全文HTML
--> --> -->本研究组在改进磁有序模型方面进行了一些尝试, 提出一个关于氧化物磁有序的O 2p巡游电子模型, 一个关于金属磁有序的新巡游电子模型和一个关于外斯分子场能量的外斯电子对模型, 发表了一系列研究论文, 相应的综述文章于2018年发表在《Physics Reports》上[6]. 此外, 基于这些磁有序模型, 还对典型磁性材料的电输运性质进行了探讨[7-9], 认为NiCu合金与典型钙钛矿结构锰氧化物在居里温度以下都存在巡游电子的自旋相关导电. 这些新模型中的价电子结构与传统模型存在显著区别. 本文首先简单介绍文献报道的一些相关典型实验结果, 然后介绍本研究组的有关工作, 最后指出这方面研究面临的挑战.
2.1.O 2p空穴和负一价氧离子的实验研究
在包括磁性材料在内的氧化物材料研究中, 传统观点认为所有的氧离子都是负二价离子, 其最外电子壳层是8个电子(2s22p6)的满壳层结构. 然而实验表明在氧化物中存在O 2p空穴, 即存在负一价氧离子(2s22p5), 其比例随材料不同而有很大差别, 可达30%或更高. 说明基于氧离子全部为负二价离子假设的传统超交换作用和双交换作用模型需要改进.3
2.1.1.关于O 2p空穴的实验研究
1988年, Nücker等[10]报道了关于超导材料YBa2Cu3O7–y的电子能量损失谱(electron energy loss spectroscopy, EELS)研究, 发现在费米能级处的电子态密度具有显著的O 2p空穴特征, 并且O 2p空穴含量随材料中氧离子空位含量y而变化, 所以认为这种超导化合物的载流子是O 2p空穴.Ju等[11]利用聚合物溶胶-凝胶技术, 在(100)面的LaAlO3 基片上制备了钙钛矿锰氧化物薄膜材料La1–xSrxMnO3 (0 ≤ x ≤ 0.7). 样品最后在空气中700 ℃退火1 h. 通过X射线衍射分析, 显示所有样品都具有单相ABO3钙钛矿结构. 测量了样品电阻率随测试温度的变化关系, 得到的结果如图1(a)所示. 曲线的变化规律与Urushibara等[12]研究单晶样品La1–xSrxMnO3(0 ≤ x ≤ 0.4)时得到的结果十分相似. 图1(b)为La1–xSrxMnO3 (x = 0, 0.3, 0.5) 样品薄膜和LaAlO3基片的O K边电子能量损失谱, 其横轴已经换算成样品发射光电子相对于费米能级的束缚能. 可以看到3个薄膜样品在529 eV 附近有一个很强的谱峰, 而LaAlO3 基片没有这个谱峰. 这个谱峰反映出样品中存在O 2p的空态, 说明La1–xSrxMnO3系列样品的导电是由于O 2p空穴造成的p型导电.
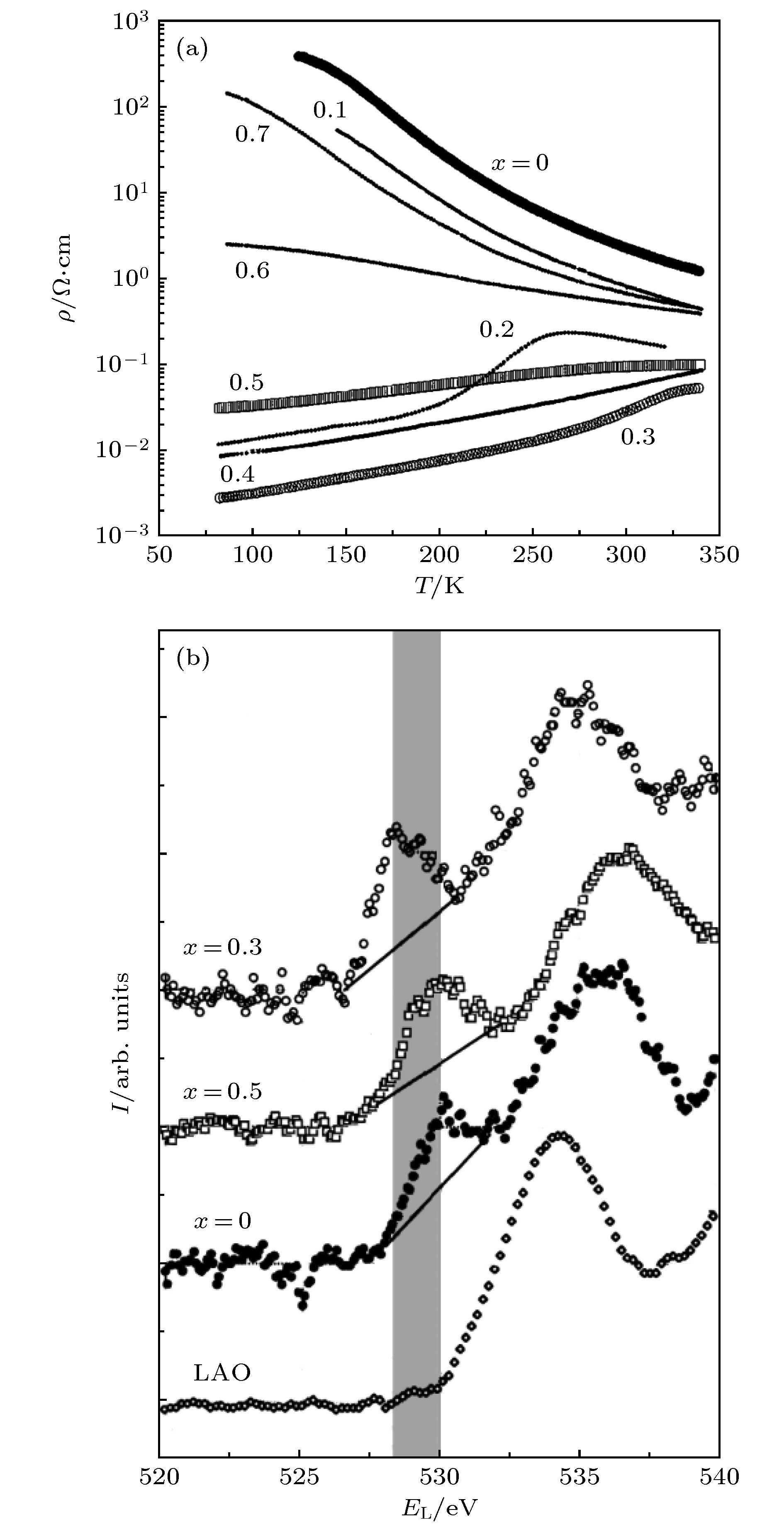 图 1 由Ju等[11]报道的La1–xSrxMnO3系列样品 (a)电阻率ρ随测试温度T的变化关系; (b)电子能量损失谱
图 1 由Ju等[11]报道的La1–xSrxMnO3系列样品 (a)电阻率ρ随测试温度T的变化关系; (b)电子能量损失谱Figure1. La1–xSrxMnO3 reported by Ju et al.[11]: (a) Curves of the resistivity ρ versus the test temperature T; (b) electron energy loss spectra.
