Key Laboratory for Radiation Physics and Technology, Ministry of Education, Institute of Nuclear Science and Technology, Sichuan University, Chengdu 610064, China
Fund Project:Project supported by the National Natural Science Foundation of China (Grant No. 51501119)
Received Date:09 May 2019
Accepted Date:14 September 2019
Available Online:27 November 2019
Published Online:01 December 2019
Abstract:Tungsten (W) alloys and W-based alloys are the primary candidate materials for plasma-facing components in future fusion reactors (e.g. ITER and CFETR). One of the critical issues still to be clarified in the design of the fusion reactor materials is the retention of hydrogen (H) isotopes in W, when the plasma-facing materials are supposed to sustain high-flux plasma and high-energy neutron. The dynamical behaviours of H in W with radiation defects (e.g. vacancy) are of serious concerns for understanding the mechanism of H capture, retention and permeation in W. In this work, a new model to extract the effective capture radius (ECR) and dissociation coefficient simultaneously is presented through coupling the trapping process and detrapping process of H in W vacancy. In the new model, the quantity ratio of vacancy to H atom in vacancy-H complex (VHx+1) in the molecular dynamics (MD) simulations is described as a function of time, while the exact occurrence time of corresponding event is not required. This new model, combined with extensive MD calculations, enables the simultaneous determining of the ECR and dissociation coefficient of H in W vacancy. It is found that the parameters are dependent not only on the event type but also on temperature. The dissociation energy of H from vacancy-H complex decreases gradually with the increase of the trapped number of H atoms in the vacancy-H complex. It is also found that the common assumption (i.e. the ECR is equal to one lattice constant and the pre-exponential factor is equal to 1013 s–1) in the long-term simulation methods (e.g. kinetic Monte Carlo and rate theory) is not always valid, since these calculated dynamical parameters are dispersive. The new model to obtain more reliable results with lower cost of computing resources can be easily extended into the other similar kinetic processes (e.g. H/He trapping and detrapping processes in other materials systems). These calculated dynamical parameters should be potentially helpful in supplying the initial input parameters for the long-term simulation methods. Keywords:capture radius/ dissociation coefficient/ molecular dynamics/ vacancy
对于解离过程, 初始超胞中只设置空位-H复合体(VHx+1), 在MD模拟过程, 会出现H的解离, 当然也可能会出现解离后的溶质H被VHx重新捕获的情况. 但根据新物理模型, 只要知道当前构型下空位-H复合体(VHx+1)的数量即可推导出相应过程的动力学参数, 而不必准确记录相应事件的首次发生时间. 图2所示是不同类型的事件在不同温度下空位-H复合体(VHx+1)的比率(${y_{{\rm{V}}{{\rm{H}}_{x + 1}}}}\left( t \right)$)随时间(t)的变化, 由图2可以发现MD模拟获得各输出时间点的VHx+1的比率(图2中点表示)能被新物理模型(图2中曲线表示)较好地拟合, 表明该模型可用于描述捕获和解离的耦合过程. 图 2 不同温度下, 空位-H复合体(VHx+1)的含量(${y_{{\rm{V}}{{\rm{H}}_{x + 1}}}}\left( t \right) = {{{N_{{\rm{V}}{{\rm{H}}_{x + 1}}}}\left( t \right)} / {{N_{\rm{b}}}}}$)与时间(t)的变化, 其中曲线由 (10)式拟合 Figure2. Ratio of VHx+1 in the simulation (${y_{{\rm{V}}{{\rm{H}}_{x + 1}}}}\left( t \right) = {{{N_{{\rm{V}}{{\rm{H}}_{x + 1}}}}\left( t \right)} / {{N_{\rm{b}}}}}$) as function of time (t), where the curves are fitted by Eq. (10).
式中, νdiss,x+1是前因子, 一般认为与温度无关; Ediss,x+1是解离能, 表示空位-H复合体(VHx+1)的H解离形成溶质H的势垒; kB是玻尔兹曼常数. 如图3所示, lnkdiss,x+1与1/T呈现较好的线性关系, 表明解离过程是个热激活过程, 因而可根据曲线截距和斜率推导出前因子和解离能. 图 3 不同空位-H复合体(VHx+1)的H解离系数(kdiss, x+1)与温度(T)的变化, 其中曲线由公式lnk = lnν – E/kB/T拟合 Figure3. Dissociation coefficients (kdiss, x+1) of H detrapping from various VHx+1 complex as functions of temperature (T), where the curves are fitted by equation lnk = lnν– E/kB/T.
