全文HTML
--> --> -->α-syn由140个氨基酸残基构成, 其N端1-95残基在磷脂膜的存在下容易形成两亲性的α-螺旋, 因而是该蛋白与膜相互作用的主要位置. 而C端(96-140)在生理条件下则携带大量的负电荷, 一般认为此区域不与磷脂膜相互作用而游离在水相中[8,9]. α-syn的C端被认为是其结合金属离子与其他配体的重要区域[10,11]. 在蛋白的中央(61-95残基)是蛋白的NAC区域, 该区域与α-syn的聚集行为密切相关[7,12]. 在水溶液中, α-syn是舒展的无规结构[13], 结合至膜上之后, N端则形成富含α-螺旋的结构[14]. 关于α-syn在膜上的构象, 有研究者认为, α-syn以37-45残基为分界, 在膜上形成中央断开的两段螺旋结构[8]; 也有研究者认为α-syn的1-95残基在膜上形成一整段较长的α-螺旋[9]. 进一步的研究指出, 这两种构象在脂质体上均存在, 而其比例可能受到膜曲率的影响[15]. α-syn与含有负电荷的磷脂双层膜(如含有PA, PS, PG等)具有较强的相互作用[16,17].
关于α-syn与磷脂膜作用的细节, 此前已有一些研究, 一般是利用中子反射、色氨酸荧光的重原子淬灭等方法进行. 一种观点认为α-syn的N端螺旋插入磷脂膜内0.9—1.4 nm处[18], 或是距离磷脂双分子层中心0.9—1.1 nm[19]; 也有研究者得出了α-syn仅位于磷脂膜头部的结论[20]. 然而这些结果均是采用系综与时间平均的方法得出的, 并未反映α-syn在膜当中的动态过程. 此前, 本课题组开发了LipoFRET方法, 在单分子层面对α-syn与膜的相互作用进行了研究[21], 结果表明, α-syn T72位点附近的区域处于膜表面附近. 而K10位点附近的区域可深入膜中, 并且在几个不同深度之间做缓慢的运动.
到目前为止, 有一些研究指出, α-syn可形成寡聚体并对膜产生破坏[1,22-24], 或者磷脂膜的存在有可能起到降低聚集的浓度阈值(可低至nM级别)的作用[25]. 这有可能是导致帕金森病发生的重要因素. 但尚未有文献报道在寡聚浓度条件下的α-syn与膜, 以及寡聚过程启动初期α-syn分子之间相互作用的单分子水平上的动态细节. 而单分子荧光方法在研究生物大分子的动态过程中具有独特的优势[26,27]. 为了探究一定浓度下α-syn在磷脂膜上的行为与单分子浓度水平上的区别, 本文采用了一种基于脂质体的单分子荧光衰减方法—LipoFRET, 以及单分子荧光成像的方法研究了溶液中存在较高浓度的α-syn时, 其与磷脂膜之间相互作用的变化. 结果表明, 在浓度50 nM (1 M = 1 mol/L)的α-syn存在时, α-syn的NAC区域可从膜表面位置脱离并进入溶液中, 而N端第一螺旋位于膜内的概率减小且深度变浅, 并有较大的概率脱离膜表面进入溶液中. 这种变化促进了α-syn从膜上的解离. 我们认为高浓度的情况下α-syn之间对磷脂膜结合区域的竞争性占据是导致高浓度促进α-syn从膜上的解离的原因.
2.1.LipoFRET方法的原理
LipoFRET是单个荧光供体对包封在脂质体中的多个淬灭剂分子(作为荧光受体)的共振能量转移[21]. 在同一时刻, 供体对任意一个受体均有发生能量转移的概率, 其速率与各自距供体的距离有关. 最终的能量转移总速率是所有能量转移速率的总和[21,28,29], 可以写成对于膜蛋白来说, 其荧光标记位点距离膜外表面越近, 或在膜中的位置越深, 标记在膜蛋白上的荧光基团距离淬灭剂越近, 能量转移效率越高, 荧光基团的相对亮度F/F0越低. LipoFRET就是利用对荧光基团亮度的测量来监测膜蛋白特定位置的垂直运动的(图1(a)).
 图 1 LipoFRET方法 (a) LipoFRET的实验体系示意图; (b) LipoFRET的距离-相对亮度曲线, 其中淬灭剂为台盼蓝, 所用的荧光分子为Alexa Fluor 555 (Alexa 555), 距离是指荧光基团到脂质体磷脂双层膜的内表面的距离, 曲线分别对应包封2.5 mM (上方曲线)及5 mM (下方曲线)台盼蓝的脂质体.