Mizoroki等[13]制备了多晶样品La1–xSrxMnO3(x = 0.1, 0.2, 0.3, 04, 0.5), 利用磁性康普顿散射实验证明, 当Sr掺杂量为0.1和0.2时, 掺杂的空穴择优进入O 2p 态. Grenier等[14]利用O K边X射线衍射, 在La7/8Sr1/8MnO3中观察到2p 电荷的有序变化, 即存在“富空穴”和“贫空穴”MnO平面的交替变化. Ibrahim等[15]制备了庞磁电阻材料Pr1–xSrxMnO3 (x = 0, 0.3), 研究了材料的X射线吸收谱和俄歇电子谱, 认为材料中存在O 2p空穴, 且其浓度随Sr掺杂量的增加而增大. Papavassiliou等[16]比较了材料La1–xCaxMnO3+δ的核磁共振结果与X射线吸收数据, 发现在O 2p轨道上形成了自旋极化的空穴.
3
2.1.2.关于负一价氧离子的实验和理论研究
Dupin等[17]研究了一些氧化物的X射线光电子谱(X-ray photoelectron spectroscopy, XPS), 提出一个观点: 在氧化物的O 1s谱中出现在527.7—530.6 eV范围内的谱峰对应于O2–离子的光电子, 在531.1—532.0 eV 范围内的谱峰对应于O–离子的光电子; 与材料表面化学吸附氧(O0)对应的谱峰出现在531.1—533.5 eV 范围内, 如图2所示.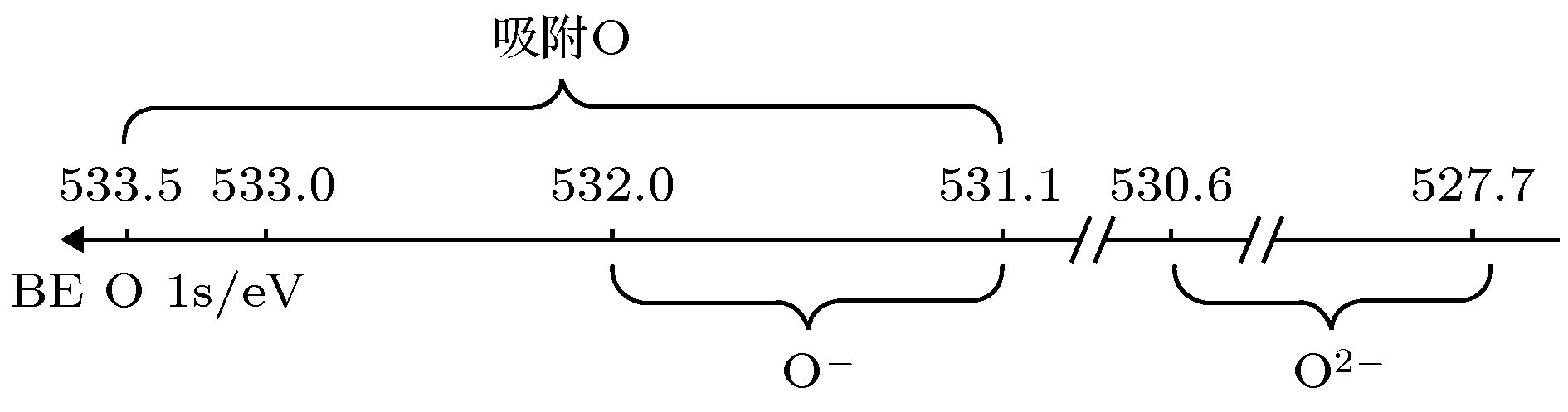 图 2 Dupin等[17]提出的O 1s谱峰所对应的氧离子价态示意图
图 2 Dupin等[17]提出的O 1s谱峰所对应的氧离子价态示意图Figure2. A binding energy scale for valence state of oxygen at the O 1s peaks, proposed by Dupin et al. [17]
Cohen等[18,19]利用密度泛函理论(density functional theory, DFT)计算了BaTiO3的价电子态密度, 计算结果给出Ba, Ti 和 O 的平均化合价分别是2.00, 2.89 和–1.63. 这个计算结果得到了XPS实验结果的证明. 分析一些氧化物的XPS, 包括BaTiO3, 一氧化物CaO, MnO, CoO, ZnO, NiO, CuO粉末样品[20], 一个SrTiO3多晶和一个SrTiO3单晶片体样品[21], 得到BaTiO3粉末、SrTiO3多晶和SrTiO3单晶样品中氧离子的平均化合价为–1.55, –1.62和–1.76, 与Cohen的计算结果–1.63接近. 此外, 发现一氧化物中氧的平均化合价绝对值随其中阳离子第二电离能(V2)的增大而减小, 即其中负二价氧离子的含量比例随V2的增大而减小[20]. 其原因在于氧的第二电子亲和能(8.08 eV)小于阳离子的第二电离能, 所以氧离子要从阳离子得到第二个电子存在不同程度的困难.
