 异质结的能带排列[76]
图 12 (a) MoS2, WS2和MoS2/WS2的吸收光谱图; (b) 布里渊区K点附近的电子能带; (c) 异质结的能带排列[76]Figure12. (a) Absorption spectra of MoS2, WS2 and MoS2/WS2; (b) electron band near the K point in the Brillouin zone; (c) band arrangement of heterostructures[76].
2018年, Gillen和Maultzsch等[77]对三种不同堆垛结构的MoSe2/WSe2异质结进行第一性原理计算. 三种不同的堆垛结构均保持六角晶格对称性, 其中, AA堆垛是WSe2层相对于MoSe2层的零度旋转; AA' 堆垛是WSe2层相对于MoSe2层旋转180°, 这种堆垛方式对应的结构是最稳定的; AB堆垛是在AA堆垛基础上将WSe2层平移

 图 13 AA, AA', AB型MoSe2/WSe2异质结的结构及能带[77]
图 13 AA, AA', AB型MoSe2/WSe2异质结的结构及能带[77]Figure13. Structure and energy band of AA, AA', AB MoSe2/WSe2 heterostructures[77].
科学家们已经预言了大量新型二维原子晶体材料, 对二维材料几何结构、基态电子结构的研究比较完善, 但其他的性质, 如光学、力学、热电、磁学和拓扑学等方面的性质还有很大的研究空间; 二维材料的应用研究还处在原理器件研究上, 距离找到具有理想性能的二维功能材料并制作出可应用的器件还有较大的差距; 一些基于新原理的新型器件可能在将来也不断涌现; 另外, 对于一些材料或材料的某些性质, 理论计算得到的结果与实验结果不能够很好地吻合, 因此, 发展更完善的理论计算体系、发掘更有效的计算方法, 也是科研人员的重要工作方向.