图 1 LipoFRET方法 (a) LipoFRET的实验体系示意图; (b) LipoFRET的距离-相对亮度曲线, 其中淬灭剂为台盼蓝, 所用的荧光分子为Alexa Fluor 555 (Alexa 555), 距离是指荧光基团到脂质体磷脂双层膜的内表面的距离, 曲线分别对应包封2.5 mM (上方曲线)及5 mM (下方曲线)台盼蓝的脂质体.Figure1. The LipoFRET method: (a) The schematic picture of LipoFRET experiment; (b) the relationship between distance from the inner surface of the liposome lipid bilayer and relative intensity F/F0. The quencher used here is trypan blue and the fluorophore is Alexa Fluor 555 (Alexa 555). The curves correspond to liposomes encapsulating 2.5 mM (above) and 5 mM (bottom) trypan blue.
实验中, 采用台盼蓝作为淬灭剂, 其与α-syn上标记的荧光Alexa Fluor 555 (Alexa 555)之间的相对亮度与Alexa 555到脂质体磷脂膜内表面上的距离之间的关系如图1(b)所示.
2
2.2.LipoFRET的实验步骤
LipoFRET实验包括制备包封台盼蓝的脂质体以及荧光观测两部分.1) 脂质体的制备过程
首先, 将所用磷脂以2 mg/mL的浓度溶解在玻璃瓶中的氯仿与甲醇的混合液当中并振荡均匀, 其中氯仿与甲醇的总体积比为2∶1. 本文中所用的脂质体磷脂组分为70% DOPC, 30% DOPA, 0.1%的16∶0 Biotinyl-cap PE (质量百分比). 接着, 将玻璃瓶敞口放置于真空干燥器中, 抽真空过夜以抽干溶剂. 此时, 磷脂应在瓶壁上形成一薄层. 然后, 以每毫升0.5—1 mg磷脂的比例, 在瓶中加入5 mM (或2.5 mM)台盼蓝溶液, 并将玻璃瓶封好在摇床上振荡1 h. 此时瓶中磷脂形成了大小不均一的脂质体悬浮液. 随后, 将液体转移至塑料的小离心管中, 放入液氮中将液体速冻, 接着转移至37 ℃的水浴中解冻, 反复冻融5个循环, 再使用挤出器(Extruder, 购于Avanti Polar Lipids)将溶液来回挤压通过孔径为100 nm的聚碳酸脂膜. 收集制备好的脂质体. 接下来, 使用填料为Sephadex G-25的脱盐柱进行凝胶过滤, 将脂质体从未包封的台盼蓝中分离出来; 或将溶液稀释10倍后, 采用规格为100 kD的超滤离心管反复进行超滤, 控制转速与离心时间, 每次留下约一半的液体, 并采用缓冲液补充至原体积, 直到将未包封的台盼蓝稀释至较低水平.
在制备包封台盼蓝的脂质体的同时, 应制备包封实验用缓冲液(10 mM PBS, pH = 7.4, 0.9% NaCl)的脂质体, 用于测定F0的值.
2) LipoFRET荧光观测的步骤
制作可供进液与排液、适合装载在全内反射荧光显微镜上的样品腔室. 其中朝向镜头的盖玻片厚度为0.17 mm. 盖玻片朝向腔室内部的一面预先修饰mPEG以防止实验中蛋白的非特异性吸附, 同时亦修饰Biotin-PEG以供脂质体向玻片表面的连接.
实验装置采用以全内反射荧光显微镜和EMCCD为基础的双通道荧光共振能量转移观测装置[30-32], 但需要将全内反射场的角度调整为“赝全内反射(Pseudo-TIRF)[33]”的程度, 以避免光强在脂质体直径尺度内的衰减而带来上下光强不一致的情况. 使用ImageJ等软件记录及分析光强数据.
实验中, 首先向腔室中注入缓冲液以润洗玻片表面. 接着, 注入浓度为0.01 mg/mL的Streptavidin溶液, 待其反应5—10 min后, 冲去多余的Streptavidin. 将包封有台盼蓝的脂质体稀释注入腔室, 反应约2 min, 再注入一些脂质体以补充反应消耗. 进而, 向腔室中注入标记Alexa 555的α-syn (α-syn T72C-Alexa 555或α-syn K10C-Alexa 555, α-syn的氨基酸序列见表1), 待视野中Alexa 555发出的荧光点数量合适, 则以50 ms/帧的曝光时间对视野进行录像, 以记录α-syn在低浓度下的单体行为. 记录足够的数据后, 向腔室中注入50 nM未标记荧光的α-syn (wt-α-syn), 以提高α-syn的总浓度, 并继续录像以记录脂质体上的标记Alexa 555的α-syn在总浓度较高的情况下的行为. 包封台盼蓝的脂质体的实验完成后, 立即进行包封缓冲液的脂质体的实验, 实验步骤与前述相同, 以记录和测定F0的值. 由于LipoFRET实验中关心的是相对亮度F/F0的值, 因此在同一样品的同组实验当中, 需要保证包封缓冲液及台盼蓝的脂质体上测试的实验条件完全一致.
| 名称 | 氨基酸序列(1—140) |
| Human α-synuclein | MDVFMKGLSKAKEGVVAAAEKTKQGVAEAAGKTKEGVL YVGSKTKEGVVHGVATVAEKTKEQVTNVGGAVVTGVTAV AQKTVEGAGSIAAATGFVKKDQLGKNEEGAPQEGILEDM PVDPDNEAYEMPSEEGYQDYEPEA |
表1α-syn的氨基酸序列
Table1.Sequences of α-syn.
