全文HTML
--> --> -->芳香族分子构成了大量生物分子的核心骨架, 在紫外光照射下, 被激发到电子激发态的芳香族分子将经历超快非绝热过程[9-11], 这种超快的非绝热过程在生命科学、环境科学、光物理和光化学过程中有至关重要的作用[12]. 作为典型的芳香族的苯分子和苯分子的衍生物[13-15]一直受到关注, 苯分子被激发到S2态之后快速内转换到S1态, S1态向S0或T1态的非绝热过程一般发生在纳秒量级, 并且S1态非绝热过程中内转换和系间窜跃过程互相竞争[16,17]; 类似的电子失活机制在苯的衍生物如甲苯[13,14]、邻二甲苯[18]、茚分子等中也有报道, 其分子S1态的寿命尺度通常在4.3—8.8 ps之间, S2态的寿命通常在100 fs内. 同时在苯的衍生物中还观察到取代基效应对电子激发态非绝热动力学的影响[13-15], 例如由于甲基的引入甲苯分子较苯分子具有更高的 S2→S1 内转换比例, 使得其S1态具有更大的振动能级密度, 更利于S1与S2态之间发生非绝热耦合[13].
苯乙炔作为苯的一个重要衍生物, 其光物理和光化学性质也引起了科研团队的广泛关注. 尽管有文献提供了苯乙炔分子的光谱信息, 但是据我们所知, 目前用时间分辨的方法对苯乙炔分子的激发态动力学进行的相关研究还较少. Stolow团队[15]用时间分辨的光电子能谱技术研究苯乙炔分析S2态的非绝热动力学过程, 认为S2态的主要衰减通道为像S1态的内转换过程, 时间尺度大概为100 fs. 但是S1态被布居以后通过什么样的衰减通道衰减呢?苯衍生物的非简并性导致了更低的对称禁戒性和更高的振动能级密度, 乙炔取代基的存在对苯衍生物的激发态非绝热动力学过程有什么样的影响, 被转换布居的S1态的衰减通道主要是向S0的内转换还是向T1态衰减的系间窜跃, 这些问题一直存在争议.
时间分辨的光电子影像技术(TRPEI)作为探测分子电子激发态超快非绝热过程的有效手段[19-21], 可以提供时间分辨的光电子能谱和光电子角分布信息. 在本文的研究中, 我们选用对称性降低的苯乙炔分子, 采用飞行时间质谱和光电子影像技术实时跟踪探测苯乙炔分子S2 态的演化过程, 研究结果表明被激发到 S2态后的苯乙炔分子将发生超快级联式的非绝热动力学过程: S2→S1→T1, 该结果进一步完善和丰富了对苯乙炔分子电子激发态的无辐射机制的研究. 此外我们还观察到苯乙炔与苯分子动力学的不同之处, 并尝试性地将其归属为取代基效应[22].
实验采用背景压力为2 atm (1 atm = 101325 Pa)的氦气为载气, 通过盛有纯度为99.9%的苯乙炔液体的样品池, 饱和蒸汽经过脉冲阀喷入真空腔形成超声分子束, 经skimmer准直进入电离区. 苯乙炔分子在电离区被泵浦探测光电离后产生光电子与光离子, 聚焦到二维位置敏感探测器上, 形成高分辨的光电子影像或离子影像. 光电子原始影像通过BASEX变换重构其三维分布, 就可以得到代表真实分布的三维重构影像 [24].