新物理模型推导出的解离能在0.58—1.9 eV之间, 与第一性原理计算结果[30]接近. 如图4(a)所示, 新模型推导的解离能(Enew)与前期单一过程物理模型给出的解离能(Eold) [26]比较接近, 表明两种模型在计算解离能时都比较合理, 另外从图4(a)还可发现空位-H复合体(VHx+1)的H解离能随捕获H的数量增加而减小, 该趋势也与前期结果[26]及第一性原理计算结果[30]一致. 图4(b)是解离前因子的比较, 可以发现二者的接近程度不如解离能, 这主要是由于经过了指数的数学变换. 事实上对lnν, 前后模型给出的数值也比较接近. 另外由图4(b)还可以发现, 前因子粗略地随捕获H数量的增加而增加, 即粗略地随解离能的减小而增加. 前因子的变化范围在6—70 ps–1, 表明在一些计算中前系数被认为是常数(1013 s–1)的假定是不合理的. 图 4 新模型推导的解离能(a)和前因子(b)与前期模型[26]计算值的比较, 其中虚线表示(Enew = Eold或νnew = νold) Figure4. (a) Dissociation energies and (b) pre-exponential factors deduced by new model (present work) and old model [26], where the dash line means Enew = Eold or νnew = νold.
23.2.捕 获 -->
3.2.捕 获
如前文所述, 在研究空位-H复合体(VHx+1)解离过程时也可能存在解离H再被捕获的情况, 而且根据(12a)式, 有效捕获半径(Rc,x)理应也能推导获得, 事实上通过解离过程拟合获得的Rc,x大多在0.5—4 ?, 与前期[26]结果相近. 然而应指出的是, 对于有效捕获半径的计算, MD模拟过程中应该保证有足够数量的捕获事件发生以满足统计学规律. 如图2(a)所示的2250 K时VH → V+H事件, 含有VH超胞的比率 (yVH)几乎随时间(t)线性下降, 表明存在较少的捕获事件, 因而该y-t曲线不宜用来推导有效捕获半径, 类似的情况还有1500 K时的VH2 → VH+H事件(图2(b))、1000 K时的VH3 →VH2+H事件(图2(c))、1000 K时的VH4 → VH3+H事件(图2(d))、500 K时的VH5 →VH4+H事件(图2(e))和700 K时的VH6 → VH5 +H事件(图2(f))等. 另外对于VH6 → VH5+H事件, 由于解离能较低, 不易发生捕获事件, 因而对于800 K和900 K也不宜用于计算有效捕获半径. 事实上, 前期事实上, 前期[26]在研究高捕获状态的空位-H复合体(如VH7)的解离过程时就发现, 在热化阶段绝大多数超胞中的(VHx+1)已发生解离, 因而在后期MD演化过程中很难发生捕获事件. 因而针对捕获过程, 在超胞中设置了VHx和溶质H原子, 增加捕获事件的发生. 同样在MD模拟过程中, 溶质H原子可能会被VHx捕获, 捕获的H也可能再解离, 那么空位-H复合体的比率(${y_{{\rm{V}}{{\rm{H}}_{x + 1}}}}\left( t \right)$)随时间(t)的变化关系仍可用(10)式来描述. 图2(g)表示的是V+H → VH事件, 图2(h)表示的是VH+H → VH2事件, 可以发现MD模拟获得比率y(t)能被新物理模型较好地拟合, 同样表明新模型的可靠性, 根据拟合参数且运用(12a)式可推导出有效捕获半径. 计算结果如图5所示, 新模型获得的有效捕获半径与单一过程物理模型[26]给出的结果比较接近. 偏离较大的主要是高捕获状态的空位-H复合体(VHx), 这是因为这些复合体的解离能较低且存在较复杂的捕获与解离过程. 另外由图5还可发现有效捕获半径在0.5—4 ?, 表明在一些计算中有效捕获半径被认为是一个晶格常数(3.14 ?)的假定可能并不合理. 图 5 新模型推导的有效捕获半径ECRnew与前期模型[26]ECRold计算值的比较, 其中虚线表示ECRnew = ECRold Figure5. Effective capture radii deduced by new model (present work) and old model [26], where the dash line means ECRnew = ECRold.