使用图1(b)中的亮度-距离曲线将分析获得的荧光亮度转换为距离信息. 如果使用不同的荧光分子与淬灭剂的组合来进行实验, 可根据该荧光分子的光谱、量子效率等参数, 参照参考文献[21]中的计算方法计算相应的亮度-距离曲线以用于荧光亮度与距离数据的转换.
2
2.3.α-syn解离过程的研究
为了研究α-syn解离速率与溶液中蛋白浓度的关系, 首先将脂质体连接在盖玻片表面上, 接着加入0.1 nM α-syn T72C-Alexa 555, 待视野中的荧光点密度足够多(200—500个为宜), 则关闭激光, 并加入一定浓度的wt-α-syn替换原溶液. 电荷耦合器件CCD的曝光时间为50 ms/帧, 总时长为5 s. 期间打开激光, 以低光强(强度可将视野中的单个分子的荧光点从背景分辨出来即可)照射约1 s即关闭, 此操作降低了光漂白导致的荧光点数量减少的效应. 每隔一定的时间重复录像-开关激光的步骤, 这样就记录了同一视野中荧光点随时间的变化. 最后, 使用ImageJ软件对图像进行分析, 统计每个录像中荧光点的个数, 即得出荧光点个数随着时间的变化趋势.3.1.α-syn中央NAC区域膜相互作用在高浓度下的变化
为研究较高浓度下α-syn与磷脂膜的相互作用特征, 用LipoFRET方法, 对溶液中存在一定浓度的α-syn时荧光标记的α-syn在磷脂膜上的动态过程进行探究.首先关注的是α-syn T72位点. 该位点位于蛋白第二螺旋中的NAC区域. NAC区域连接了α-syn的N端与C端, 且此区域在体外有聚集的特性, 因而被认为是介导聚集的关键区域[7]. 亦即是说, 该区域是高浓度下参与α-syn多体相互作用的可能区域. 在实验中, 首先将包封5 mM台盼蓝的脂质体连接在玻片表面上. 接着, 将0.1 nM α-syn T72C-Alexa 555加入样品腔室. 待视野中的荧光点密度足够后, 进行录像, 以记录样品在浓度较低时与磷脂膜相互作用的特征. 在记录足够数据之后, 再在其中加入50 nM的未标记荧光的α-synuclein (wt-α-syn), 继续进行录像, 以记录单个α-syn T72C-Alexa 555在蛋白总浓度较高时的行为特征. 实验结果如图2所示.
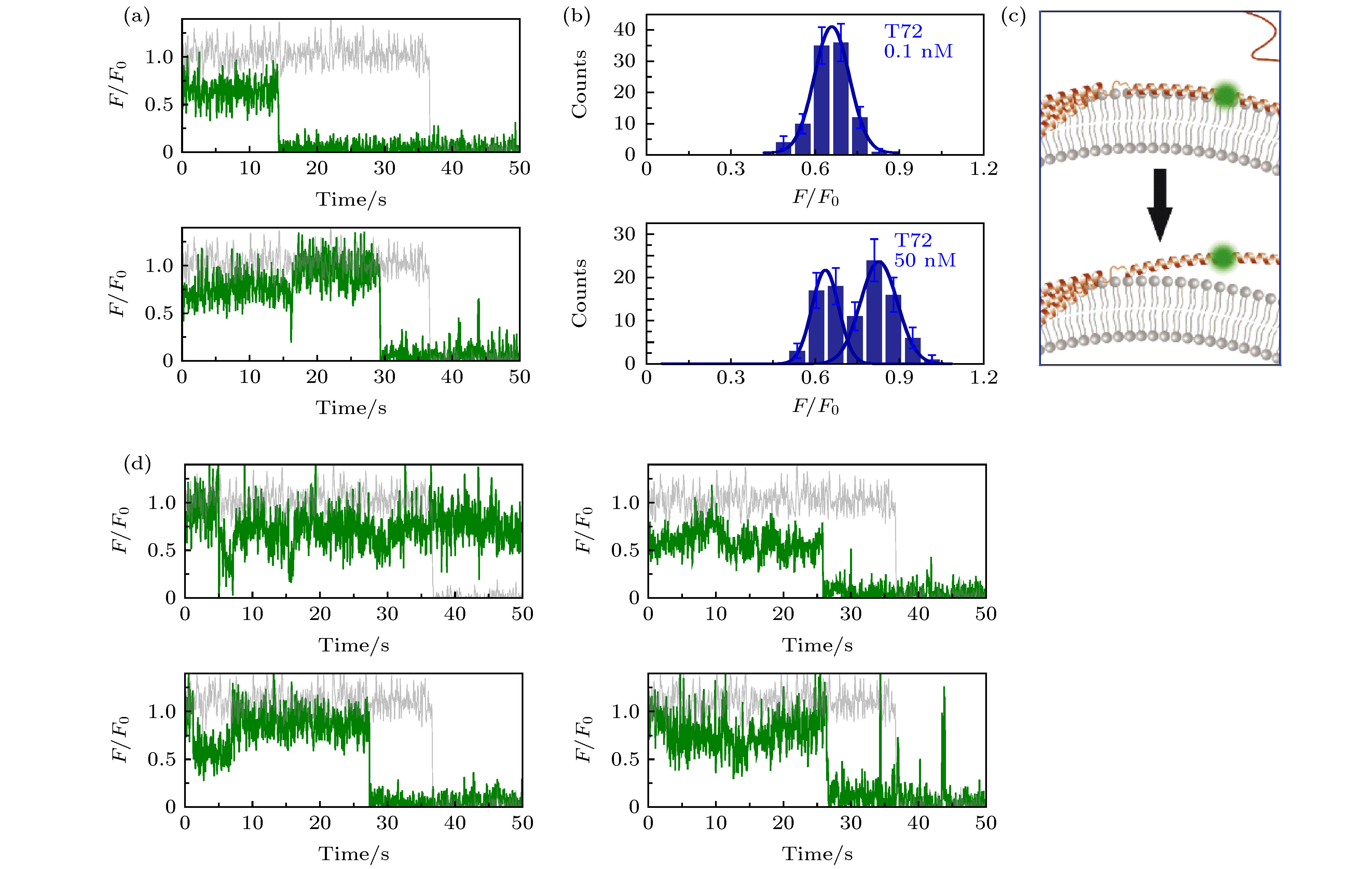 图 2 溶液中存在低蛋白浓度或高蛋白浓度时, α-syn T72C-Alexa 555在包封5 mM台盼蓝的脂质体上的动态特征 (a)蛋白总浓度为0.1 nM (上图)与50 nM (下图)时的典型亮度曲线(其中灰色曲线与绿色曲线分别为包封缓冲液脂质体与包封台盼蓝脂质体上的相对亮度曲线(F/F0)); (b)蛋白总浓度为0.1 nM (上图)与50 nM (下图)时的亮度统计(误差线代表统计偏差); (c)低浓度(上部)与高浓度(下部)时, 在磷脂膜上的位置特征示意图; (d)蛋白总浓度为50 nM时更多的典型亮度曲线