2
2.2.金属的价电子结构研究
实验表明, 在3d过渡金属Fe和Co从自由原子形成金属晶体的过程中, 一部分4s电子进入3d轨道, 变成了3d电子; 绝大部分3d电子是局域电子. 说明用能带论处理金属磁有序问题时, 把全部3d和4s都看成巡游电子的假设[1]需要改进.1995年, 美国****Chen等[22]在超高真空条件下制备了厚度为50—70 ?的Fe和Co金属薄膜, 原位测量了样品的L2,3吸收边的X射线吸收谱(X-ray absorption spectra, XAS)和X射线圆二色谱(X-ray magnetic circular dichroism, XMCD). Chen等的分析给出两个值得注意的信息: 1) Fe和Co薄膜中平均每个原子的自旋磁矩分别为1.98μB和1.55μB, 轨道磁矩分别为0.086μB和0.153μB. 轨道磁矩分别为自旋磁矩的4.3%和9.9%, 说明轨道磁矩的贡献很小; 2) 分析过程中, 对于Fe和Co的3d电子平均数目, 采用了理论计算结果6.61和7.51[23,24], 明显多于自由原子的3d电子数目6和7, 说明在从自由原子形成金属的过程中, 一部分4s电子进入3d轨道, 变成了3d电子.
2007年, 德国****Jauch和Reehuis[25]使用316.5 keV的γ射线, 在295 K下测量了α-Fe单晶的高精度结构因子. 通过对这个结构因子的分析, 认为在α-Fe中的价电子组态应为3d74s1, 而不是自由Fe原子的3d64s2.
2015年, 瑞士****Pacchioni等[26]研究了吸附在Cu(111)面上的Fe单原子和小团簇的磁学性质. 在一个真空腔中, 利用氩离子溅射制备了(111)面的Cu单晶. 然后, 利用电子束在原位蒸发Fe. 在蒸发过程中, Cu基片的温度为3.5 K, 以保证Fe原子是吸附在Cu(111)面上, 而不存在与Cu基片的较强相互作用. 与制备样品的真空腔连接有扫描隧道显微镜, 用以原位检测Fe层的厚度, 并且可在不破坏真空的条件下, 把样品转移至XAS和XMCD测量室进行测量. 他们制备了系列样品, Fe的覆盖范围在0.007—0.145单层(ML)之间. 其中ML定义为每个Cu 晶胞的(111)面上有一个Fe原子. 他们首先研究了0.007 ML的样品, 这相当于Fe原子为吸附在Cu(111)面上的孤立原子. XAS测量在温度T = 2.5 K 和磁场 B = 6.8 T 条件下进行. 通过对XAS和XMCD实验结果的分析, 得到吸附在Cu(111)面上的孤立Fe原子3d次壳层的3d空穴数, h d = 3.03, 价电子结构近似为3d74s1. 相对于自由的Fe原子, 相当于有一个4s电子进入了3d轨道. 对于吸附在Cu(111)面上的Fe原子团簇, 随着团簇中平均原子数目逐渐增加, 平均每个原子的轨道磁矩迅速减小, 从孤立Fe原子时的0.66μB减小到5个Fe原子时的0.2μB. 而由电子自旋形成的平均原子磁矩在5个Fe原子时为2.4μB, 与公认的块体金属Fe的平均原子磁矩2.22μB非常接近. 平均轨道磁矩与自旋磁矩比值为8.3%.
2
3.1.O 2p空穴在高温超导和钙钛矿结构锰氧化物研究中的应用
基于Nücker等[10]的实验结果以及相应的其他一些实验结果, 在高温超导性质研究方面, 已经考虑到O 2p空穴的影响. 1998年韩汝珊[27]所著《高温超导物理》一书在介绍用固体能带论研究超导体氧化物时, 给出一个Hubbard模型三带形式的能量表达式, 其中就包含了与O 2p空穴相应的能量项.对于钙钛矿锰氧化物La1–xSrxMnO3 (0 ≤ x ≤ 0.4), 传统的磁有序理论用双交换作用(DE)模型和超交换作用(SE)模型解释其中Sr含量从0到0.4变化过程中电输运性质的变化, 认为其中所有氧离子都处于负二价, 在三价和四价Mn离子之间跃迁的Mn 3d电子是其中电流载流子的来源[5,12]. 然而基于上述电子能量损失谱等实验结果[10-16], Alexandrov等[28,29]指出, 传统的磁耦合DE模型与这些实验结果相冲突, 这些实验结果清楚地表明铁磁锰氧化物中的电流载流子是氧的p空穴而不是d电子. 从而提出一个完全不同于DE模型的O 2p空穴载流子模型, 用于解释钙钛矿锰氧化物的导电性质.
