摘要: 应用完全活动基自洽场方法, 结合
N 电子价态微扰近似(NEVPT2), 对TiAl金属二聚体的基态和若干最低电子激发态的势能曲线进行了计算. 完全活动空间由Al的3个价电子(3s
2 3p
1 )轨道和Ti的4个价电子(3d
2 4s
2 )轨道构成, 计算基组选用Karlsruhe group的价分裂全电子基组def2-
n ZVPP(
n = T, Q). 在确认TiAl的基态为四重态的基础上, 在核间距
R = 0.200—0.500 nm范围内, 扫描获得了TiAl基态和最低二个激发态的完整势能曲线, 并对电子态进行了标识, 发现在0.255 nm附近存在电子态结构的“突变”. 在
R > 0.255 nm区域, 基态和两个激发态分别为X
4 Δ, A
4 Π和B
4 Γ; 在
R < 0.255 nm区域, 基态仍为X
4 Δ, 但两个激发态变为A'
4 Φ和B'
4 Π, 且存在激发态简并解除的现象. 基于NEVPT2修正后的势能曲线, 获得了TiAl电子态的平衡核间距、束缚能、激发能、跃迁偶极矩等特征参数, 并解释了实验上观测不到TiAl电子跃迁光谱的原因. 电子激发态存在“突变”的结构特征, 可为分析理解TiAl合金在室温下的脆性问题提供参考.
关键词: TiAl /
激发态 /
势能曲线 /
完全活动基自洽场 /
N 电子价态微扰近似English Abstract Ab initio calculation of electronic state structure of TiAlZhang Shu-Dong Wang Chuan-Hang Tang Wei Sun Yang Sun Ning-Ze Sun Zhao-Yu Xu Hui School of Physics and Physical Engineering, Qufu Normal University, Qufu 273165, China Fund Project: Project supported by the National Natural Science Foundation of China (Grant No. 11705101)Received Date: 05 September 2019Accepted Date: 23 October 2019Available Online: 27 November 2019Published Online: 01 December 2019Abstract: The potential energy curves (PECs) of the low-lying electronic states of TiAl are calculated with the complete active space self-consistent field (CASSCF) method combined with the N -electron valence perturbation theory (NEVPT2) approximation. The complete active space is mainly composed of the (3s2 3p1 ) valence orbital of Al and (3d2 4s2 ) valence orbital of Ti. Moreover, the valence splitting all-electron basis set def2-n ZVPP (n = T, Q) proposed by Karlsruhe group is used in the calculation. On the basis of confirming that the ground state of TiAl is a quadruple state, the PECs of the ground state and the lowest two excited states of TiAl are obtained in a range of nuclear distance R of 0.200–0.500 nm, and the electronic states are identified. It is found that there is a “break” of the electronic structure near R = 0.255 nm. In the R > 0.255 nm region, the ground state and the two excited states are X4 Δ, A4 Π and B4 Γ respectively; in the R < 0.255 nm region, the ground state is still X4 Δ, but the two excited states become A'4 Φ and B'4 Π, and the degeneracy of the excited state tends to be eliminated. Based on the PECs of TiAl obtained by the dynamic correlation correction with NEVPT2, the characteristic parameters of three low-lying quadruple electronic states (such as equilibrium nuclear distance, binding energy, adiabatic excitation energy) and transition dipole moment, are obtained, and these parameters are used to explain the reason why the electronic transition spectrum of TiAl is not observed experimentally. The characteristic of “break” in the electronic state structure also provides a meaningful reference for analyzing and understanding the brittleness of TiAl alloy at room temperature.Keywords: TiAl /excited state /potential energy curve /complet active space self-consistent field /N electronic valence perturbation theory approximation 全文HTML --> --> --> 1.