图 2 溶液中存在低蛋白浓度或高蛋白浓度时, α-syn T72C-Alexa 555在包封5 mM台盼蓝的脂质体上的动态特征 (a)蛋白总浓度为0.1 nM (上图)与50 nM (下图)时的典型亮度曲线(其中灰色曲线与绿色曲线分别为包封缓冲液脂质体与包封台盼蓝脂质体上的相对亮度曲线(F/F0)); (b)蛋白总浓度为0.1 nM (上图)与50 nM (下图)时的亮度统计(误差线代表统计偏差); (c)低浓度(上部)与高浓度(下部)时, 在磷脂膜上的位置特征示意图; (d)蛋白总浓度为50 nM时更多的典型亮度曲线Figure2. Dynamic of α-syn T72C-Alexa 555 on liposome encapsulating 5 mM trypan blue in the presence of low and high protein concentration in solution: (a) Typical intensity traces of α-syn T72C-Alexa 555 under 0.1 nM (upper panel) or 50 nM wt-α-syn (lower panel) in solution (the grey and green line correspond to the intensity traces on liposomes entrapping buffer (F0) and on liposomes entrapping trypan blue (F), respectively); (b) the intensity histograms of the states under 0.1 nM (upper panel) or 50 nM wt-α-syn (lower panel) in solution (the error bars on the histograms represent the statistical error in the bins); (c) the scheme of the position change of α-syn T72 on the membrane with increased protein concentration in the solution; (d) more intensity traces of α-syn T72C-Alexa 555 in the presence of 50 nM wt-α-syn in solution.