2
3.2.磁有序新模型探索
基于上述氧化物和金属磁性材料的实验和理论研究结果, 针对传统铁磁学在解释一些典型磁性材料磁有序问题时遇到的困难, 提出了一个关于氧化物磁有序的O 2p巡游电子(itinerant electron model for magnetic oxides, IEO)模型[6,30,31]、一个解释金属中平均原子磁矩与电阻率关系的金属磁有序巡游电子(itinerant electron model for magnetic metals, IEM)模型[6,32]、以及一个关于解释氧化物和金属磁有序能来源的外斯电子对(WEP)模型[6,33].3
3.2.1.IEO模型
1) 磁性氧化物中同时存在O2–(2s22p6)离子和O–(2s22p5)离子, O2–离子的外电子壳层为满壳层结构, O–离子的外电子壳层存在一个2p空穴. O2–离子的2p电子有一定的几率以阳离子为媒介跃迁到邻近O–离子外层轨道的2p空穴上, 成为巡游电子, 巡游电子在跃迁过程中自旋方向保持不变.2) 由于每个O2–离子的外层轨道存在自旋方向相反的两个2p电子, 造成一个O2–离子周围的阳离子分成两个磁性子晶格, 在每个子晶格中巡游电子的自旋方向相同, 但是两个磁性子晶格中巡游电子的自旋方向相反. 例如尖晶石结构铁氧体中八面体位和四面体位的阳离子分别处于(A)和[B]子晶格. 在每个子晶格中巡游电子的自旋方向相同, 但是(A)和[B]子晶格中巡游电子的自旋方向相反.
3) 在同一子晶格中, 由于巡游电子在跃迁过程中自旋的方向保持不变, 每个离子的电子自旋方向(包括局域电子和巡游电子)都必须遵守洪特(Hund)定则. 因此, 在同一子晶格中, 如果两个3d过渡金属离子的3d电子数目(nd)同时满足nd ≥ 5 (或它们都满足nd ≤ 4), 则它们的磁矩平行排列; 如果一个阳离子的nd ≥ 5, 另一个阳离子的nd ≤ 4, 则它们的磁矩反平行排列.
应用IEO模型替代传统的超交换和双交换作用模型, 研究了多系列尖晶石结构[30,34-45]和钙钛矿结构[8,9,31,46-49]磁性氧化物的磁有序问题, 包括应用传统模型难以给出合理解释的Cr掺杂尖晶石铁氧体的磁有序问题[34-39]、CrFe2O4的反常红外光谱[40]、Ti掺杂导致尖晶石铁氧体出现附加反铁磁相[41,42]、钙钛矿结构锰氧化物La1–xSrxMnO3的磁矩随Sr掺杂量变化的关系[46]等典型问题.
3
3.2.2.IEM模型
1) 基于γ射线衍射等研究结果, 认为在3d过渡族原子(除Cu和Zn外)结合成金属的过程中, 由于受到原子间电子的泡利排斥力的挤压作用, 原子的大部分4s电子进入3d轨道, 变成3d电子, 剩余的4s电子作为自由电子.2) 处于费米能级附近的3d电子有一定几率在邻近原子的外层轨道间发生跃迁, 形成巡游电子, 其余的3d电子都是局域电子.
3) 金属的电阻率随自由电子含量的增加而减小. 自由电子的迁移过程受到晶格的弱周期性势场影响, 不受任何离子外层轨道的束缚, 所以自由电子的自旋对材料磁矩没有贡献. 在居里温度以下巡游电子跃迁属于自旋相关跃迁, 当温度接近居里温度时, 其跃迁几率迅速减小; 温度越低, 巡游电子跃迁对电导率的贡献越大.
应用IEM模型, 解释了Fe, Ni, Co, Cu金属[32]以及NiCu合金[7]的电阻率与磁矩的关系.
3
3.2.3.WEP模型