引 言 过渡金属铝化物合金属于金属间合金, 是开发在恶劣环境中仍具有高强度和优异抗氧化性材料的重要候选物之一, 对其性质的研究正受到实验和理论计算的广泛关注[1 -9 ] . 作为钛铝合金材料的基本单元, 对TiAl双原子体系几何结构和电子态结构的认识, 为了解该合金的性质提供有意义的重要参考. 钛铝合金在室温下的脆性问题一直困扰着人们, 也限制了其作为结构材料的使用[10 -13 ] . Behm等[14 -16 ] 通过共振双光子电离光谱技术对第一周期过渡金属铝化物进行了实验研究, 但唯独没有观测到ScAl, TiAl和FeAl的光谱. 就是TiAl的基态, 也仅仅是参照MnAl的实验结果加以推断, 得出TiAl的基态可能是4 Δ态. 理论研究方面, 目前多采用密度泛函方法计算合金的性质[17 -20 ] , 但对TiAl双原子体系的研究较少. Ouyang等[21 ] 通过B3LYP密度泛函理论研究了3d金属铝化物的基态, 在6-311+G(2d)(Al)+Lan12DZ (过渡金属原子)基组下计算了键长、谐振动频率和解离能D 0 . 其中对TiAl的计算中, 在默认TiAl的基态为4 Δ态下获得该态的键长、谐振动频率和解离能D 0 分别是0.277 nm, 234.4 cm–1 和1.42 eV. 尚未见对TiAl激发态计算研究的报道.N 电子价态微扰近似(NEVPT2)的基础上, 获得了TiAl最低3个四重电子态的特征参数, 分析讨论实验上观测不到TiAl电子光谱的原因.2.计算方法 所有计算是使用ORCA程序包(版本4.1.1)在计算服务器上完成的[22 ] . 该程序包的突出优点之一就是提供了丰富的激发态计算手段. 使用该程序包, 在完全活动基自洽场(CASSCF)计算[23 ,24 ] 的基础上, 通过非收缩的多参考组态相互作用(uncontracted MRCI)方法, 完成了对ZnAl最低几个电子态的势能曲线扫描计算[25 ] . 由于过渡金属Zn中的d轨道已被电子占满, 故活动空间的选取并未包含d轨道. 但最近在进行ScAl的计算时, 仅仅因为d轨道加入活动空间, 就因为遇到分子轨道收敛困难使得MRCI计算无法顺利进行. 这里报道对TiAl的计算, 也遇到类似的问题, 即用MRCI方法进行动态相关修正计算时, 存在收敛困难或者存在势能曲线不光滑. 为此, 尝试采用强收缩的N 电子价态微扰理论(strong contracted N -electron valence state perturbation theory), 即SC-NEVPT2 (简称NEVPT2), 对CAS的计算结果进行动态相关修正计算, 获得了较为理想的势能曲线图像. 同时也发现, 采用Weigend 和Ahlrichs[26 ] 提出的Karlsruhe全电子分裂价基组, 例如def2-TZVP, def2-TZVPP和def2-QZVPP, 均能完成势能曲线的扫描计算, 但def2-TZVP的计算精度明显不够, 能够用来分析讨论的结果至少需要在def2-TZVPP及以上的基组.[27 ] . 本文的计算中, 首先用密度泛函方法对基态构型进行了优化, 即采用B3LYP/def2-TZVPP优化基态构型, 获得基态的平衡核间距R e . 在保持该核间距不变的条件下, 用MP2方法进行单点能计算并对分子轨道进行分析, 生成自然轨道. 将该自然轨道作为CAS计算中初始轨道猜测输入. 分析CAS计算的活动基轨道组成, 最终确定选用了主要由Al和Ti的10个价电子所在轨道构成的活动空间, 即CAS (7, 10). 在该空间下, 用态平均尝试计算最低的四重态能级, 根据能级分布特征, 选择了最低3个具有二重简并特征的电子态作为势能曲线扫描计算对象. 在顺利完成CAS扫描计算的基础上, 进而选择NEVPT2修正计算.3.计算结果与讨论 23.1.TiAl的可能电子态 3.1.TiAl的可能电子态 由原子数据库数据[28 ] 可知, Al原子的基电子组态和基电子态分别是KL3s2 3p1 和2 Pu , Ti原子的基电子组态及基电子态为KL3s2 3p6 3d2 4s2 和a3 Fg . 按照分子理论[29 ] , 原子群表示分解为异核双原子${C_{\infty \upsilon }}$ 点群表示的对应关系, Pu →$\Sigma _{\rm{u}}^ + + {\Pi _{\rm{u}}}$ , Fg →$\Sigma _{\rm g}^ - \!+ {\Pi _{\rm{g}}} \!+ {\Delta _{\rm{g}}} \!+ {\Phi _{\rm{g}}}$ , 通过$\Sigma _{\rm{u}}^{\rm{ + }} + {\Pi _{\rm{u}}}$ 和$\Sigma _{\rm{g}}^ - + {\Pi _{\rm{g}}} + $ ${\Delta _{\rm{g}}} + {\Phi _{\rm{g}}} $ 的直积可推导出对应最低离解极限Al(2 Pu )+Ti(3 Fg ), TiAl可能的电子态有${}^{2, 4}\Sigma _{\rm{u}}^{\rm{ + }} + $ $ {}^{2, 4}\Sigma _{\rm{u}}^ - (2) + {}^{2, 4}{\Pi _{\rm{u}}}(3) \!+\! {}^{2, 4}{\Delta _{\rm{u}}}(2) + {}^{2, 4}{\Phi _{\rm{u}}}(2) \!