在实验中, 发现在蛋白总浓度较低(0.1 nM)时, 亮度曲线仍呈现较为平稳的状态, 其相对亮度的值为0.65 ± 0.07 (中心值 ± 半峰宽, 下同). 膜的厚度约4.5 nm[34], 此结果确认了T72位点位于脂质体磷脂膜的表面附近. 而当加入50 nM的wt-α-syn之后, 部分荧光亮度有所增强, 并且亮度曲线出现了明显的上下起伏(图2(a)和图2(d)). 对亮度曲线中较明显状态的亮度进行了统计, 以确定亮度状态的存在与分布. 结果显示, 在较高浓度的α-syn存在时, α-syn T72C-Alexa 555的亮度呈现两个峰的分布(图2(b)). 亮度较低峰的中心值与低浓度下的α-syn亮度基本一致, 为F/F0 = 0.63 ± 0.06; 而亮度较高峰的为F/F0 = 0.83 ± 0.09, 对照图1(b)中的曲线, 其中心值对应于膜外2.1 nm的位置. 这显示α-syn浓度较高时, 一部分蛋白的NAC中央区域脱离膜表面, 进入了溶液中(图2(c)), 且可在膜表面和溶液中的状态之间互相转换.
接着, 对α-syn K10位点的动态过程进行研究. K10位点所在的N端第一螺旋是α-syn与膜相互作用较强的区域. 第一螺旋较第二螺旋多出3个净的正电荷, 并含有更高比例的疏水残基侧链. 此前的报道中揭示了单个α-syn分子K10位点在膜中有较明显的动态过程[21], 在这里探索较高浓度α-syn下, K10位点与膜的相互作用. 由于该位点在膜中位置较深, 为避免荧光信号过弱, 采用2.5 mM台盼蓝包封于脂质体中对其进行研究.
图3的实验结果显示, 在蛋白总浓度较高时, α-syn N端在膜中的动态过程也会有所变化. 在腔室中的蛋白浓度为0.1 nM时, 观察到3个主要的状态, 其亮度为F/F0 = 0.47 ± 0.08, 0.68 ± 0.06, 以及0.83 ± 0.05, 分别对应膜以内3.6 nm, 1.4 nm, 以及膜表面附近的位置(图3(a)和图3(b)). 而当在溶液中增加了50 nM的wt-α-syn之后, N端的动态过程发生了变化. 从典型的亮度曲线(图3(d))来看, K10位点有更大概率跳变到更高亮度的位置. 而从亮度统计来看, 较高浓度的α-syn存在时, 仍然有3个峰的位置, 前两个峰与低浓度的情况相比, 中心位置向更亮处移动, 分别为F/F0 = 0.54 ± 0.07, 0.71 ± 0.06. 这两个峰对应膜以内2.9, 1.1 nm的位置. 亮度最高的峰为F/F0 = 0.86 ± 0.06, 其中心对应膜以外0.9 nm的位置. 相比于低浓度下的情况, 除了位置更高之外, 最高亮度的峰所占比例明显升高, 而亮度较低的峰所占比例明显降低. 这提示增加了α-syn的溶液中浓度之后, K10位点所在的第一螺旋更倾向于处在溶液中略高于膜表面的位置(图3(c)). 从实验结果来看, 溶液中更高浓度的α-syn的加入, 一方面促使T72位点脱离膜表面进入溶液中, 另一方面使K10位点倾向于从膜内切换至更浅甚至较膜表面更高的位置, 结果表明, α-syn溶液中浓度的升高很有可能带来单个蛋白与膜相互作用的减弱.
