全文HTML
--> --> -->众所周知, 化学和材料科学是现代科学技术和国民经济发展的重要基石, 基于第一性原理的分子、材料模拟是研究化学反应和材料物性最重要的方法之一, 为目标功能导向的材料设计提供了可能. 第一性原理计算方法在材料的性质研究和预测方面发挥着越来越大的作用, 已经成为继实验和理论分析之外的最为重要的研究方法和手段之一. 尤其是对于新型的拓扑材料的预测和研究, 第一性原理方法发挥了无可替代的作用.
当前我国在材料计算、反应模拟领域仍然缺乏核心的竞争力和影响力, 一个重要的原因就是成熟的第一性原理系列计算软件的缺失, 仍处于利用国外成熟商业软件, 如: GAUSSIAN[1], VASP[2] (Vienna Ab-initio Simulation Package, 维也纳从头算模拟程序包), 进行研究工作的下游. 这也导致相关基础人才储备不足、后劲缺乏, 影响到计算研究进一步的发展. 虽然目前国内的许多研究小组已经在该领域做出了出色的工作, 也有越来越多的相关程序包发布(如BDF[3], ABACUS[4], BSTATE[5]等), 但是目前有关这方面的研究积累仍然不足, 尤其是国产的适用于大型计算集群、支持异构框架的大规模并行软件仍然缺失. 这极大限制了可计算模拟的材料尺度或者分子大小, 进而制约了相关材料和化学科学的发展.
BSTATE (Beijing Simulation Tool for Atom Technology), 中文名为北京原子技术模拟工具包, 是基于密度泛函理论和平面波赝势(包括模守恒赝势[6]和超软赝势[7,8])方法的第一性原理计算程序包, 主体程序在20世纪90年代由中国科学院物理所方忠研究员及其合作者共同开发. BSTATE主要用于周期性晶体结构下能量、电子密度、能带结构等的计算模拟, 进而得到物质的电、磁、光等其他特性. 除了可以很好地处理一般的基态电子结构计算, 该程序包的长处在于能比较好的处理各种磁有序、轨道序、强自旋轨道耦合体系, 包含了独创的处理强关联体系的LDA+Gutzwiller[9-11]方法, 大幅提高了计算效率[5]. 基于BSTATE程序包, 以物理所凝聚态研究组为主体的研究人员针对Bi2Te3, Bi2Se3和Sb2Te3等拓扑材料进行了第一性原理的计算, 其研究结果表明这些体系是首次发现的三维拓扑绝缘体, 并且在其二维磁性掺杂薄膜中能实现量子化的反常霍尔效应. 这也为极低能量耗散的自旋电子器件设计指出了一个新的发展方向[12-15].
虽然BSTATE程序包已经在拓扑绝缘体的计算中取得了很好的成绩, 但是BSTATE的使用主要局限在少数课题组内, 有必要进行进一步的推广. 当前BSTATE局限性的主要在于用户门槛较高; 泛函数量较为单一, 不好满足特定材料的计算要求; 以及暂时没有考虑流行的硬件框架. 针对上述问题, 我们拟对中科院物理所的第一性原理材料计算软件包BSTATE进行重构. 以期提高软件的易用性和适用范围、降低用户门槛、优化其对流行计算框架的支持.
2
2.1.Schr?dinger方程与DFT理论
波恩-奥本海默近似下, 体系中电子的定态可以由满足非含时Schr?dinger方程的N电子波函数













2
2.2.KS-DFT与泛函近似
KS理论下









为了校正电子密度分布不均匀带来的误差, 将表示电子密度不均匀性的密度梯度(







除了LDA和GGA泛函, meta-GGA和hybrid-GGA[23,24]以及双杂化泛函都是重要的泛函形式. 当前BSTATE暂时没有用到后三种形式的泛函. 泛函确定后, 体系的波函数需要通过求解


2
2.3.BSTATE程序框架
作为第一性原理计算程序包, BSTATE程序包的核心部分是对交换关联势做了LDA或者GGA近似的KS方程的自洽求解. 其基本流程示意图如图1所示. 图 1 BSTATE计算流程示意图
图 1 BSTATE计算流程示意图Figure1. Flowchart of BSTATE package.
以上数值求解的过程中必须把连续量离散化, 如系统实空间原胞划分成足够细的网格, k空间的布里渊区的k点离散化. 在迭代求解KS方程的过程中需要初始化电荷密度


3.1.编译系统
重构前的BSTATE仅能采用GNU make进行程序构建和管理. Makefile文件描述了整个工程的编译、连接等规则. 使用Makefile时, BSTATE的编译步骤是:1)通过预编译指令(GNU的cpp或者intel的fpp), 生成编译时使用的Fortran代码. 值得注意的是, BSTATE的mlwf(Maximally Localised Wannier Functions)部分需要使用自由格式的预编译; 其余需要使用固定格式的预编译. 此外, 预编译后实际生成的代码会受条件编译的控制.
