全文HTML
--> --> -->迄今为止, 研究者相继开发了掺锑氧化锡(ATO)[1]、掺铟氧化锡(ITO)[2]、稀土六硼化物(ReB6)[3,4]、氮化钛(TiN)[5]、二氧化钒(VO2)[6]、钨青铜类化合物MxWO3[7-15] (M=Tl, Na, K, Rb, Cs; 0 < x ≤ 0.33)等透明隔热窗用近红外吸收材料, 但上述材料各有其优缺点. 纳米ATO[1]只能屏蔽光波长1500—2500 nm的近红外辐射, 纳米ITO[2]只能屏蔽光波长1200—2500 nm的近红外辐射, 但对光波长780—1200 nm的高能近红外辐射无能为力. 氮化钛[5]是非计量化合物, 它的组成范围为 TiN0.6—TiN1.16, 但其光学性能受N/Ti比率的影响很大. 纳米VO2[6]虽然存在从高温金属相到低温绝缘体相的转换特性可用于智能温控窗, 但其透明隔热性能较差, 无法达到高性能窗用透明隔热材料的要求. 纳米六硼化物ReB6, 特别是LaB6, 虽然可屏蔽掉光波长780—1300 nm的高能近红外辐射[3,4], 但对光波长1300 nm以上的近红外辐射屏蔽性能较差. 在诸如此类透明隔热窗用近红外吸收材料中, 由于MxWO3具有其它材料无法比拟的可见光高透明、近红外辐射强吸收屏蔽性能, 研究者对金属离子掺杂六方相三氧化钨(MxWO3)的透明隔热性能进行了大量的实验研究[7-15], 而理论研究较少. Yang等[16,17]采用HSE杂化密度泛函计算了碱金属单掺杂和共掺杂六方相三氧化钨的电子结构和光学性质, 但掺杂含量太低(0.083 %), 远少于实验掺杂含量(0.15%— 0.33%); Lee等[18]也采用HSE杂化密度泛函计算了Na0.33WO3 和 K0.33WO3的光学性质, 但与实验值相差甚远; Yoshio和Adachi[19]采用密度泛函理论(DFT)+U方法研究了铯掺杂六方相三氧化钨的光学性质. Xu等[20]采用广义密度梯度近似(GGA)研究了Cs0.33WO3的光学性质, 理论研究结果与实验结果基本一致.