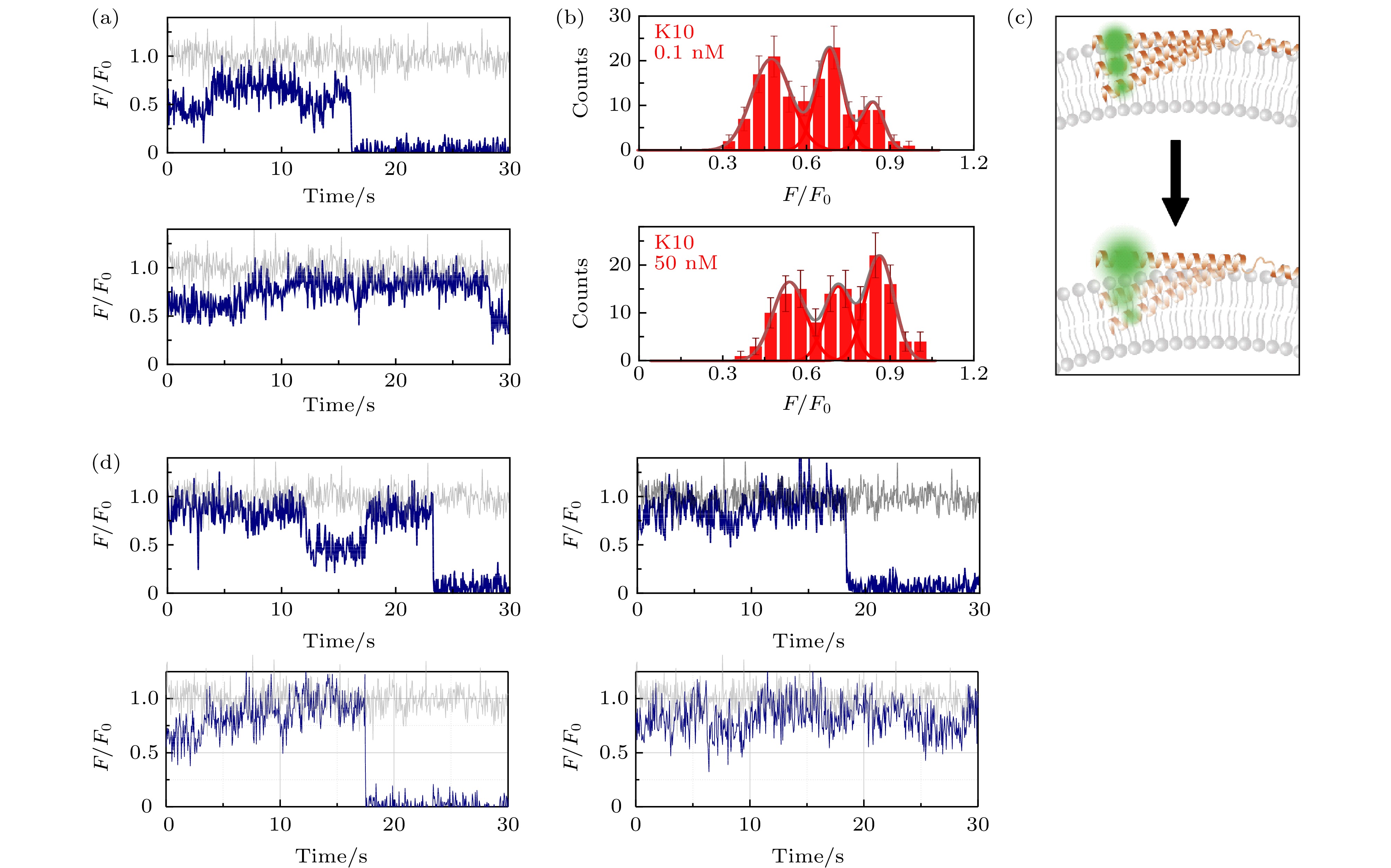 图 3 包封2.5 mM台盼蓝的脂质体上的α-syn K10C-Alexa 555在溶液中存在低浓度与高浓度wt-50 nM wt-α-syn时的动态特征(a)蛋白总浓度为0.1 nM (上图)与50 nM (下图)时的典型亮度曲线(其中灰色曲线与深蓝色曲线分别为包封缓冲液脂质体与包封台盼蓝脂质体上的相对亮度曲线(F/F0)); (b)蛋白总浓度为0.1 nM (上图)与50 nM (下图)时的亮度统计(误差线代表统计偏差); (c)蛋白低浓度(上部)与高浓度(下部)时, 在磷脂膜上的位置特征示意图; (d)蛋白总浓度为50 nM时更多的典型亮度曲线
图 3 包封2.5 mM台盼蓝的脂质体上的α-syn K10C-Alexa 555在溶液中存在低浓度与高浓度wt-50 nM wt-α-syn时的动态特征(a)蛋白总浓度为0.1 nM (上图)与50 nM (下图)时的典型亮度曲线(其中灰色曲线与深蓝色曲线分别为包封缓冲液脂质体与包封台盼蓝脂质体上的相对亮度曲线(F/F0)); (b)蛋白总浓度为0.1 nM (上图)与50 nM (下图)时的亮度统计(误差线代表统计偏差); (c)蛋白低浓度(上部)与高浓度(下部)时, 在磷脂膜上的位置特征示意图; (d)蛋白总浓度为50 nM时更多的典型亮度曲线Figure3. Dynamic of α-syn K10C-Alexa 555 on liposome encapsulating 2.5 mM trypan blue in the presence of low and high protein concentration in solution: (a) Typical intensity traces of α-syn K10 C-Alexa 555 under 0.1 nM (upper panel) or 50 nM wt-α-syn (lower panel) in solution (the grey and dark blue line correspond to the intensity traces on liposomes entrapping buffer (F0) and on liposomes entrapping trypan blue (F), respectively); (b) the intensity histograms of the states under 0.1 nM (upper panel) or 50 nM wt-α-syn (lower panel) in solution (the error bars on the histograms represent the statistical error in the bins); (c) the scheme of the position change of α-syn K10 on the membrane with increased protein concentration in the solution; (d) more intensity traces in the presence of 50 nM wt-α-syn in solution.