+\! {}^{2, 4}{\Gamma _{\rm{u}}}$ , 共11个二重态和11个四重态, 考虑态的简并性, 计算中需要各计算19个二重态和四重态的根. 显然, 要完整地获得如此众多的电子态的信息是困难的, 为此围绕基态和最低的若干激发态开展分析计算.[21 ] 用6-311+G(2d)(Al)+Lan12DZ(Ti)基组的计算值0.277 nm基本一致. 下面的计算围绕四重态进行.3.2.活动基空间选择 -->3.2.活动基空间选择 Al原子有3个价电子(3s2 3p1 ), Ti原子有4个价电子(3d2 4s2 ), 这7个价电子共涉及10个原子轨道. 由群论分析可知, 在${C_{\infty \upsilon }}$ 点群下, Ti的4s, $(3{{\rm{d}}_{xz}}, 3{{\rm{d}}_{yz}})$ , ($3{{\rm{d}}_{{x^2} - {y^2}}}, 3{{\rm{d}}_{xy}}$ ), $(3{{\rm{d}}_{{z^2}}})$ 分别构成${C_{\infty \upsilon }}$ 点群的${\Sigma ^ + }, \;\Pi,\;\Delta, \;{\Sigma ^ + }$ 不可约表示基函数, 即将Ti原子的4s, 3d轨道对称性匹配为$\text{σ}$ (4s), ${\text{π}} (3{{\rm{d}}_{xz}}, $ $3{{\rm{d}}_{yz}}), \text{δ} (3{{\rm{d}}_{{x^2} - {y^2}}}, 3{{\rm{d}}_{xy}}), \sigma (3{{\rm{d}}_{{z^2}}})$ 轨道; 同样, Al的3s和3p轨道分别对称性匹配为${\rm{\text{σ} (3 s), {\text{π}} (3}}{{\rm{p}}_x}, {\rm{3}}{{\rm{p}}_y}), $ $\text{σ} (3{{\rm{p}}_z}) $ 轨道. 由对称性相同的轨道进行组合而得到TiAl的分子轨道. 由这10个价电子原子轨道构成的10个分子轨道为${\rm{\text{σ} (4) + {\text{π}} (2) + \text{δ} }}$ , 其中${\rm{{\text{π}}, \text{δ} }}$ 分别为二重简并轨道.R = 0.27206 nm下, 计算得到的活动基轨道分布如表1 所列.MO No. 14 15 16 17 18 19 20 21 22 23 Energy/Eh –0.4035 –0.1838 0.0029 –0.0159 –0.0159 0.0743 0.0743 0.0439 0.1714 0.1714 Number of occupied electron 1.957 1.772 0.669 0.595 0.595 0.494 0.494 0.222 0.099 0.099 Symbol σ σ σ π π δ δ σ π π Ti s σ 12.1 47.5 7.7 0 0 0 0 13.4 0 0 Ti pz σ 7.5 2.6 1.2 0 0 0 0 38.2 0 0 Ti px π 0 0 0 4.9 0 0 0 0 0 0 Ti py 0 0 0 1.8 0 0 0 0 0 0 Ti dz 2 σ 7 1.7 86.1 0 0 0 0 10.4 0 0 Ti dxz π 0 0 0 31.4 11.4 0 0 0 34.6 31.2 Ti dyz 0 0 0 11.4 31.4 0 0 0 31.2 34.6 Ti dx 2 y 2 δ 0 0 0 0 0 82.6 17.2 0 0 0 Ti dxy 0 0 0 0 0 17.2 82.6 0 0 0 Al s σ 72.9 7.8 1.2 0 0 0 0 0 0 0 Al pz σ 0.5 39.2 3.2 0 0 0 0 36.6 0 0 Al px π 0 0 0 34.4 12.5 0 0 0 16.7 15.1 Al py 0 0 0 12.5 34.4 0 0 0 15.1 16.7
表1 CAS (7, 10)/def2-TZVP计算的活动基分子轨道(MO14?MO23)系数(E h = 2625.5 kJ/mol)Table1. Coefficients of the CAS orbital (MO14?MO23) calculated by CAS (7, 10)/def2-TZVP.表1 可见, 这10个分子活动基轨道编号为MO14-MO23. 注意轨道的次序是按照轨道上电子占居数的多少排列的, 而不是按照轨道的能量高低. 这10个分子轨道恰好均主要由Al和Ti原子的10个价电子对应的原子轨道构成. 按照轨道能量由低到高的次序, 这10个分子轨道依次为σσππσσδδππ, 为方便讨论, 将其依次编号为(1σ)(2σ)(1π)(2π)(3σ)(4σ)(1δ)(2δ)(3π)(4π). 但需要指出的是: 1)${\rm{(1{\text{π}} )}}$ 和${\rm{(2{\text{π}} )}}$ 为简并轨道, 类似的简并轨道还有(1δ)和(2δ), ${\rm{(3{\text{π}} )}}$ 和${\rm{(4{\text{π}} )}}$ ; 2)在势能曲线的扫描计算过程中, 随着核间距的变化, 这10个分子轨道的能级次序可能发生变化, 需要注意观察每个核间距下分子轨道的排序.R = 0.27206 nm下CAS (7, 10)的波函数作为TiAl基态及激发态势能曲线扫描计算的初始值, 进行势能曲线的扫描计算. 扫描区间设定为R = 0.200—0.500 nm. 首先进行态平均自洽场计算, 即SA-CAS (7, 10)的扫描计算, 在此基础上进行强收缩-N 电子价态微扰(SC-NEVPT2)的动态相关的修正计算和非收缩-多参考组态相互作用的修正计算. 但MRCI修正计算的势能曲线存在明显的跳变现象, 这里仅给出SC-NEVPT2修正计算的结果.3.3.TiAl的势能曲线扫描计算 -->3.3.TiAl的势能曲线扫描计算 33.3.1.对势能曲线的CASSCF扫描计算 -->3.3.1.对势能曲线的CASSCF扫描计算 经多次尝试, 分别在基组为def2-TZVP, def2-TZVPP, def2-QZVPP下完成了对TiAl最低6条四重态势能曲线的态平均CASSCF扫描计算, 即SA-CAS (7, 10)计算, 结果如图1 所示, 结果可归结如下.