1) 假设电子在一个离子外壳层中高速运动时自旋方向不变, 由于相邻离子的最外层轨道十分接近, 其电子分别有一定几率处于图3(a), (b), (c)所示的状态.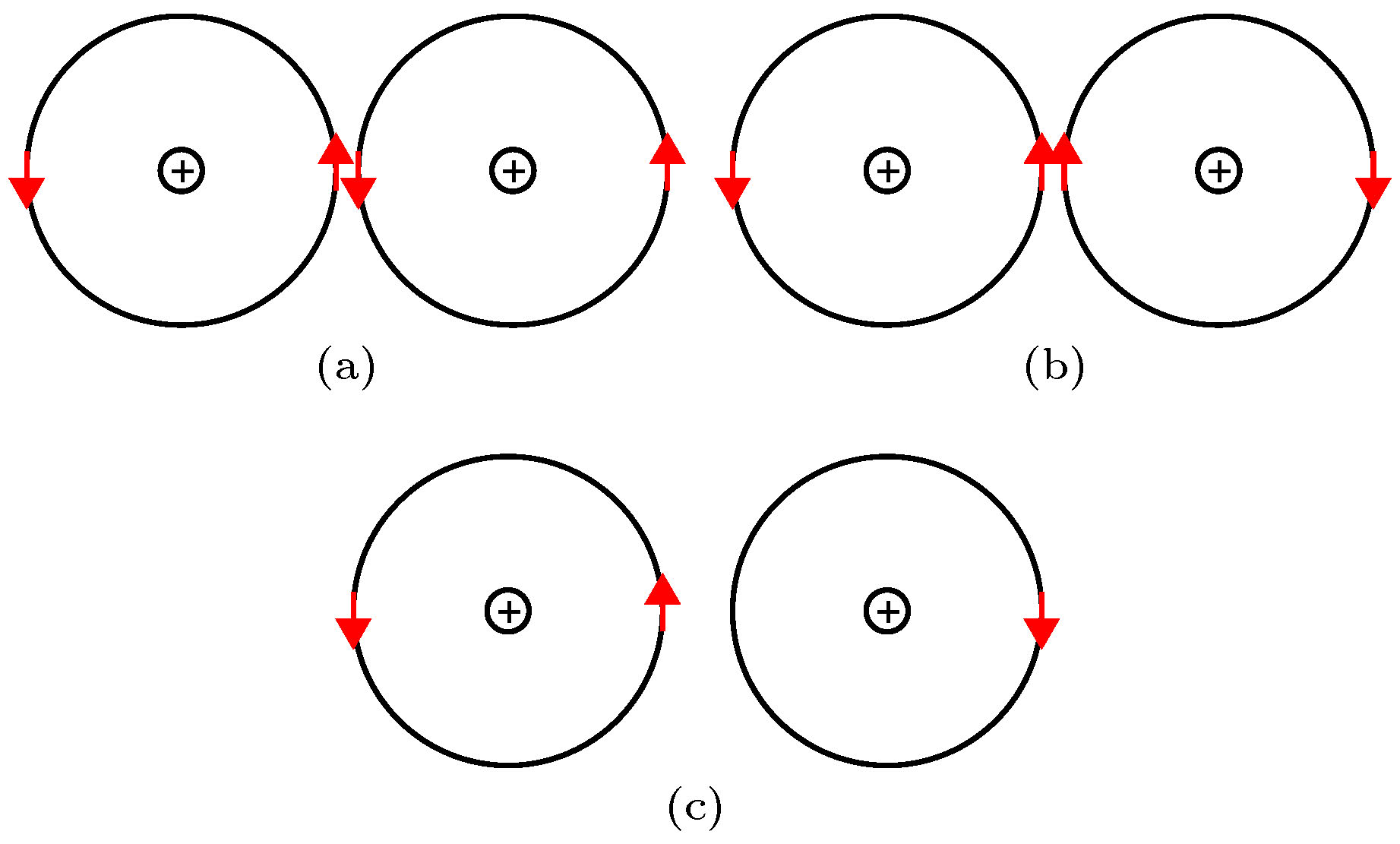 图 3 近邻离子外层电子轨道的(a)外斯电子对和(b), (c)巡游电子示意图[33]
图 3 近邻离子外层电子轨道的(a)外斯电子对和(b), (c)巡游电子示意图[33]Figure3. Illustrations of (a) a Weiss electron pair and (b) and (c) itinerant electrons in the outer orbits of adjacent ions[33].
2) 当两离子处于图3(a)的状态时, 离子间的两电子自旋磁矩反平行. 由于最外层轨道不能同时容纳自旋方向相同的两个电子, 所以电子不能在两离子间交换, 产生静磁吸引能, 同时也存在泡利排斥能, 从而可处于吸引能和排斥能的短暂平衡态, 具有确定的平衡间距和寿命, 称为外斯电子对(WEP). 本研究小组认为WEP的能量是磁性材料磁有序能的来源.
3) 当两个离子处于图3(b)所示状态时, 两个电子的自旋磁矩平行, 容易发生互相交换, 交换前后电子的自旋方向保持不变, 并且这种状态的两个电子间存在磁性排斥能; 当一个离子外层轨道有两个电子, 其相邻的离子外层轨道只有一个电子, 且处于图3(c)的状态时, 中间的电子可以跃迁到右侧离子上, 并且保持自旋方向不变. 图3(b)和(c)所示的跃迁统称为巡游电子的跃迁.
上面分析解释了在磁性材料中巡游电子的自旋方向保持不变的原因, 如果自旋方向不同, 就不能发生图3(b)所示的交换或图3(c)所示的跃迁.
综合应用IEO, IEM和WEP模型, 可以解释为什么Co, Fe, Ni, NiCu合金, Fe3O4和La1–xSrxMnO3具有不同的居里温度[7,50].
2
3.3.新模型中价电子结构与传统模型的主要区别
33.3.1.是否考虑氧化物中的负一价氧离子, 导致不同的磁有序机制
在传统的超交换和双交换作用模型中, 假设磁性氧化物中的所有氧离子都是负二价离子, 具有2s22p6的满壳层结构, 因而阳离子也都具有传统的化合价数值. 例如, 在钙钛矿结构锰氧化物La1–xSrxMnO3中, 当Sr含量为0时, 所有Mn离子为Mn3+, 认为样品的反铁磁性源于Mn3+与Mn3+离子间的超交换作用; 当掺杂Sr时, 出现Mn4+, 并且Mn4+含量与Sr含量x相同, 认为在Mn3+和Mn4+间存在双交换作用, 导致铁磁耦合[5,12].基于前述电子谱的实验结果, 氧化物中同时存在负二价和负一价氧离子, 其平均化合价的绝对值低于传统观点的数值, 随阴/阳离子含量比的增大而减小. 例如, 二氧化锰中氧离子平均化合价的绝对值小于一氧化锰中的数值. 这是由于锰离子的第三、四电离能分别为33.67和51.2 eV, 即使在二氧化锰中, 锰离子也几乎不会失去第四个电子[6]. 这直接导致利用IEO模型对于钙钛矿结构锰氧化物La1–xSrxMnO3磁有序问题的解释不同于传统模型[46]: 当Sr含量x小于0.15时, 样品的反铁磁性源于Mn2+与Mn3+离子间的反铁磁耦合, 样品磁矩随x的增加而增大, 是由于Mn3+/Mn2+离子含量比随x的增加而增大; 当0.15 < x < 0.40时, 所有Mn离子为Mn3+离子, 样品磁矩随x的增加而减小, 源于Mn3+离子间为倾角铁磁耦合, 并且倾角随x的增加而增大.