鉴于实验报道Tl掺杂h-WO3形成的Tl0.33WO3也具有太阳辐射屏蔽性能[7], 但其理论研究鲜有报道. 因此, 本文采用DFT-GGA方法对Tl0.33WO3的能带结构、形成能、态密度和光学性质进行理论研究, 在此基础上预测其太阳辐射屏蔽性能.
2.1.晶体结构
六方相三氧化钨(h-WO3)属于六方晶系, 空间群为P6/mmm (No. 191), 具有WO6氧八面体六元环和三元环的ab平面内的沿c轴堆积, 形成平行c轴的六边形和三边形的准一维孔道[21]. 六方相三氧化钨中W原子位于(0.5, 0, 0), O原子位于(0.5, 0, 0.5)和(0.212, 0.424, 0). Tl离子嵌入在WO6八面体六元环内, 位置为(0, 0, 0)[22] (如图1所示).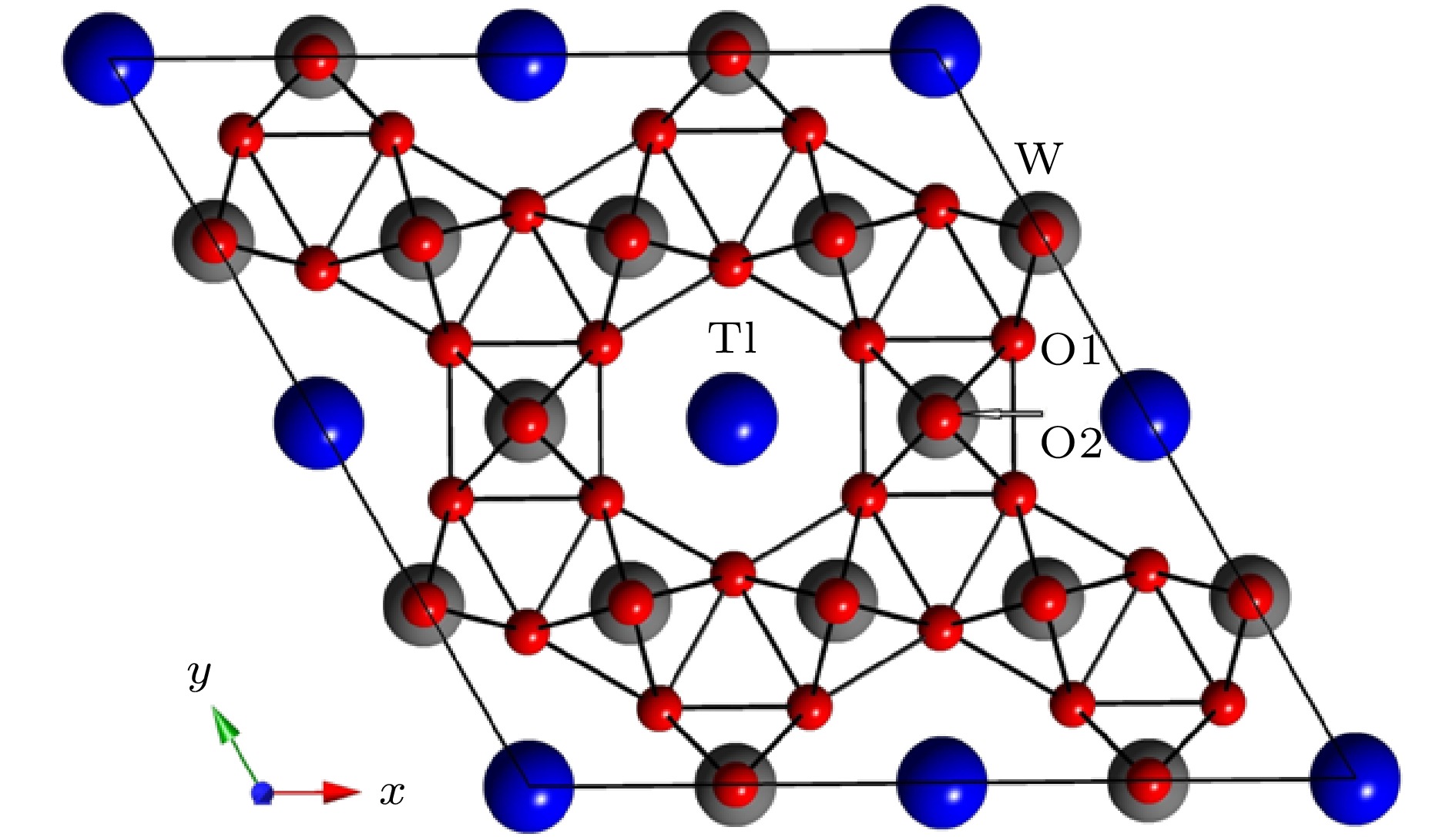 图 1 Tl掺杂h-WO3的2 × 2 × 1超晶胞俯视图
图 1 Tl掺杂h-WO3的2 × 2 × 1超晶胞俯视图Figure1. Top view of the 2 × 2 × 1 supercell of Tl-doped h-WO3
2
2.2.计算方法
本文所进行的第一性原理计算是采用基于密度泛函理论的CASTEP软件[23]. 计算首先采用BFGS算法对六方相三氧化物(h-WO3)的晶体结构进行几何结构优化, 然后建立2 × 2 × 1的超晶胞, 几何结构优化后, Tl原子掺杂在WO6八面体六元环内, 构成Tl0.33WO3晶体结构. 在对其进行几何结构优化得到稳定晶体体系的基础上, 进行能带结构、态密度和光学性质的计算. 交换关联势采用广义梯度近似(GGA)[24]中的PBE[25] 提出的形式, 描述离子实与价电子之间的相互作用势采用超软赝势进行. 计算中Tl, W和O各原子的电子价态分别为6s26p1, 5p65d46s2, 2s2p4, 其它轨道电子作为芯电子进行计算. 