3.1.S2态的衰减动力学
苯乙炔S2态的带源位于5.2 eV附近[25], 因此235 nm泵浦光(5.28 eV)可以将苯乙炔分子从电子基态激发到 S2态, 苯乙炔的电离能为8.8 eV[25], 要将被激发的S2态分子电离至少还需要2个400 nm的探测光子. 实验中调节探测光和泵浦光的光强, 保证在双光作用下的飞行时间质谱中母体离子
图1显示了在235 nm泵浦、400 nm探测条件下的母体离子时间衰减曲线. 本实验中泵浦光是235 nm, 泵浦光激发苯乙炔的S2态, S2态被激发后很有可能内转换到S1态或S0态, 考虑到这样的一个动力学过程, 用双指数函数与高斯函数将母体离子的时间分辨质谱信号进行卷积拟合, 拟合得到两个明显不同的时间组分, 分别为τ1 = 116 fs和τ2 = 106 ps. 前人的研究中指出, 苯乙炔S2态很有可能快速内转换到S1态[15], 时间尺度为几十飞秒, 因此得出快速组分116 fs很有可能是S2态到S1态内转换的时间, 较慢的组分为S1态的衰减时间.
 图 1 235 nm泵浦、400 nm探测条件下获得的母体离子时间衰减曲线, 圆圈代表实验结果, 实线代表拟合结果
图 1 235 nm泵浦、400 nm探测条件下获得的母体离子时间衰减曲线, 圆圈代表实验结果, 实线代表拟合结果Figure1. Time-resolved total ion signals of parent ion as a function of delay time between the pump pulse at 235 nm and the probe pulse at 400 nm. The circles are the experimental results, and solid lines are the fitting results.
为了得到S2态向S1态内转换的更多证据, 实验中采集了不同泵浦探测时间延迟下的光电子影像, 用BASEX程序[26]对光电子影像进行变换得到电子影像的三维空间分布. 为了更清楚地探究不同能态之间的能量转移过程, 我们通过光电子影像得到了光电子能谱. 图2给出了0时刻和163 fs时的光电子能谱, 从图中我们可以看到有明显的3个带, 第1个带为0.4—0.7 eV左右的比较宽的带, 第2个带为比较强比较尖的0.7—1.3 eV带, 第3个带为1.3—2.5 eV的比较宽的带, 依次被标记为 1, 2, 3, 从能谱中可以看到随着泵浦-探测延迟时间不同, 光电子能谱有明显的变化. 随着时间的演化, 第2和第3光电子能带在衰减的同时, 第1光电子能带在上升, 这个很有可能表明存在两个态之间的耦合. 根据文献[15]苯乙炔的电离能IP为8.83 eV, S2态的0振动态的能量为5.2 eV, S1 态0振动态的能量为4.45 eV, S2态和S1态能量差为0.75 eV, 235 nmm双光探测光电子最大动能Epump+Eprobe – IP为2.65 eV. 我们泵浦光泵浦S2态带源附近, 因此第3光电子能带来自于S2态电离. 图中第2个光电子能带的衰减趋势和第3个光电子能带相似, 随着时间延迟, 第2个和第3个光电子能带都在衰减, 因此我们认为第2光电子能带来自于S1态的电离. 第2个和第3个光电子能带衰减的同时, 第1个光电子能带在增加, 而且第1个光电子带和第2个光电子带的能量差为0.7 eV, 与S2态和S1态能量差为0.75 eV吻合, 因此, 第1个光电子能带很有可能来自与S1态的电离. 第2和第3光电子能带衰减的同时第1光电子能带增加, 很有可能反映的是S2到S1内转换过程. 从母体离子时间分辨质谱中得到的快速衰减时间116 fs很有可能是S2态内转换时间.