图 1 SA-CAS (7, 10)对TiAl最低6条电子态势能曲线的扫描计算, 其中采用的基组分别是(a) def2-TZVP; (b) def2-TZVPP, (c) def2-QZVPPFigure1. The lowest 6 potential energy curves of TiAl calculated by SA-CAS (7, 10) with basis set of (a) def2-TZVP; (b) def2-TZVPP; (c) def2-QZVPP.R = 0.37 nm附近出现明显的不正常起伏, 这是波函数不稳定造成的. 但基组增大到def2-TZVPP及以后, 函数的稳定性已能满足计算要求. Karlsruhe group提供的这套基组非常适合非相对论条件下对含过渡金属原子分子体系的计算, 而且def2-QZVPP给出最精确的能量计算值. 在本计算中, 基组def2-TZVP, def2-TZVPP, def2-QZVPP所用的收缩后的基函数数目分别为82, 106, 163. 为了得到相对可靠的计算结果, 后续的计算均采用def2-TZVPP基组或更大的def2-QZVPP基组.R = 0.255 nm附近存在跳变. 在def2-TZVP基组下这种跳变极为明显, 随着基组的加大, 这种跳变仍无法消除. 怀疑在该核间距处存在电子轨道的跳变, 后面对活动基轨道能量及其轨道电子占居数随核间距的变化进行分析, 也说明这一推断.$\Pi, \Delta, \Phi $ 等, 而不会是$\Sigma $ 态. 具体电子态的指认通过电子组态及跃迁偶极矩加以分析.$ R\!= $ 0.500 nm处各势能曲线并未完全重合, 但能量差仅为270 cm–1 (def2-TZVP), 220 cm–1 (def2-TZVPP), 200 cm–1 (def2-QZVPP).–1 . 更精确的值应由后面考虑动态相关计算后加以确定.R e 分别为0.285 nm和0.286 nm.3.3.2.活动基轨道能级及轨道上电子占居数分析 -->3.3.2.活动基轨道能级及轨道上电子占居数分析 为了分析图1 中势能曲线在R = 0.255 nm附近的“突变”, 下面以图1(b) 中def2-TZVPP基组下的计算结果为例, 从SA-CAS (7, 10)计算输出文件中逐点提取出10个活动基轨道在各核间距下的轨道能量分布次序以及各轨道上的电子占居数, 结果如图2 和图3 所示.图 2 活动基轨道能量随核间距的变化Figure2. Variation of CAS orbital energy with nuclear distance.图 3 活动基轨道上电子占居数随核间距的变化Figure3. Variation of occupied electrons in the CAS orbital with nuclear distance.图2 和图3 所示的结果可以看出, 在R = 0.255 nm前后, 轨道能级以及轨道上的电子占居数均发生“剧烈”变化.R = 0.200—0.255 nm之间: 除了能量接近的轨道${\rm{(1\text{σ} )}}$ 和${\rm{(1{\text{π}} )(2{\text{π}} )}}$ 在R = 0.215 nm处出现交叉外, 其余轨道能级次序均保持不变, 且电子占居数在该核间距范围内变化不大, 如在R = 0.210 nm和R = 0.240 nm处的电子占居数分布为:${\rm{(1{\text{π}} )(2{\text{π}} )}}$ 和${\rm{(3{\text{π}} )(4{\text{π}} )}}$ 之间. ${\rm{(1{\text{π}} )(2{\text{π}} )}}$ 的轨道能随核间距增大在线性增加, 而${\rm{(3{\text{π}} )(4{\text{π}} )}}$ 的轨道在线性降低; 轨道${\rm{(1{\text{π}} )(2{\text{π}} )}}$ 上的电子占居数从1.24减小为1.18, 而轨道${\rm{(3{\text{π}} )(4{\text{π}} )}}$ 上的电子数却从0.10增加为0.15. 考虑到${\rm{(1{\text{π}} )(2{\text{π}} )}}$ 等为简并轨道, 上面的电子占居分布可简化或等效为:$R = {\rm{ }}0.210\;{\rm{ nm}}, {\left( \text{σ} \right)^{1.93}}{\left( {\text{π}} \right)^{2.5}}{\left(\text{σ} \right)^{1.23}}{\left(\text{δ} \right)^1}$ , 近似为${\left(\text{σ} \right)^2}{\left( {\text{π}} \right)^3}{\left( \text{σ} \right)^1}{\left(\text{δ} \right)^1}$ ;$R = {\rm{ }}0.240\;{\rm{ nm}}, {\left( \text{σ}\right)^{1.93}}{\left( \text{σ} \right)^{1.23}}{\left( {\text{π}} \right)^{2.36}}{\left( \text{δ} \right)^1}$ , 近似为${\left(\text{σ}\right)^2}{\left(\text{σ} \right)^1}{\left( {\text{π}} \right)^3}{\left(\text{δ}\right)^1}$ .R = 0.255 nm前后选取了4个特征点, 即R = 0.200, 0.240, 0.280, 0.490 nm, 从计算输出文件中提取出这4个点处这两组${\text{π}}$ 轨道的主要原子轨道构成, 如表2 所列, 与前面理论分析一样, 这两组π 轨道均主要由${\rm{Ti(3}}{{\rm{d}}_{xz}}, 3{{\rm{d}}_{yz}}{\rm{)}}$ 和${\rm{Al}}(3{{\rm{p}}_x}, 3{{\rm{p}}_y})$ 构成. 