平面波截止能和布里渊区k点的设置分别为500 eV和9 × 9 × 15. 迭代过程中的自洽收敛精度为5.0 × 10–6 eV/atom, 晶体内应力收敛标准为0.02 GPa, 最大位移为5.0 × 10–4 ?以及原子间相互作用力收敛标准为小于0.01 eV/?. h-WO3的电子能带结构和光学性质计算均采用了剪刀算符修正带隙值. h-WO3的带隙计算值为0.62 eV, 而实验值为2.6—3.25 eV[26-29], 因此, 剪刀算符设为2.0 eV.3.1.几何结构优化结果
表1为采用基于第一性原理的广义密度梯度近似方法, 对h-WO3和Tl0.33WO3的进行结构优化及能带结构计算得到的晶格参数和能带间隙. 其中, Tl0.33WO3的晶格参数为Tl空位掺杂六方相三氧化钨(h-WO3) 的超晶胞结构几何优化后, 折合成单晶胞的结构参数. 计算得到的纯h-WO3的晶格参数和带隙值的计算误差均小于2.0 %, 与实验和理论值基本一致. Tl掺杂后晶格体积比纯h-WO3晶体稍有增大, 这主要是六元环的半径为0.163 nm[30], 而Tl的离子半径为0.184 nm[31], 当嵌入的离子半径大于六元环的半径时, 造成晶格畸变, 从而使晶格体积增大.| a/? | b/? | c/? | Eg/eV | Ef/eV | |
| h-WO3 (Expt.) | 7.298[1] | 7.298[1] | 3.899[32] | ||
| h-WO3 (Calc.) | 7.4403 | 7.4403 | 3.8240 | 0.62 | |
| 7.438[2] | 7.438[2] | 3.827[33] | 0.66 | ||
| Tl0.33WO3(Calc.) | 7.4673 | 7.4673 | 3.8220 | 0 | –2.359 |
表1Tl掺杂六方相WO3前、后的晶格参数、带隙与形成能
Table1.Lattice parameters, band gap and formation energy of pure h-WO3 before and after Tl-doped