3
3.3.2.新旧模型的巡游电子定义不同
在传统理论中, 用巡游电子模型解释关于金属磁性的直接交换作用. 例如, 对于磁性金属Fe, Co, Ni, 把3d和4s电子都看成可以在晶格中自由巡游的巡游电子, 处于自旋向上和自旋向下的两个能带中[1].改进后的模型认为, 在Fe, Co, Ni从自由原子形成磁性金属的过程中, 一部分4s电子进入3d轨道, 变为3d电子, 因而价电子分为自由电子、巡游电子和局域电子. 自由电子不受离子实外层电子轨道的束缚, 对样品磁矩没有贡献; 巡游电子指的是离子实的最外层轨道电子有一定几率在最近邻离子间跃迁, 因而对样品磁矩有贡献; 除巡游电子外的3d电子都是局域电子. 由于价电子的轨道磁矩对样品磁矩的贡献很小[22,26], 故可以把金属和合金中平均每个原子磁矩的实验值作为估算3d电子数目的依据[6,7,32].
3
3.3.3.新旧模型中对钙钛矿锰氧化物导电机制的理解不同
传统的双交换作用模型认为La1–xSrxMnO3的巡游电子源于Mn离子的3d电子, 在居里温度以下为金属导电性, 在居里温度以上为半导体导电性, 在居里温度附近发生导电性的“金属-半导体转变”[12].根据IEO模型, 对于钙钛矿结构锰氧化物La1–xSrxMnO3, 在居里温度以下巡游电子的电输运行为属于自旋相关电输运, 在居里温度以上的半导体导电性属于自旋无关电输运[8], 在居里温度附近, 巡游电子发生从自旋相关输运到自旋无关输运的转变.
3
3.3.4.新旧模型中对磁性金属和合金导电机制的理解不同
在传统模型中, 没有考虑金属中巡游电子和自由电子导电机制的区别. 根据IEM模型, 对于NiCu合金, 在居里温度以上, 为自由电子导电, 巡游电子的电导远小于自由电子的电导; 在居里温度以下, 是巡游电子与自由电子共同导电, 温度越低, 巡游电子的电导越大[7].可见, 在NiCu合金和钙钛矿结构锰氧化物La1–xSrxMnO3中, 巡游电子的自旋相关跃迁对电输运性质具有相似的影响[7,8]: 在居里温度以下, 巡游电子的电输运行为都属于自旋相关输运, 在居里温度附近, 巡游电子发生从自旋相关输运到自旋无关输运的转变.
3
3.3.5.新旧模型对磁有序能来源的解释不同
对于外斯分子场能量的来源, 传统观点认为完全是一种量子效应[1], 在密度泛函理论中归结在交换关联能中. 而交换关联能不仅包括与磁有序相关的能量, 还包括在泛函表达式中没有明确给出的所有能量[51], 并且迄今为止, 还一直没能给出交换关联能的函数表达式, 对于不同的材料, 只能用不同模型进行拟合.根据外斯电子对模型, 外斯分子场能量来源于在相邻离子间形成的自旋方向相反的外斯电子对, 这种处于激发态的电子对可在一定条件下形成, 具有一定的形成几率和一定的寿命[6,7,33,50]. 利用这个观点, 可成功解释几种典型的磁性材料为什么具有不同的居里温度[6,7].
2
3.4.自由原子电离能和电子亲和能对晶体中价电子结构的影响