但随着核间距的变化, 原子轨道所占比例发生明显变化. 比如${\rm{(1{\text{π}} )(2{\text{π}} )}}$ 轨道, 在R = 0.200和0.240 nm处, ${\rm{Ti}}(3{{\rm{d}}_{xz}}, 3{{\rm{d}}_{yz}})$ 的占比近似为${\rm{Al}}(3{{\rm{p}}_x}, 3{{\rm{p}}_y})$ 的2倍. 而且无论${\rm{(1{\text{π}} )(2{\text{π}} )}}$ 还是${\rm{(3{\text{π}} )(4{\text{π}} )}}$ , 轨道占比以${\rm{Ti}}(3{{\rm{d}}_{xz}}, 3{{\rm{d}}_{yz}})$ 为主.Orbital R = 0.200 nmR = 0.240 nmR = 0.280 nmR = 0.490 nm${\rm{(1{\text{π}})(2{\text{π}})}}$ $\begin{aligned}& {\rm{Ti(}}3{{\rm{p}}_x}, {\rm{ }}3{{\rm{p}}_y}{\rm{) }}7{\rm{\% }} \\& {\rm{Ti(}}3{{\rm{d}}_{xz}}, 3{{\rm{d}}_{yz}}{\rm{) }}60{\rm{\% }} \\ &{\rm{Al(}}3{{\rm{p}}_x}, 3{{\rm{p}}_y}{\rm{) }}28{\rm{\% }}\end{aligned} $ $\begin{aligned}& {\rm{Ti(3}}{{\rm{p}}_x}, {\rm{ 3}}{{\rm{p}}_y}{\rm{) }}7{\rm{\% }} \\& {\rm{Ti(3}}{{\rm{d}}_{xz}}, 3{{\rm{d}}_{yz}}{\rm{) }}5{\rm{7\% }} \\& {\rm{Al(3}}{{\rm{p}}_x}, 3{{\rm{p}}_y}{\rm{) }}3{\rm{2\% }}\end{aligned} $ $\begin{aligned}& {\rm{Ti(3}}{{\rm{p}}_x}, {\rm{ }}3{{\rm{p}}_y}{\rm{) 3\% }} \\ &{\rm{Ti(3}}{{\rm{d}}_{xz}}, 3{{\rm{d}}_{yz}}{\rm{) 73\% }} \\& {\rm{Al(3}}{{\rm{p}}_x}, 3{{\rm{p}}_y}{\rm{) 21\% }}\end{aligned} $ ${\rm{Ti}}(3{{\rm{d}}_{xz}}, 3{{\rm{d}}_{yz}}){\rm{ }}1{\rm{00}}\% $ ${\rm{(3{\text{π}})(4{\text{π}})}}$ $\begin{aligned} &{\rm{Ti(3}}{{\rm{d}}_{xz}}{\rm{, 3}}{{\rm{d}}_{yz}}{\rm{) 52\% }} \\ &{\rm{Al(3}}{{\rm{p}}_x}{\rm{, 3}}{{\rm{p}}_y}{\rm{) 36\% }}\end{aligned} $ $\begin{aligned} &{\rm{Ti(3}}{{\rm{d}}_{xz}}, 3{{\rm{d}}_{yz}}{\rm{) 52\% }} \\& {\rm{Al(3}}{{\rm{p}}_x}, 3{{\rm{p}}_y}{\rm{) 40\% }}\end{aligned} $ $\begin{aligned}& {\rm{Ti(3}}{{\rm{p}}_x}, {\rm{ }}3{{\rm{p}}_y}{\rm{) 12\% }} \\ &{\rm{Ti(3}}{{\rm{d}}_{xz}}, 3{{\rm{d}}_{yz}}{\rm{) 34\% }} \\ &{\rm{Al(3}}{{\rm{p}}_x}, 3{{\rm{p}}_y}{\rm{) 52\% }}\end{aligned} $ ${\rm{Al(3}}{{\rm{p}}_x}, 3{{\rm{p}}_y}{\rm{) 99\% }}$
表2 两组${\text{π}}$ 轨道的组成分析Table2. Composition analysis of two π orbitsR = 0.255—0.500 nm之间: 由图2 可见, 轨道能级次序有多处交叉, 首先是0.255 nm处的${\rm{(3{\text{π}} )(4{\text{π}} )}}$ 与${\rm{(1\text{δ})(2\text{δ} )}}$ 交叉, 然后是0.300 nm附近的${\rm{(3{\text{π}} )(4{\text{π}} )}}$ 分别与${\rm{(1{\text{π}} )(2{\text{π}} )}}$ 和${\rm{(3\text{σ} )}}$ 交叉. 从图3 的电子占居数分析, ${\rm{(2\text{σ} )}}$ 轨道占居数明显增加, 且逐渐接近电子双占居. 而${\rm{(1{\text{π}} )(2{\text{π}} )}}$ 电子占居数在R = 0.255 nm前后发生了急剧变化, 电子占居数几乎减半. 同时, ${\rm{(3{\text{π}} )(4{\text{π}} )}}$ 与${\rm{(1{\text{π}} )(2{\text{π}} )}}$ 恰好相反, 电子占居数急剧增加, 相当于轨道${\rm{(1{\text{π}} )(2{\text{π}} )}}$ 上的电子有一半转移至${\rm{(3{\text{π}} )(4{\text{π}} )}}$ 轨道上. 总之, 在该核间距段, ${\rm{(1{\text{π}} )(2{\text{π}} )}}$ , ${\rm{(3{\text{π}} )(4{\text{π}} )}}$ , ${\rm{(1\text{δ})(2\text{δ} )}}$ 上的电子占居数差距变小, 均趋向于占居数0.5. 