2
3.2.形成能分析
通过计算Tl掺杂纯h-WO3的形成能可分析其相对稳定性[34,35], 计算公式为2
3.3.能带结构和态密度分析
33.3.1.纯h-WO3能带结构和态密度分析
为方便比较和分析空位掺杂对纯h-WO3能带结构和态密度的影响, 先计算了纯h-WO3的能带结构和态密度如图2所示. 从图2(a)的能带结构计算结果可知, 纯h-WO3带隙较大, 导带底和价带顶分别位于Brillouin区高对称点Γ和点A处, 为间接带隙半导体. 其间接带隙计算值为0.62 eV, 经剪刀算符2.0 eV修剪后间接带隙值为2.62 eV. 由图2纯h-WO3的总态密度可知, 价带和价带顶态密度则主要由O-2p组成, 导带和导带底的态密度主要是W-5d态构成. 从分态密度可知, 纯h-WO3的价带和导带都主要由局域化的O-2p和W-5d轨道电子构成并发生杂化, 表明O原子与W原子之间共价键比较明显, 这一特性与文献的计算结果一致.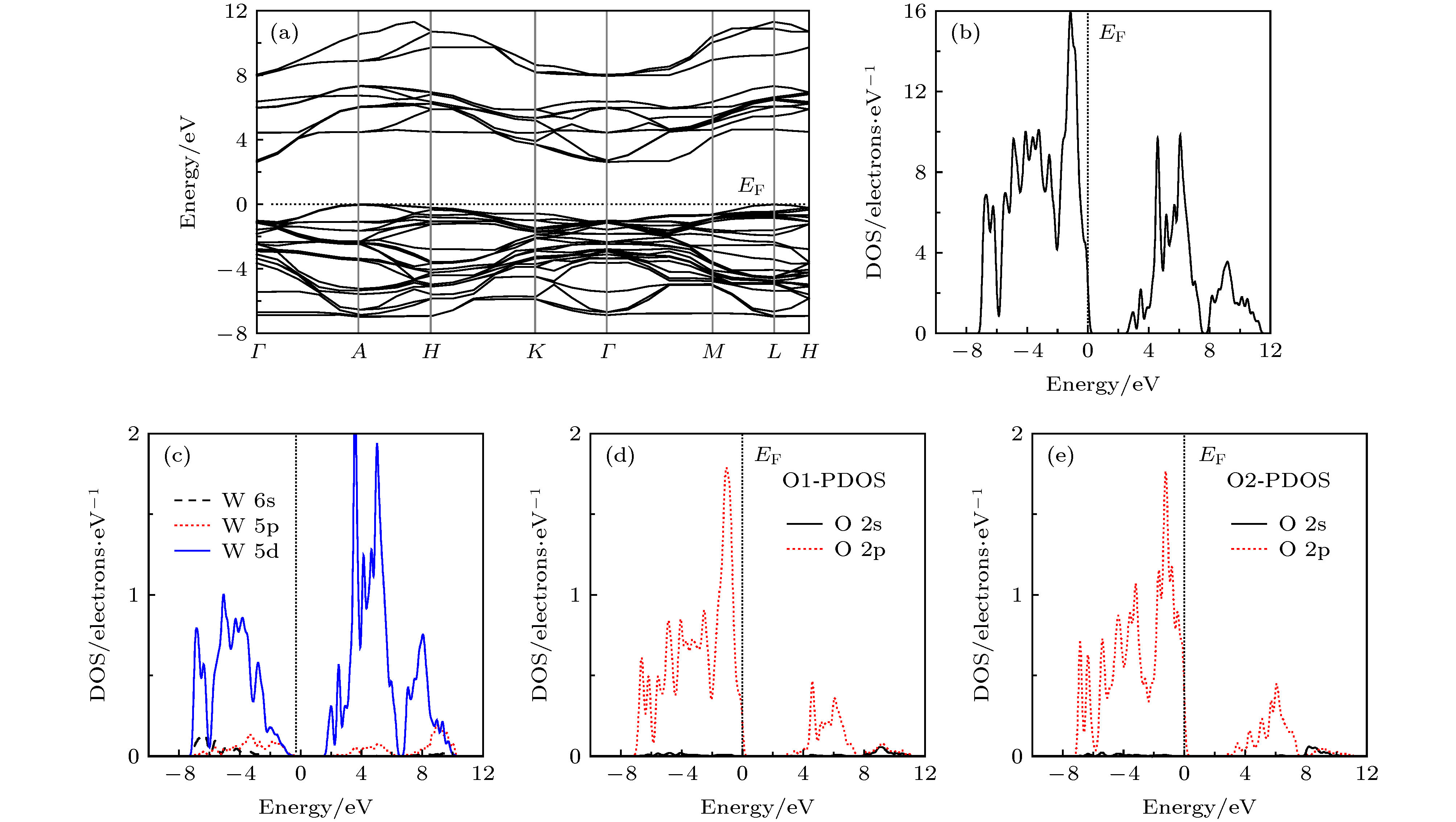 图 2 纯h-WO3的能带结构和态密度: (a)能带结构; (b)总态密度; (c) W的分态密度; (d) O1的态密度; (e) O2的分态密度
图 2 纯h-WO3的能带结构和态密度: (a)能带结构; (b)总态密度; (c) W的分态密度; (d) O1的态密度; (e) O2的分态密度Figure2. Energy band structure and DOS of pure h-WO3: (a) Energy band structure; (b) TDOS of h-WO3; (c) PDOS of W; (d) PDOS of O1; (e) PDOS of O2
3
3.3.2.Tl0.33WO3能带结构和态密度分析