对于上述改进的价电子结构模型的合理性, 可利用自由原子的电离能和电子亲和能进行分析.3
3.4.1.晶体中电子的束缚能与自由原子电离能的关系
自由原子的电离能[52]是自由原子中电子离开所在能级到达(相对于离子实作用场)无穷远处所需要的能量. 晶体中离子对其电子的束缚能数据可以从XPS手册[53]查到, 这种束缚能是电子从所在能级到达费米能级所需的能量. 图4给出晶体中离子对1s电子的束缚能Eb和自由原子中1s电子的电离能VN随原子序数的变化关系. 可见, 尽管Eb小于VN, 但二者都随原子序数的增大而迅速增大, 也可理解为Eb随VN的增大而增大. Eb与VN的这种关系是利用自由原子电离能分析材料中电子结构的依据.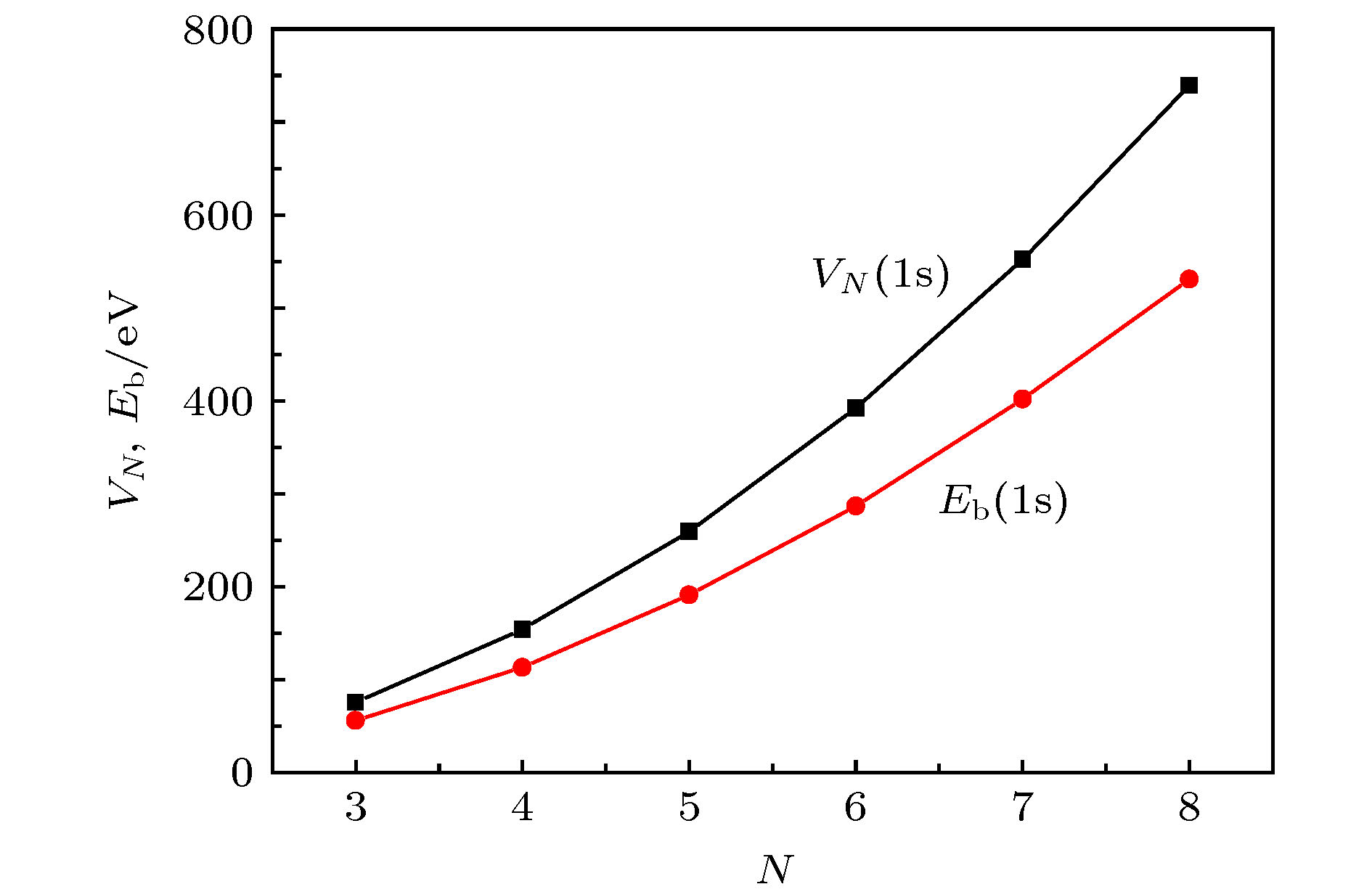 图 4 晶体中离子对1s电子的束缚能Eb和自由原子中1s电子的电离能VN随原子序数N的变化[52,53]
图 4 晶体中离子对1s电子的束缚能Eb和自由原子中1s电子的电离能VN随原子序数N的变化[52,53]Figure4. Dependences on the atom number (N) of the binding energy (Eb) of 1 s electron in a crystal and the ionization energy (VN) of 1 s electron in an free atom[52,53]
利用XPS还可得到晶体的价电子谱. Ley 等[54]给出了从Sc到Fe金属的价带谱, 表明这些金属的价电子分布在费米能级以下约12 eV的范围内. 研究CaO, ZnO, MnFe2O4, ZnFe2O4的价带谱, 这些氧化物的价电子也分布在费米能级以下约12 eV的范围内(图5)[55]. 从图5还可以看到一个有趣的结果, 在ZnO和ZnFe2O4中, Zn 3d 电子集中分布在费米能级以下约8.2—11.5 eV范围内, 这是因为其中的Zn2+离子有10个 3d 电子, 具有3d满壳层结构, 自由Zn原子的第三电离能为39.72 eV, 极难失去第三个电子. 这些价电子谱实验结果说明绝大多数价电子是局域电子, 只有费米能级附近很小范围的电子形成巡游电子. 这也说明上述关于巡游电子和局域电子的定义是合理的.
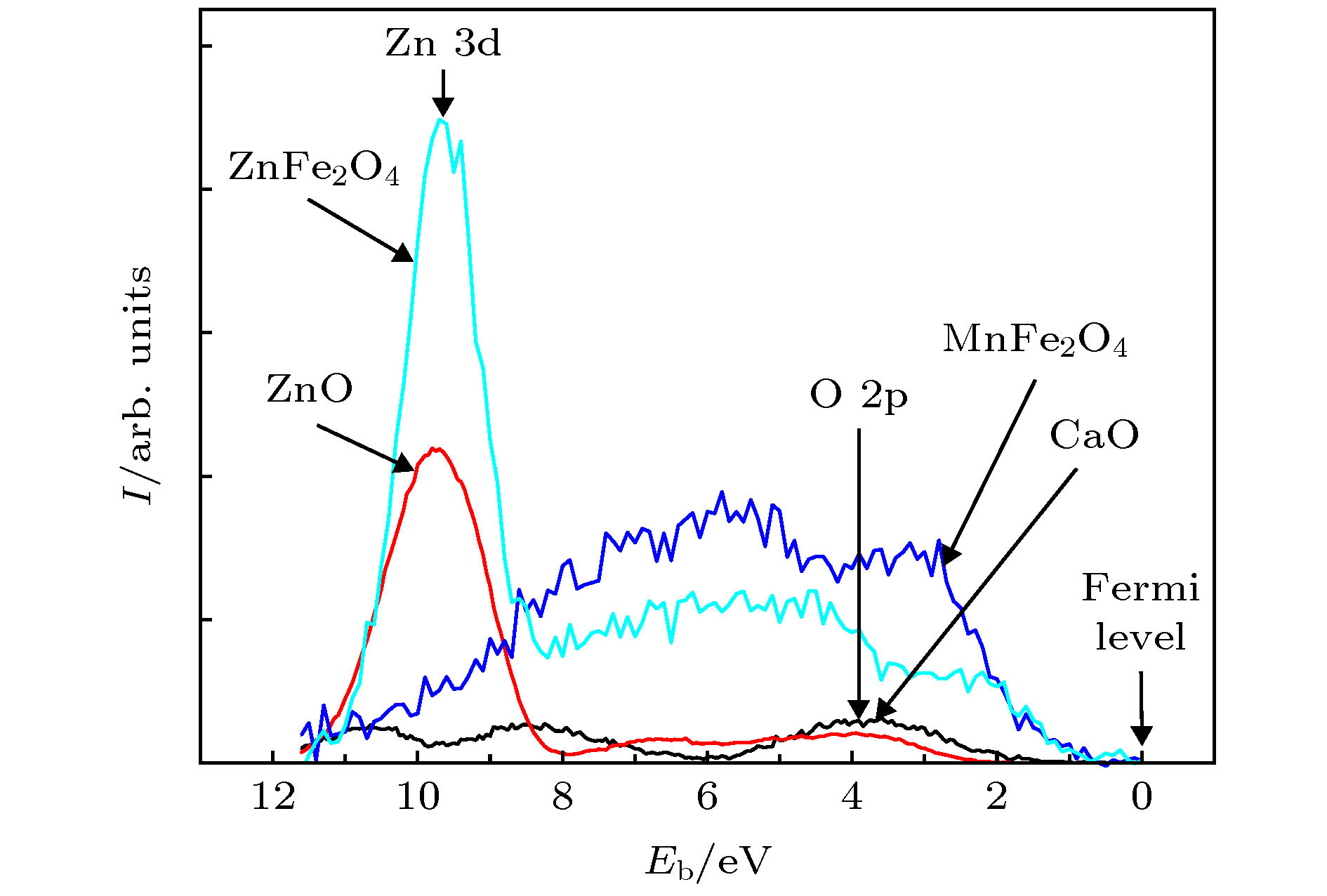 图 5 CaO, ZnO, MnFe2O4, ZnFe2O4的价带光电子谱[55]
图 5 CaO, ZnO, MnFe2O4, ZnFe2O4的价带光电子谱[55]Figure5. Valence band photoelectron spectra of samples CaO, ZnO, MnFe2O4 and ZnFe2O4[55]
3
3.4.2.晶体中的价电子状态与自由原子电离能和电子亲和能的关系