为便于观察比较, 下面列出R = 0.280 nm和R = 0.490 nm处的活动基分子轨道次序以及电子占居分布:$(1{\text{π}} )(2{\text{π}} )$ 等为简并轨道, 上面的电子占居分布可简化或等效为:$R \!=\!0.280\;{\rm{ nm}}, {\left( \text{σ} \right)^{1.95}}{\left( \text{σ} \right)^{1.68}}{\left( {\text{π}} \right)^{1.35}}{\left( \text{σ} \right)^{0.32}}{\left( {\text{π}} \right)^{0.80}}{\left( \text{δ} \right)^{0.88}}$ , 近似为${\left( \text{σ} \right)^2}{\left( \text{σ} \right)^2}{\left( {\text{π}} \right)^1}{\left( {\text{π}} \right)^1}{\left( \text{δ}\right)^1};$ $R \!=\!0.490\;{\rm{ nm}}, {\left( \text{σ} \right)^{1.92}}{\left( \text{σ} \right)^{1.90}}{\left( {\text{π}} \right)^{1.0}}{\left( {\text{π}} \right)^{1.18}}{\left( {1\text{δ} } \right)^{0.82}}$ , 近似为${\left( \text{σ} \right)^2}{\left( \text{σ} \right)^2}{\left( {\text{π}} \right)^1}{\left( {\text{π}} \right)^1}{\left( \text{δ} \right)^1}$ .R = 0.490 nm处的活动基轨道组成, 见表3 , 其中编号14—23为活动基分子轨道, 主要电子占居分布可近似表示为(3sAl )1.92 (4sTi )1.90 (3dTi )0.62 (3dTi )0.56 (3pAl )0.52 (3pAl )0.52 (3dTi )0.41 (3dTi )0.41 , 即(3sAl )1.92 (3pAl )0.93 , (4sTi )1.90 (3dTi )2.00 , 近似为(3sAl )2 (3pAl )1 , (4sTi )2 (3dTi )2 , 可见, 此时TiAl基本分解为两个中性原子, 这也恰好符合前面对TiAl最低离解的分析.Orbital No 12 13 14 15 16 17 Energy/Hartree –1.79778 –1.7976 –0.37486 –0.21513 0.02001 0.03822 Occupied electron 2.00000 2.0000 1.91976 1.89900 0.62463 0.55980 Ti s 0 0 2.8 94.5 0 0 Ti pz 0 99.8 0.5 0 0 0 Ti px 55.7 0.1 0 0 0 0 Ti py 44.3 0 0 0 0 0 Ti dxz 0 0 0 0 55.8 43.6 Ti dyz 0 0 0 0 44.1 55.2 Al s 0 0 95.8 2.8 0 0 Orbital No 18 19 20 21 22 23 Energy/Hartree –0.00791 –0.00682 0.0885 0.08979 0.05651 0.1227 Occupied electron 0.51786 0.51750 0.41058 0.40645 0.10159 0.04283 Ti pz 0 0 0 0.0 91.4 5.3 Ti dx 2 y 2 0 0 1.4 98.6 0 0 Ti dxy 0 0 98.4 1.4 0 0 Al pz 0 0 0 0 7 92.7 Al px 55.3 43.3 0 0 0 0 Al py 43.7 54.8 0 0 0 0
表3 R = 0.490 nm处活动基分子轨道MO14-MO23组成Table3. Composition of CAS orbitals MO14-MO23 at R = 0.490 nm.R = 0.255 nm附近的“突变”现象, 可大致归结为电子占居轨道的突变, 即由${\left( \text{σ} \right)^2}{\left( {\text{π}} \right)^3}{\left( \text{σ} \right)^1}{\left( \text{δ} \right)^1}$ 跳变为${\left( \text{σ} \right)^2}{\left( \text{σ} \right)^2}{\left( {\text{π}} \right)^1}{\left( {\text{π}} \right)^1}{\left( \text{δ} \right)^1}$ , 因而R = 0.255 nm两边的能态是不同的, 下面对能态进行分析标识.3.3.3.电子态的标识 -->3.3.3.电子态的标识 由图1(b) 和图1(c) 的势能曲线可得出, 在“突变”点R = 0.255 nm前后的电子态是不同的, 为此, 以def2-TZVPP基组下的计算结果为例, 即从图1(b) 所示的计算输出文件中分别提取出R = 0.240 nm和R = 0.285 nm时各电子态的相应的组态以及由基态向激发态的跃迁偶极矩, 结果如表4 所列.R /nmstate Main configuration Excitation energy/cm–1 Transition dipole moment T 2 /Debye2 Possible quartet state Idetified state 0.285 Ground state ${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{2}}}{{\text{π}}^{\rm{2}}}{{\rm{\text{δ} }}^{\rm{1}}}{{\text{π}}^{\rm{0}}}$ 0 4 ΔX4 Δ 1st excited state ${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{2}}}{{\text{π}}^{\rm{2}}}{{\rm{\text{δ} }}^{\rm{0}}}{{\text{π}}^{\rm{1}}}$ 3212 0.034 4 ΠA4 Π 2nd excited state ${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{2}}}{{\text{π}}^{\rm{1}}}{{\rm{\text{δ} }}^{\rm{1}}}{{\text{π}}^{\rm{1}}}$ 3462 0 4 Σ, 4 Δ(2), 4 ΓB4 Γ 0.240 Ground state ${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{2}}}{{\text{π}}^{\rm{2}}}{{\rm{\text{δ} }}^{\rm{1}}}$ 0 4 ΔX4 Δ 1st excited state ${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{1}}}{{\text{π}}^{\rm{3}}}{{\rm{\text{δ} }}^{\rm{1}}}$ 4140 0.00824 4 Π, 4 ΦA'4 Φ 2nd excited state ${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{1}}}{{\text{π}}^{\rm{3}}}{{\rm{\text{δ} }}^{\rm{1}}}$ 4727 0.00869 4 Π, 4 ΦB'4 Π 3rd excited state ${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{1}}}{{\text{π}}^{\rm{3}}}{{\rm{\text{δ} }}^{\rm{1}}}$ 5074 0.00551 4 Π, 4 ΦB'4 Π
表4 基态及最低激发态的组态及跃迁偶极矩Table4. Configuration and transition dipole moment of the ground state and the lowest excited stateR = 0.285 nm${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{2}}}{{\text{π}}^{\rm{2}}}{{\rm{\text{δ} }}^{\rm{1}}}{{\text{π}}^{\rm{0}}}$ 可以推算出可能的四重态只有一个, 即4 Δ, 即基态为X4 Δ. 对于第一激发态的组态${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{2}}}{{\text{π}}^{\rm{2}}}{{\rm{\text{δ} }}^{\rm{0}}}{{\text{π}}^{\rm{1}}}$ , 可能的四重态也只有一个, 即4 Π, 将其标记为A4 Π. 对于第二激发态的组态${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{2}}}{{\text{π}}^{\rm{1}}}{{\rm{\text{δ} }}^{\rm{1}}}{{\text{π}}^{\rm{1}}}$ , 可能的四重态有四个, 即4 Σ, 4 Δ(2), 4 Γ, 考虑到势能曲线已经表明该态为二重简并态, 按照Hund定则, 4 Γ的能量应该最低, 故第二激发态应该为4 Γ态, 且基态X4 Δ到4 Γ态的跃迁不符合跃迁规则, 这也符合表4 中该跃迁偶极矩为0的计算结果, 将其标识为B 4 Γ. 图4 给出了SA-CAS (7, 10)计算下的标识.图 4 TiAl最低四重电子态的标识(SA-CAS计算结果)Figure4. Identification of the lowest quadruple electronic state of TiAl (SA-CAS calculation results).R = 0.240 nmR = 0.285 nm时一样, 故基态为X4 Δ. 对于简并的第一激发态, 其主要组态为${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{1}}}{{\text{π}}^{\rm{3}}}{{\rm{\text{δ} }}^{\rm{1}}}$ , 可能的四重态只有2个, 即4 Π, 4 Φ. 按照Hund定则, 4 Φ的能量应该最低, 故该激发态应该为4 Φ态, 且基态X4 Δ到4 Φ态的跃迁符合跃迁规则, 这也符合表4 中该跃迁偶极矩不为0的计算结果, 将其标识为A'4 Φ. 对于第二和第三激发态, 虽然势能曲线没有能够重合, 但它们的主要电子组态和第一激发态的一样, 均为${{\rm{\text{σ} }}^{\rm{2}}}{{\rm{\text{σ} }}^{\rm{1}}}{{\text{π}}^{\rm{3}}}{{\rm{\text{δ} }}^{\rm{1}}}{\rm A}$ , 可能的四重态也只能为4 Π和4 Φ, 且基态X4 Δ到它们的跃迁偶极矩也不为0, 将其标识为B'4 Π. 同时注意到, 基态X4 Δ到A'4 Φ和B'4 Π的跃迁偶极矩远小于X4 Δ到A4 Π的值.3.3.4.动态相关修正计算 -->3.3.4.动态相关修正计算 基于上面的CAS计算及其态的标识, 可将TiAl最低的三个四重态明确标识出来, 并在CAS(7, 10)计算的基础上, 进行了SC-NEVPT2动态相关修正计算, 图5 给出了在Karlsruhe提供的最大基组def2-QZVPP下的计算结果. 为方便显示, 取基态势能曲线的最小值为能量参考点, 势能的相对能量用波数表示.