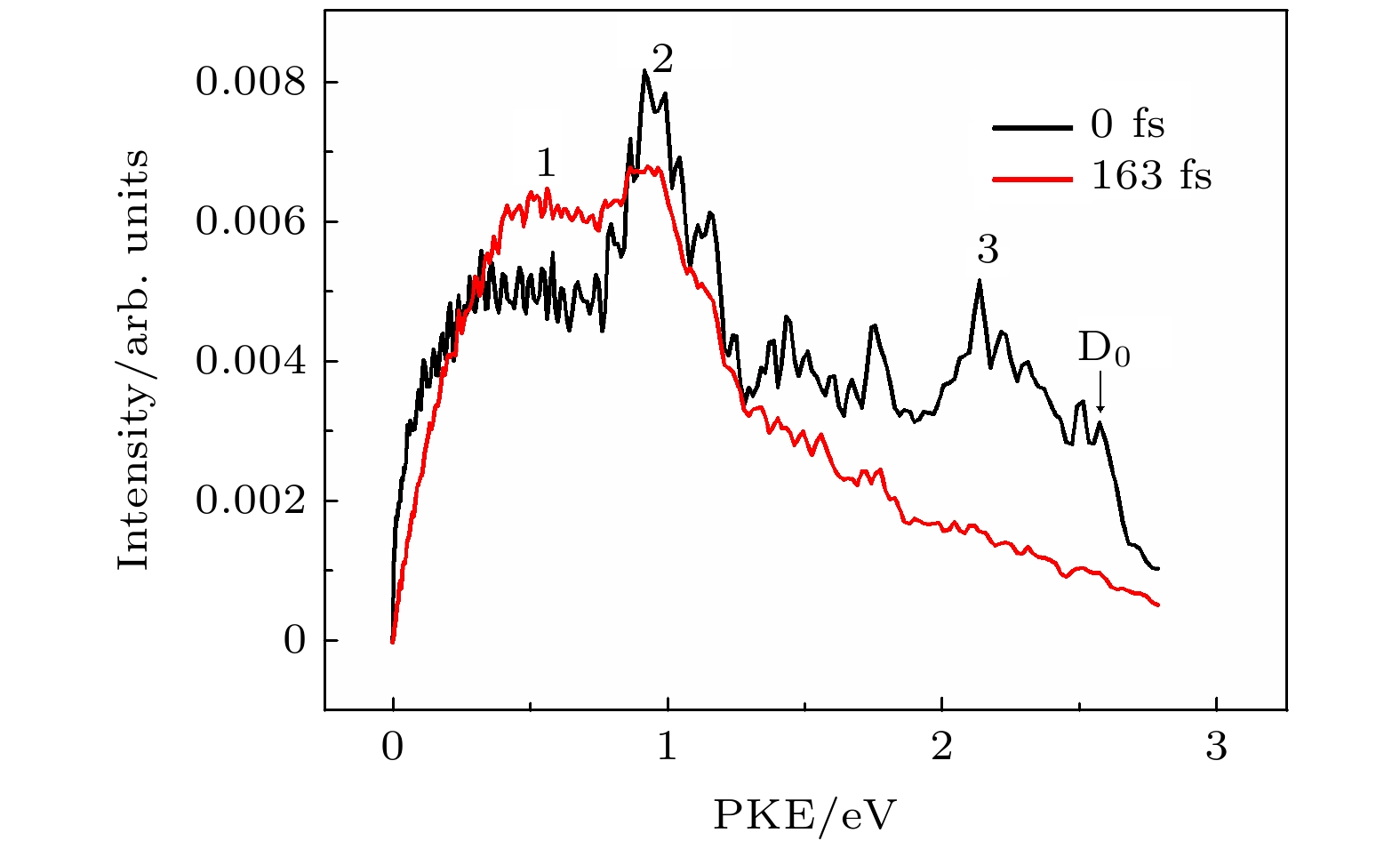 图 2 从Δt = 0 fs和Δt = 163 fs的影像中提取得到的光电子动能分布, 位于D0处的箭头表示(1+2')电离机制下最大的可资用能
图 2 从Δt = 0 fs和Δt = 163 fs的影像中提取得到的光电子动能分布, 位于D0处的箭头表示(1+2')电离机制下最大的可资用能Figure2. Photoelectron kinetic energy distributions at Δt = 0 ps and Δt = 92 ps. The arrow at D0 indicates the maximum electron energy by two-photon absorption of probe beam at 400 nm after one-photon excitation of pump at 235 nm.