Tl空位掺杂h-WO3能带结构和态密度如图3所示. 对比图3与图2的分态密度可知, Tl0.33WO3价带主要由O-2p、W-5d和Tl-6s态组成, 导带主要W-5d和Tl-6p态组成, 尤为重要的是W-5d越过费米能级EF进入价带, 价带顶和导带底均由W-5d态构成. 这表明Tl掺杂h-WO3主要扮演着捐献电子的角色. 从图3(a)可知, 价带顶和导带底的位置没有改变; 但价带整体下移, 导带底越过费米能级EF形成简并态, 并且导带和价带之间仍存在一定的间隙, 这表明Tl空位掺杂构成的Tl0.33WO3晶体仍保持n型电导率. h-WO3掺杂前后费米能级的变化表明晶体从半导体向导体转变, 这必将导致材料的光学性质的改变[36,37]. 后文中的光学性质的计算结果将进一步印证这一结论. 图 3 Tl掺杂h-WO3(Tl0.33WO3)能带结构和态密度: (a)能带结构; (b)总态密度; (c) Tl的分态密度; (d) W的分态密度; (e) O1的态密度; (f) O2的分态密度
图 3 Tl掺杂h-WO3(Tl0.33WO3)能带结构和态密度: (a)能带结构; (b)总态密度; (c) Tl的分态密度; (d) W的分态密度; (e) O1的态密度; (f) O2的分态密度Figure3. Energy band structure and DOS of Tl0.33WO3: (a) Energy band structure; (b) TDOS of Tl0.33WO3; (c) PDOS of Tl; (d) PDOS of W; (d) PDOS of O1; (e) PDOS of O2
2
3.4.光学性质
为研究Tl掺杂前后h-WO3的光学性质, 对Tl空位掺杂前后六方相h-WO3的介电函数进行了计算研究. 在计算h-WO3的光学性质时, 首先采用剪刀算符对其电子结构进行了修正, 计算光学性质时也采用相同剪刀算符进行修正, 本文h-WO3的电子结构和光学性质计算的剪刀算符取值为2.0 eV. 介电函数表达式为 ε(ω) = ε1(ω) + iε2(ω), 相关计算公式如下[38]:
图4 (a)为Tl掺杂h-WO3前后的介电函数虚部ε2(ω), 虚部与带间跃迁有关, 取决于导带与价带的跃迁, 可通过能带结构与态密度解释. 从图4 (a)可以看出, h-WO3的介电函数阈能约为2.6 eV, 这是h-WO3的基本吸收边, 当光子能量高于此值时, 虚部曲线迅速上升. 纯h-WO3的介电函数有两个特征峰, 峰1和峰2分别对应光子能量为5.8 eV和11.4 eV. 峰1对应O-2p态电子向W-5d态直接跃迁所致; 峰2是下价带能级电子向上导带能级跃迁所致. Tl原子空位掺杂后, Tl0.33WO3在低能0.1 eV处增加新的最大特征峰, 这是价带顶W-5d态向导带底W-5d态跃迁所致, 其它两个特征峰向低能方向移动.
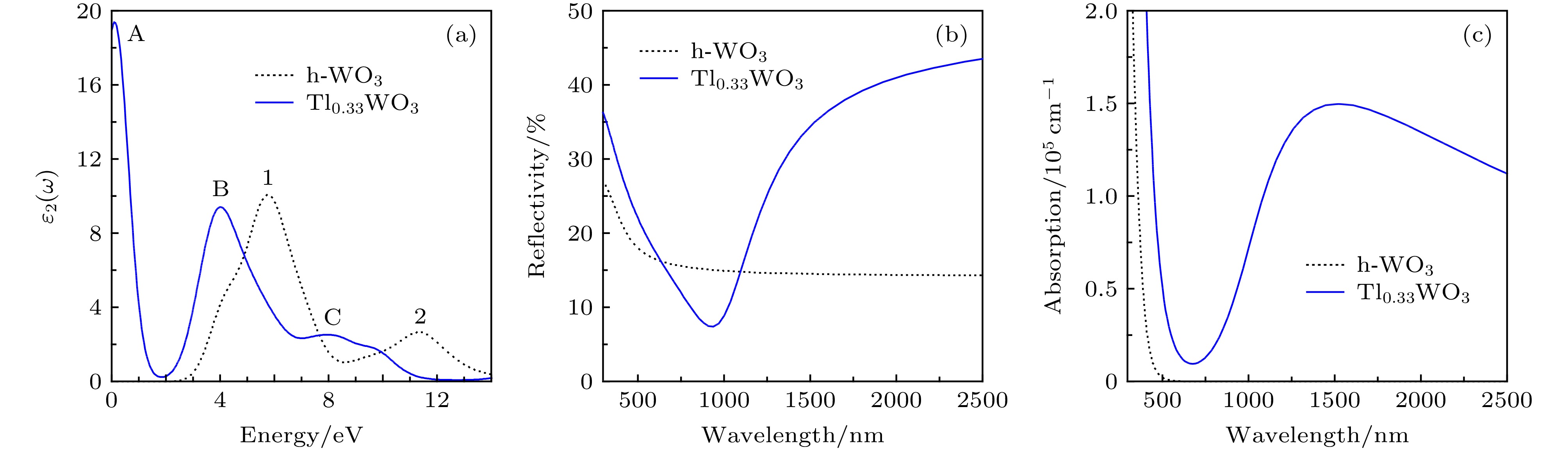 图 4 h-WO3和Tl0.33WO3的光学性质: (a)介电函数虚部; (b)反射谱; (c)吸收谱
图 4 h-WO3和Tl0.33WO3的光学性质: (a)介电函数虚部; (b)反射谱; (c)吸收谱Figure4. Optical performance of h-WO3 and Tl0.33WO3: (a) Imaginary part of the dielectric function; (b) reflectivity; (c) absorption spectrum
采用第一性原理计算出来的材料的反射谱R(ω)和吸收谱α(ω)代入公式[39]:
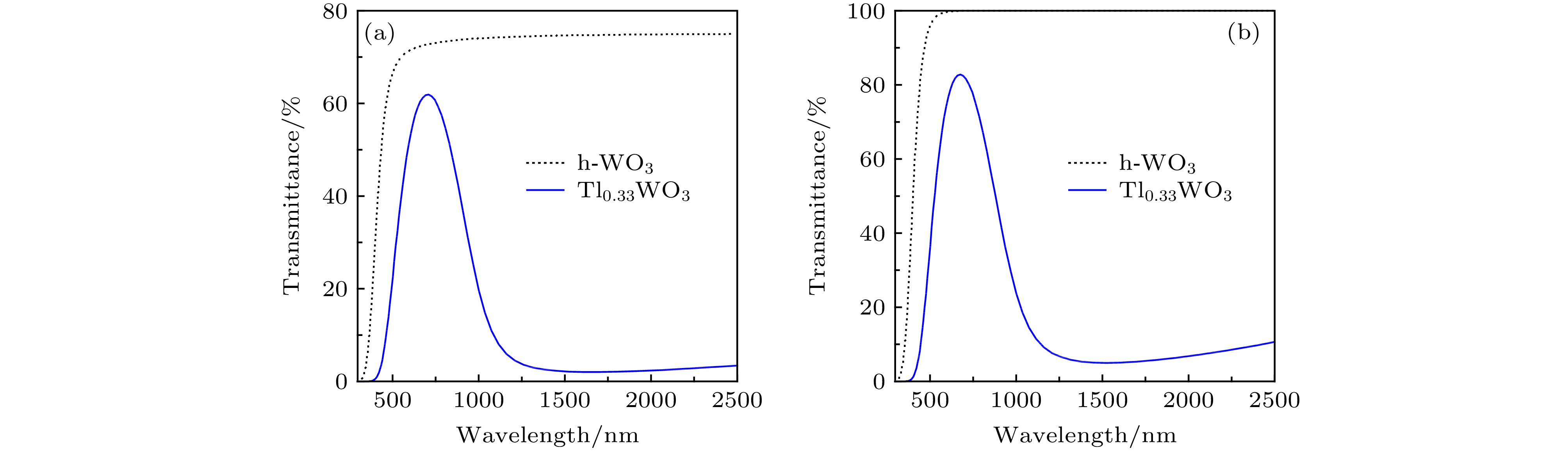 图 5 h-WO3和Tl0.33WO3薄膜的理论透过率: (a)致密薄膜; (b)涂层薄膜
图 5 h-WO3和Tl0.33WO3薄膜的理论透过率: (a)致密薄膜; (b)涂层薄膜Figure5. Theoretical transmittance of h-WO3 and Tl0.33WO3 films: (a) The compacted film; (b) the coated film