图 5 TiAl电子态势能曲线的SC-NEVPT2动态相关修正计算Figure5. Dynamic correlation correction calculation of SC-NEVPT2 for TiAl electronic potential energy curve.4 Π和B4 Γ而言, CAS计算显示只在“突变”点R = 0.255 nm右方, 即R = 0.26—0.50 nm之间出现势阱, 而SC-NEVPT2修正后, 在该点前后均存在极小值, 即出现双势阱, 两势阱之间的势垒高度大约为2700 cm–1 .4 Δ的平衡核间距分别为0.288 nm和0.266 nm, 即NEVPT2修正后核间距明显减小了. 激发态的情形也类似, 如突变点右方的势阱, A4 Π的R e 由0.320 nm (CAS)减小为0.296 nm (NEVPT2), B4 Γ态的相应变化为0.324 nm (CAS)和0.306 nm (NEVPT2).R = 0.500 nm处的能量作为参照, 基态X4 Δ的束缚能由3016 cm–1 (CAS)增大为8151 cm–1 (NEVPT2). 激发态A4 Π和B4 Γ态的势阱深度也相应增加, 具体数值见表5 .State R e /nmD e /cm–1 CAS NEVPT2 CAS NEVPT2 X4 Δ 0.288 0.266 3016 8151 A4 Π 0.320 $\left\{\begin{aligned}& {0.248} \\ & {0.296} \end{aligned} \right.$ 796 $\left\{\begin{aligned}& {3845} \\ & {3406} \end{aligned} \right.$ B4 Γ 0.324 $\left\{\begin{aligned}& {0.248} \\ & {0.306} \end{aligned} \right.$ 711 $\left\{\begin{aligned}& {2884} \\ & {3406} \end{aligned} \right.$
表5 TiAl最低3个四重态的结构参数Table5. Structural parameters of the lowest three quadruple states of TiAl.R < 0.255 nm区域, A'4 Φ和B'4 Π态的简并特征趋向于退简并. 这可能是由于核间距的缩小, 电子自旋与电子轨道的耦合得以加强, 使得简并趋于消除.3.4.实验上观测不到TiAl光谱的解释 -->3.4.实验上观测不到TiAl光谱的解释 如果图5 所示的势能曲线最为接近真实情况, 那么可以根据这一计算结果来解释为什么实验上没有能够观测到TiAl的光谱. TiAl基态X4 Δ的平衡核间距为R e = 0.266 nm (考虑NEVPT2修正), 而结构的“突变”点在0.255 nm附近, 非常靠近R e . 因而TiAl由基态的光跃迁可能存在不稳定, 或者吸收系数很小. 同时, 基态X4 Δ与共振激发态A4 Π的平衡核间距差距也比较大, 即分别为0.266 nm和0.296 nm, 因而Franck-Conden因子也应该偏小. 这些因素最终决定了实验上很难观测到TiAl的光谱. 同时从电子态的束缚能分析(仅依据NEVPT2修正后的计算结果), 如表5 所列, TiAl基态的束缚能D e 仅为8151 cm–1 (约1.01 eV). TiAl由基态X4 Δ (R e = 0.266 nm)到共振激发态A4 Π的垂直激发能和绝热激发能分别为6001 cm–1 (0.74 eV)和4908 cm–1 (0.61 eV), 而A4 Π的束缚能仅为3406 cm–1 (0.42 eV). 如果TiAl被垂直激发到激发态A4 Π, 其过剩的动能很容易跨越激发态A4 Π的浅势阱, 从而导致TiAl离解. TiAl电子结构上的这一特殊情形, 也可为理解TiAl合金表现出的脆性提供参考.4.结 论 采用CASSCF+NEVPT2方法, 在CAS (7, 10)空间下完成了TiAl基态及最低两个激发态的势能曲线扫描计算. 这几个态均为束缚态, 且完全趋向于中性原子Ti+Al的离解极限. 根据电子组态特征、跃迁偶极矩大小等信息, 对这几个电子态进行了标识, 得出基态为4 Δ态, 并发现R = 0.255 nm附近可能存在一个电子态结构的“突变”点. 由于电子在分子轨道中的布局在该点处发生跳变, 导致该点前后的激发态不同. 在R > 0.255 nm区域, 基态和两个激发态分别为X4 Δ, A4 Π和B4 Γ; 在R < 0.255 nm区域, 三个电子态变为X4 Δ, A'4 Φ和B'4 Π, 且存在激发态简并解除的现象. 通过对电子态平衡核间距、激发能、束缚能等特征参数的分析, 得出实验上观测不到TiAl电子跃迁光谱可能与激发态结构存在“突变”有关, 且由于Franck-Conden因子偏小、激发态势阱太浅, 均容易导致TiAl被激发后容易发生解离. 
























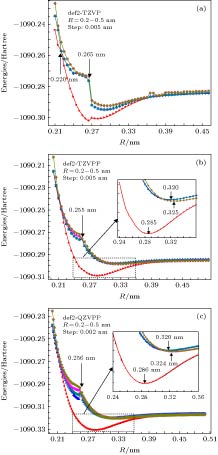 图 1 SA-CAS (7, 10)对TiAl最低6条电子态势能曲线的扫描计算, 其中采用的基组分别是(a) def2-TZVP; (b) def2-TZVPP, (c) def2-QZVPP
图 1 SA-CAS (7, 10)对TiAl最低6条电子态势能曲线的扫描计算, 其中采用的基组分别是(a) def2-TZVP; (b) def2-TZVPP, (c) def2-QZVPP


 图 2 活动基轨道能量随核间距的变化
图 2 活动基轨道能量随核间距的变化 图 3 活动基轨道上电子占居数随核间距的变化
图 3 活动基轨道上电子占居数随核间距的变化














































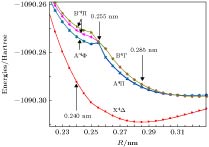 图 4 TiAl最低四重电子态的标识(SA-CAS计算结果)
图 4 TiAl最低四重电子态的标识(SA-CAS计算结果)

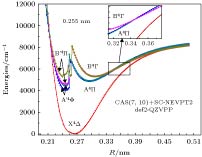 图 5 TiAl电子态势能曲线的SC-NEVPT2动态相关修正计算
图 5 TiAl电子态势能曲线的SC-NEVPT2动态相关修正计算