2
3.2.S1态的衰减动力学
上面的研究中我们观察到了S2态被泵浦光布局后内转换到S1态, 为了研究S1态的衰减过程, 在实验中我们采集了长时间延迟下的时间分辨离子信号, 同时也采集了长时间延迟下的光电子影像, 图3为长时间延迟下的时间分辨离子信号. 考虑到S1态有可能发生的动力学过程, 我们把离子信号用一个衰减函数、一个上升函数和高斯函数(半高宽为200 fs)的卷积来拟合. 与前面的讨论相结合, 我们认为衰减寿命106 ps (不确定度为 ±2 ps)应该是S1态的寿命, 上升时间60 ps (不确定度为 ±3 ps)很有可能是三重态T1态布局的时间. 图 3 长时间延迟的235 nm泵浦、400 nm探测条件下获得的母体离子时间衰减曲线, 圆圈代表实验结果, 实线代表拟合结果
图 3 长时间延迟的235 nm泵浦、400 nm探测条件下获得的母体离子时间衰减曲线, 圆圈代表实验结果, 实线代表拟合结果Figure3. Time-resolved total ion signals of parent ion as a function of delay time between the pump pulse at 235 nm and the probe pulse at 400 nm. The circles are the experimental results, and solid lines are the fitting results.
实验中我们采集了长时间泵浦-探测延迟下的光电子影像, 图4为从光电子影像中得到的光电子能谱. 从能谱中可以看到随着泵浦探测延迟时间的增加第2和第3光电子能带快速衰减, 而第1个光电子能带向低的光电子动能方向移动, 在553 ps时我们观察到了明显的光电子信号. 在553 ps时S1态的衰减已经结束, 因此0—0.4 eV的这个光电子能带很有可能来自于三重态T1态的电离, 从母体离子时间分辨质谱中拟合得到的60 ps的上升时间就是三重态布局的时间. 实验中观察到的S1态的衰减和T1态的布局, 是内转换布局的S1态继续通过系间交叉衰减到T1态的过程. S1态衰减可能的另外一个衰减通道为内转换到S0态, 但由于我们的探测光没办法探测来自于S0态的信号, 因此不能排除S1向S0内转换的衰减通道, S1到S0的内转换很有可能是S1态衰减的另外一个很重要的通道.