为了探究高浓度下α-syn的存在所导致的微观层面蛋白与膜相互作用的变化是否会对α-syn在膜上的解离性质有所影响, 用不含台盼蓝的脂质体做了α-syn在DOPC/DOPA膜上的解离测试, 实验结果如图4所示. 从图4可以看出, 加入wt-α-syn后, 随着时间的推移, 原本结合于脂质体上的α-syn T72C-Alexa 555逐渐减少. 并且, 溶液中α-syn的浓度越高, 荧光点减少的速度越快. 此过程表明荧光标记的α-syn从脂质体上逐渐解离. 这种现象与一般的结合-解离过程不同. 一般而言, 生物大分子从作用位点上解离由一个解离常数决定, 这个解离常数通常是一个恒定的值, 不随溶液中生物大分子浓度的变化而变化. 而α-syn的解离则明显地受浓度调控, 该现象在以往关于α-syn的文献中并未报道过. 溶液中的α-syn使得已经结合在膜上的蛋白更容易从膜上脱落, 因此, LipoFRET得到的高浓度状态下α-syn在膜上垂直位置的变化, 确实导致α-syn与磷脂膜相互作用的减弱, 促进了α-syn的解离.
 图 4 溶液中含有1 nM, 10 nM, 100 nM的wt-α-syn时, 视野中脂质体上的α-syn T72C-Alexa 555数量相对于原始数量的比值随时间变化趋势(误差线代表统计偏差)
图 4 溶液中含有1 nM, 10 nM, 100 nM的wt-α-syn时, 视野中脂质体上的α-syn T72C-Alexa 555数量相对于原始数量的比值随时间变化趋势(误差线代表统计偏差)Figure4. Fraction of the remaining fluorescence spots of α-syn T72C-Alexa 555 on the liposomes versus time. The solution contains 1 nM, 10 nM, or 100 nM wt-α-syn. The error bars on the histograms represent the statistical error in the bins.
根据在实验中从微观与宏观层面得到的结果, 溶液中存在高浓度的α-syn时, 应当存在一个包含多个蛋白与磷脂膜形成复合物的过程(图5). 对于低浓度下的单个α-syn来说, K10位点有较大概率深入磷脂膜的较深处, 而T72位点基本稳定在磷脂膜的表面上. 当溶液中的α-syn浓度较高时, 两个α-syn有一定的概率充分靠近, 从而发生一个α-syn K10位点所在的第一螺旋占据另外一个α-syn的T72位点所在的第二螺旋与膜作用区域的情况. 此时, 作用位点被占据的α-syn第二螺旋与膜的作用减弱, 有一定概率离开磷脂膜的表面. 此变化导致同一蛋白的第一螺旋切换至膜的浅表位置. 此时, α-syn与磷脂膜的相互作用很不稳定, 当第一螺旋与第二螺旋同时处于膜外时, 最终使得其与膜的作用位点容易从膜上脱落下来, 从而导致了宏观上溶液中蛋白浓度升高反而促进蛋白从膜上解离的现象. 需要注意的是, 蛋白之间互相占据膜作用位点的情况是有可能发生的, 因为α-syn的第一螺旋带有3个正的净电荷, 而C端带有12个净的负电荷. 其静电作用可能倾向于使α-syn的第一螺旋从另外一个α-syn的第二螺旋方向靠近并促使替代过程发生. 该过程与Kamar等[35]揭示的转录因子从DNA上解离的动力学过程具有类似的机理, 因为从微观上看, α-syn与磷脂膜之间同样涉及两个α-螺旋导致的多位点相互作用.