2)预编译后的源代码(.f90, .for)被统一为.f后缀的Fortran代码. BSTATE编译的过程中首先会编译整个程序所需要的模块函数(module), 之后继续编译和连接所有的程序文件.
针对不同的计算机构架和编译环境, BSTATE有着不同的Makefile模板. 并支持Intel MKL和FFTW2等一系列的函数库. 针对不同的编译环境, 用户需要重新修改所有的编译选项. 这对于非计算机专业的用户存在不低的使用门槛, 同时使得跨平台和交叉编译复杂化.
重构后的BSTATE在保留旧版Makefile编译流程的基础上, 提供全新的CMake编译系统支持[25]. CMake全称是“cross platform make”, 是一个开源的跨平台自动化构建系统. 新版的BSTATE使用指定名为CMakeLists.txt的配置文件来控制软件的预编译、编译、连接等流程. 与此同时, CMake天生跨平台的特征, 使得仅需要对于CMakeList.txt进行简单配置, 原则上即可以生成不同目标平台(如Linux, Windows, Mac)的程序文件. 这可以大大简化跨平台和交叉编译方面的工作. 重构后的BSTATE编译系统的特点主要有:
1)保留原版Makefile的编译流程. 使用cmake或者ccmake时可以指定预编译环境(cpp、fpp). 预编译时可以指定条件编译的选项, 生成编译BSTATE时使用的有效代码.
2)内置了不同优化等级的预编译和编译方案(如debug, release等); 支持多线程编译.
3)提供Libxc、FFTW2/3、CUFFT/CUFFTW等外接函数库开关选项; 支持不同语言的混合编译.
4)通过cmake-curses-gui提供图形交互式(GUI)的预编译和编译支持.
5)通过对“MATH_ROOT”或类似关键环境变量的查找, 提供对于常见数学函数库的自动搜索和支持功能. 该功能默认开启, 并按照“MKL, ESSL, OPENBLAS, ATLAS, ACML, SYSTEM_NATIVE”的默认顺序进行数学函数库的自动配置.
通过以上的调整, 重构后的BSTATE可以通过CMake实现自动和快速编译. 比如终端中键入FC = mpif90 ccmake [BSTATE_CMakeList.txt], 即可开启预编译和编译的GUI, 进行自动配置或按提示修改编译选项.
在表1中我们也总结了BSTATE新老编译系统的对比. 可以看出重构后的编译系统相对于旧版, 无论是在代码维护、扩展性、用户使用等等方面均有长足的进步.
| 项目 | 重构前 | 重构后 |
| 编译系统 | GNUMake | CMake |
| 图形GUI | 不支持 | 支持 |
| 跨平台 | 手工修改Makefile文件提供支持 | 原生支持 |
| 数学库 | 手工配置 | 自动配置 |
| 外置函数库 | 手工配置 | 支持自动配置 |
| 异构支持 | 否 | 是 |
| 高级编译选项 | 手工配置 | 支持GUI配置 |
| 多线程编译 | 不支持 | 支持 |
| 用户门槛 | 高 | 低 |
表1重构前后BSTATE编译系统的对比
Table1.Comparison of BSTATE compilation system.
2
3.2.接口升级、函数库整合
重构前的BSTATE程序已经可以调用多种函数库来实现额外的功能或加速计算过程, 例如傅里叶变换FFTW函数库, 但其仅支持老旧的FFTW2接口. 因此, 首先在BSTATE里面增加FFTW3[26]的调用接口, 以便使用常用的SSE2和AVX指令集, 加速运算. 同时, 也初步接入了基于CUDA的傅里叶变换函数库(CUFFTW和CUFFT), 已可取得同CPU版本一致的计算结果, 但是效率仍有很大的提升空间.重构前的BSTATE已经支持LDA, GGA, LDA+U, GGA+U等计算. 但其仅通过固定的代码支持ldapw91和ggapbe两个泛函. 因此将Libxc库[27]接入BSTATE, 使其可以支持流行的各种泛函. Libxc是当前最为全面的交换关联泛函库. 该泛函库由ETSF(European Theoretical Spectroscopy Facility)维护, 并几乎收录了主流期刊公开报道的所有交换关联泛函.
Libxc内部函数相互依赖, 调用需要编译为库文件后才能在子程序中调用. 因此重构时主要通过预编译的方式在多个子程序中配置, 然后再进行调用. 在重构的BSTATE程序中, LDA和GGA泛函通过新添加的extern_functionals.f90接口程序接入. 对于LDA泛函, 势能仅仅依赖于电子的密度(即



作为测试, 使用砷化镓体系(GaAs, 见图2)和石墨烯片段作为测试的算例. 测试的结果表明重构后的BSTATE在DFT计算中性能可以与之前持平(表2和表3), 无论是单核、多核还是MPI并行计算, 性能均没有明显差距. 此外, 受益于FFTW3对于SSE2和AVX系列指令集的支持, 在Intel异构众核平台(MIC)的计算结果会有一定的提升.