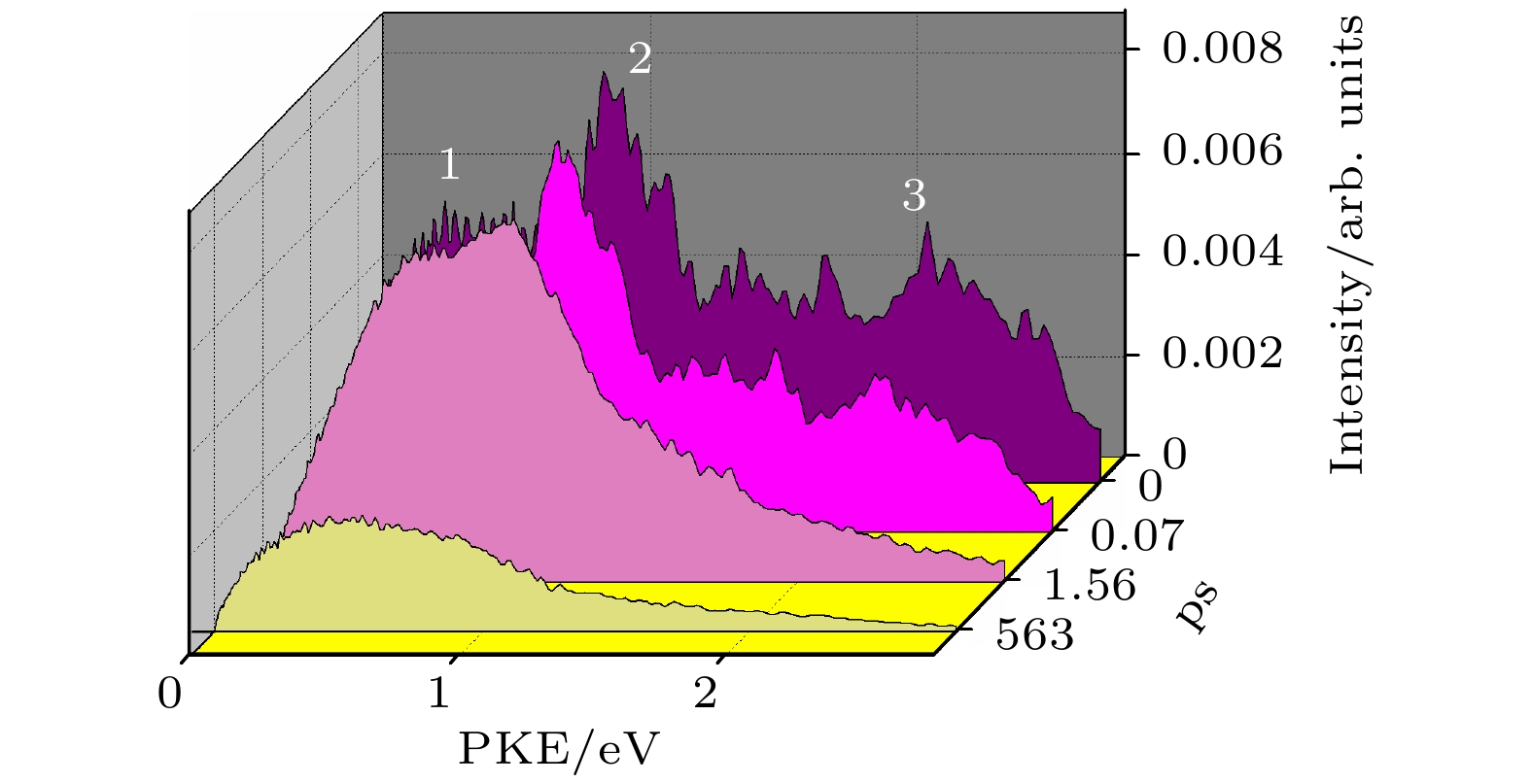 图 4 在235 nm泵浦、400 nm探测不同泵浦-探测延迟下的光电子能谱
图 4 在235 nm泵浦、400 nm探测不同泵浦-探测延迟下的光电子能谱Figure4. Photoelectron kinetic energy distributions (PKE) at different time delay observed at 235 nm pump and 400 nm probe.
由S2内转换的 S1 态具有短寿命为106 ps, 而带源附近的 S1 态寿命却长达纳秒量级, 由此表明苯乙炔 S1 态的超快无辐射过程极有可能是通过 “channel three”[23]进行的. 当苯乙炔分子被激发至 S1 态带源大约为 3500 cm–1 以上的能量区间时 “channel three”打开, 这与“channel three”大约位于S1态带源3000 cm–1以上能量区间的苯分子基本是一致的. 由于苯乙炔分子由内转换S2到S1态之后具有较大的振动能(大约0.75 eV, 6049 cm–1), 因此非常稳定地处于“channel three”内, 随后发生的快速非绝热弛豫过程中, 对应S1态寿命为106 ps, 我们把106 ps归属为二次布居态S1态到三重态T1的系间窜跃时间. 在氟苯胺、苯胺等分子中观察到了通过内转换被布局的S1态向T1态的ISC过程, 氟苯胺分子的ISC时间尺度为379 ps, 苯胺的时间尺度为 > 1 ns和600 ps[23]; 在我们的苯乙炔实验中, 观察到S1向T1态的系间窜跃过程时间为106 ps, 这个可能是由于苯乙炔中乙炔取代基导致其激发态超快动力学过程与苯的其他衍生物差别较大, 表现出取代基效应, 加快了体系发生系间交叉过程的速率.