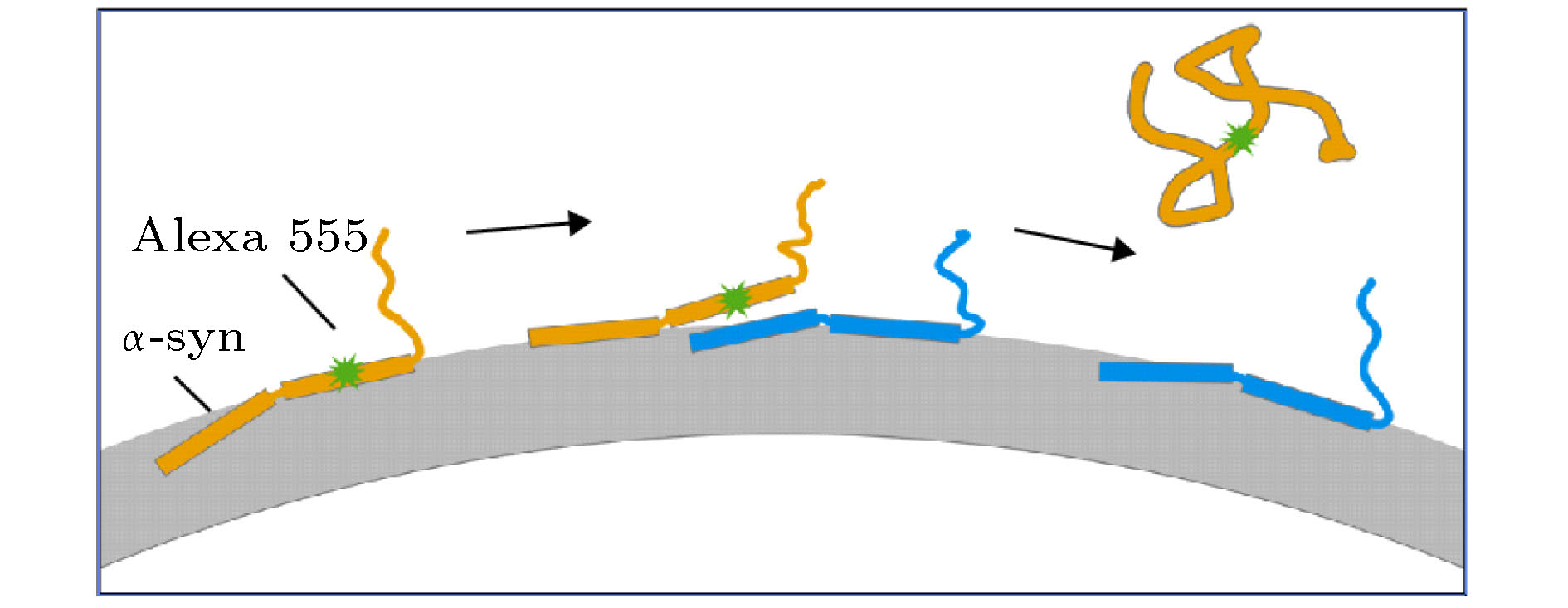 图 5 溶液中高浓度的α-syn促进标记Alexa 555的α-syn从膜上解离的过程模型
图 5 溶液中高浓度的α-syn促进标记Alexa 555的α-syn从膜上解离的过程模型Figure5. Model corresponding to the dissociation of α-syn from the membrane which was promoted by increased protein concentration.
综上所述, 实验揭示了α-syn在高浓度下的行为. 溶液中高浓度的α-syn使T72位点从膜上解离至溶液中, 而K10位点从膜内转移至膜表面甚至溶液中, 从而在宏观上促进了α-syn从磷脂膜上的解离. 这与一些研究中的α-syn寡聚体在膜上聚集的结果并不一致, 因为如果α-syn更易于从膜上解离, 其导致的结果是在膜上的聚集体更难以形成. 但是, 按照LipoFRET实验的时间尺度(仅关注了单分子水平上α-syn在与膜相互作用后短时间内的动态变化), 可以认为α-syn在磷脂膜存在下的聚集是一个包含解离与成核竞争的复杂动态过程, 这也解释了一些研究中α-syn聚集体的生成至少需要数小时的时间尺度的原因[22]. 高浓度的α-syn促进其自身从磷脂膜上解离的性质, 很可能是α-syn在体内调控其聚集过程的重要方式, 因为高浓度下α-syn从膜上解离的速率更快则限制了聚集体大小的增长速率. 帕金森病的致病机理有可能和α-syn的解离与聚集性质的失调有关. 另外, 此前文献[36]报道了α-syn与膜作用的不同位点分别连接两个脂质体、介导脂质体之间聚集的模型假设. 此研究中α-syn的状态与本文中LipoFRET得出的高浓度下α-syn在膜上的状态较为吻合. 该现象的发生与一定浓度下α-syn发生部分位点从脂质体膜上解离从而与另一个脂质体发生相互作用有关, 即使该相互作用可能是瞬态的. 从另一方面来看, 蛋白浓度是调节α-syn与膜的相互作用的重要参数. 在低浓度时, 存在的是较长时间的持续相互作用; 而高浓度时, 则主要为瞬态的相互作用. 在解读和比较研究α-syn与膜相互作用的不同研究结果时, 蛋白浓度应作为重要影响因素被纳入考虑.
在实验中发现, 当溶液中α-syn的浓度提高时, 磷脂膜上的α-syn T72位点所在的NAC附近区域可从磷脂膜上解离至溶液中. 而K10位点附近区域处在膜中的概率减小, 深度变浅, 并有很大概率脱离膜表面进入溶液中. 这提示在此情况下α-syn与磷脂膜的相互作用可能会减弱. 进一步地, 采用单分子荧光成像的方法对α-syn与脂质体的解离过程进行了研究. 结果表明, α-syn从脂质体上的解离速率确实随着溶液中蛋白浓度的提高而加快, 这与LipoFRET的单分子实验的结果是自洽的. 针对这样的现象, 本文提出了α-syn蛋白浓度升高导致膜上的α-syn与膜的作用位点互相竞争, 进而导致α-syn的膜作用位点从膜上解离, 并最终导致α-syn与磷脂膜的相互作用减弱从而从膜上解离的模型. α-syn浓度调控解离的性质是调节其在膜上寡聚过程的重要一环, 该寡聚过程很可能涉及聚集与解离的竞争.
此外, 本文的结果也证明了LipoFRET方法在探测膜蛋白动力学过程中的适用性. 期待将来在膜蛋白功能与结构的研究中, LipoFRET能得到更广泛的应用.