 图 2 闪锌矿结构的GaAs双原子化合物
图 2 闪锌矿结构的GaAs双原子化合物Figure2. GaAs compound with zinc blende structure.
| 项目 | 单核/s | 多核/s |
| BSTATE | 42.4 | 22.6 |
| BSTATE+Libxc | 43.6 | 23.4 |
| 性能比 | 0.97 | 0.97 |
| * 测试机器为AMD A10 PRO-7800 B R7 (4核); GaAs体系 | ||
表2Libxc版本与原始版本性能比较
Table2.Benchmarks between BSTATEs with/without Libxc.
| 项目 | CPU平台/s | MIC平台/s |
| FFTW2 | 1181 | 1717 |
| FFTW3+Libxc | 1179 | 1593 |
| 性能比 | 1.00 | 1.08 |
| * CPU平台为Intel至强E7-4830v3 (56核); 石墨烯体系 * MIC平台为Intel Phi-7210 (64核); 石墨烯体系 | ||
表3FFTW3+Libxc版本与原始FFTW2版本性能比较
Table3.Benchmarks between V.FFTW2 and V.FFTW3+ Libxc.
使用整合Libxc和FFTW3的BSTATE版本, 我们在原有的lpw91(LDA)和ggapbe(GGA)泛函的基础上使用相对论(lda_x_rel[28])和自适应(lda_x_rae[29])的LDA泛函计算了GaAs的态密度. 其计算结果列于图3, 可以看出对于该体系不同的泛函会得到整体相似的态密度结果, 主要区别在–6 eV、0—4 eV能量区间.
 图 3 多种泛函计算的GaAs的态密度 (a) (b) (c) (d)
图 3 多种泛函计算的GaAs的态密度 (a) (b) (c) (d)Figure3. Calculated density of states (DOS): (a); (b); (c); (d)
2
3.3.并行分析和测试
提高第一性原理计算模拟软件的计算效率一直是计算科学领域的一大挑战. 其主要原因是电子的强相互作用导致能量和波函数只能在哈密顿构造完之后才可以求解; 而且构造和求解的过程中也涉及各种数据结构或者数值代数方面的问题. BSTATE软件在设计之初就已经很好地考虑这些方面的问题, 并已经可以通过MPI模块按照k点进行分布式求解. 所谓 k点并行化, 即将k点均匀分配给(并行环境中的)多个 CPU, CPU 之间的数据交换较少, 计算效率高.为了详细分析BSTATE的并行性能, 我们选用GaAs晶体和石墨烯片段分别进行小规模和大规模的测试计算. 对于较小体系如GaAs晶体, 使用个人PC在多核(4线程)计算, 分析过程中的各个大类模块所占时间比例统计于图4中(编译采用-O2优化). 可以看出, 傅里叶变换时间占比接近58%, 紧随其后为特征值求解, 时间占比约为12%左右, 态密度、球贝塞尔函数计算部分合计占比均为15%左右. 对于较大体系如石墨烯片段的计算(图4), 我们使用56核MPI并行的集群进行了测试, 测试结果表明特征值求解部分占用最多的时间比例, 约为65%, 同时傅里叶变换时间占比下降到18%左右.
 图 4 个人PC (左图)及集群MPI (右图)测试中BSTATE各模块的时间占比统计
图 4 个人PC (左图)及集群MPI (右图)测试中BSTATE各模块的时间占比统计Figure4. Time percentages of subroutines in BSTATE using PC (left) and Cluster (right).
在图4中集群MPI的并行模式下面, 我们也通过使用TAU ( Tuning and Analysis Utilities; 重构的BSTATE通过CMake可以整合TAU模块) 分析了BSTATE并行时各个线程的执行情况, 并示于图5中. 可以看出在按照现有的基于k点的并行策略, BSTATE各个线程有着较好的负载均衡, 各个计算核心的计算能力都得到很好的发挥.