 图 1 含有空位-H复合体(VH2)的初始超胞, 其中红色点表示W原子, 蓝色球表示H原子
图 1 含有空位-H复合体(VH2)的初始超胞, 其中红色点表示W原子, 蓝色球表示H原子
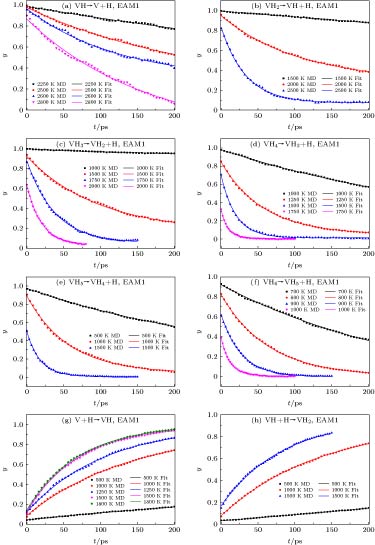 图 2 不同温度下, 空位-H复合体(VHx+1)的含量(
图 2 不同温度下, 空位-H复合体(VHx+1)的含量(

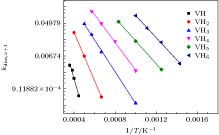 图 3 不同空位-H复合体(VHx+1)的H解离系数(kdiss, x+1)与温度(T)的变化, 其中曲线由公式lnk = lnν – E/kB/T拟合
图 3 不同空位-H复合体(VHx+1)的H解离系数(kdiss, x+1)与温度(T)的变化, 其中曲线由公式lnk = lnν – E/kB/T拟合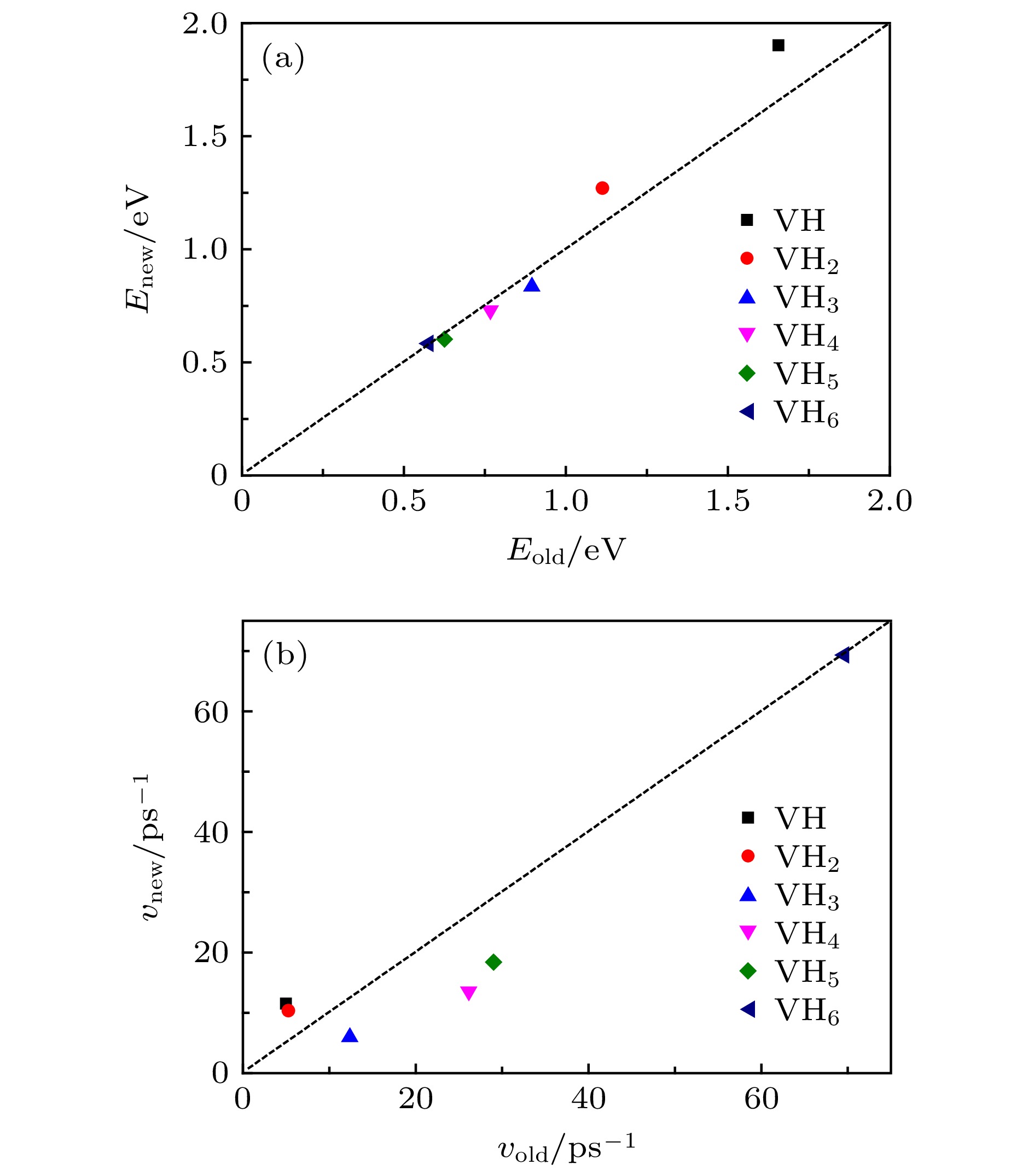 图 4 新模型推导的解离能(a)和前因子(b)与前期模型[26]计算值的比较, 其中虚线表示(Enew = Eold或νnew = νold)
图 4 新模型推导的解离能(a)和前因子(b)与前期模型[26]计算值的比较, 其中虚线表示(Enew = Eold或νnew = νold)
 图 5 新模型推导的有效捕获半径ECRnew与前期模型[26] ECRold计算值的比较, 其中虚线表示ECRnew = ECRold
图 5 新模型推导的有效捕获半径ECRnew与前期模型[26] ECRold计算值的比较, 其中虚线表示ECRnew = ECRold