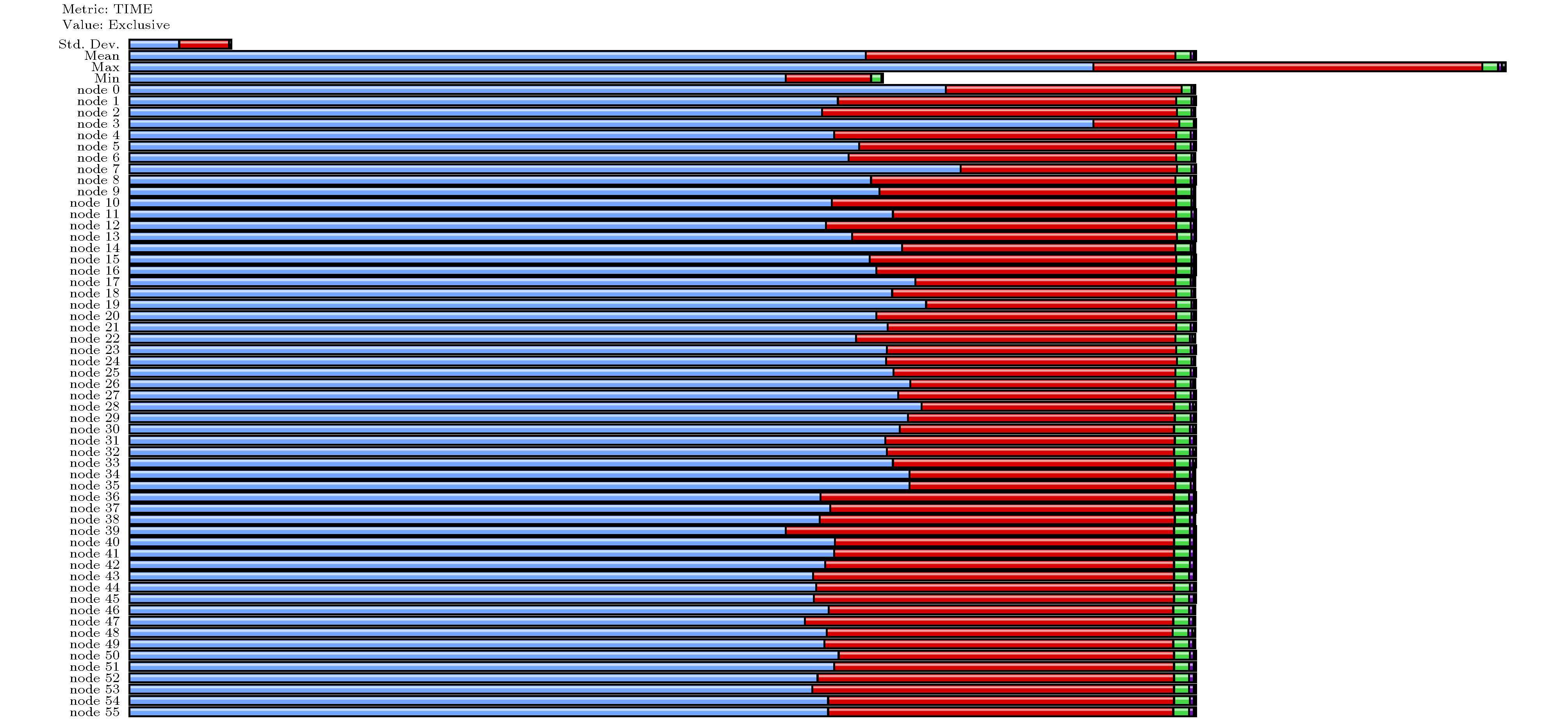 图 5 BSTATE的MPI多线程并行分析; 其中蓝色、红色、黄色、黑色分别代表application(BSTATE), MPI_barrier, MPI_reduce以及MPI_allreduce.
图 5 BSTATE的MPI多线程并行分析; 其中蓝色、红色、黄色、黑色分别代表application(BSTATE), MPI_barrier, MPI_reduce以及MPI_allreduce.Figure5. Analysis via TAU performance system framework for BSTATE
最后, 我们使用重构的BSTATE版本(即包含CMake+TAU+FFTW3+Libxc的版本), 在中国科学院的“元”集群上对石墨烯片段体系进行了更大规模的测试, 以验证新的版本可以稳定的执行计算任务. 该测试平台基于曙光TC4600 E刀片平台, 采用Intel Xeon E5 2680 V3处理器(2.5 GHz、12核). 测试的结果示于图6中. 可以看出, 重构的BSTATE相对于重构前基于FFTW2的版本在跨节点并行方面有着一定的性能提升(0—17%左右的加速). 这应该得益于新的FFTW3调用接口以及基于Libxc的泛函求解方案. 同时我们也注意到当前基于k点的并行求解方案在核心过多的情况下效率下降很快, 这也是实际计算中特别要注意的方面.
 图 6 重构前后的BSTATE跨节点并行测试
图 6 重构前后的BSTATE跨节点并行测试Figure6. Benchmark of MPI parallel for BSTATE with and without refactoring.
在重构的进程中, 也注意到BSTATE仍有较大的改进空间. 比如在原理层面上的哈密顿的构造、子空间迭代等自洽迭代改进方案, 以及技术层面上的CPU-GPU协同计算方面. 这些需要更多的第一性原理算法上的积累和深入, 也是我们后面需要进一步考虑的方面.
感谢徐顺博士关于程序优化方面的指导和讨论.