1) 卤碱化合物是典型的离子晶体Li, Na, K, Rb, Cs的第一电离能在5.39—3.89 eV之间, F, Cl, Br, I的电子亲和能在3.614—3.061 eV 之间. 由于这些碱金属的第一电离能与卤素的电子亲和能比较接近, 当它们结合成卤碱化合物时, 卤素原子容易从碱金属得到一个电子, 形成负一价离子, 同时碱金属原子形成正一价离子, 所以卤碱化合物是典型的离子晶体.
2) 氧化物的平均化合价绝对值小于传统观点的数值
首先考虑一氧化物, 如果按照传统观点, 在氧化物中每个氧离子都得到2个电子, 形成负二价离子. 注意到氧的第二电子亲和能为8.08 eV, 碱土离子Mg, Ca, Sr, Ba的第二电离能在15.04—10.00 eV 之间, 3d过渡金属离子的第二电离能在12.80—20.29 eV 之间. 如果这些金属离子与氧离子形成氧化物, 氧离子要得到第二个电子, 存在不同程度的困难, 因而有一部分氧离子不能得到第二个电子, 只能形成负一价氧离子. 如前所述, 阳离子的第二电离能越大, 形成负一价氧离子的几率越大, 平均化合价的绝对值越小.
当从Ti到Cu的3d过渡金属与氧形成比例为3/4, 2/3, 1/2的氧化物时, 由于氧离子的比例逐渐增大, 按传统观点应出现三价和四价阳离子. 但是, 因为这些金属离子的第三电离能在27.49—36.83 eV 之间, 第四电离能在43.27—55.2 eV 之间, 难于形成四价阳离子, 出现三价阳离子的比例也小于传统观点的值. 所以氧化物的平均化合价绝对值随其中氧离子含量比的增加而减小. 例如, 按照传统观点, 在BaTiO3和SrTiO3中Ti离子应为正四价, 而按照Cohen的计算[18,19]和本研究组的实验结果[20,21], 在BaTiO3和SrTiO3中绝大部分Ti离子为正三价, 少量为正二价, 没有正四价的Ti离子. 按照Dupin等[17]的XPS分析结果, 在TiO2和ZrO2中, Ti和Zr离子的平均化合价分别为+2.3和+2.36, 而不是+4.0.
3) 金属中的价电子绝大部分是局域电子
在金属中存在自由电子, 这是因为金属原子的第一电离能一般为几个电子伏特, 例如Fe, Co, Ni, Cu的第一电离能在7.87—7.73 eV 之间. 对于作为良导体的金属Cu, 理论和实验研究表明, 其自由电子浓度为平均每个Cu原子贡献一个自由电子. 除自由电子外, 其余的价电子受到原子核的束缚, 形成离子实. 这种离子实与离子晶体中的离子非常相似, 只有最外层轨道的电子具有一定几率在相邻离子实间跃迁形成巡游电子(金属Cu的离子实具有3d10价电子态, 不易形成巡游电子), 其他电子都是局域电子, 所以金属中的绝大部分价电子是局域电子.
4.1.密度泛函理论研究方法的改进
利用密度泛函做材料模拟计算, 对许多材料的预测取得了很好的效果, 但是对于磁性材料的模拟和预测还存在很大困难, 其主要原因在于把磁有序能与所有未知能量都包含在交换关联能中, 而交换关联能至今还是未知函数, 对于不同材料交换关联能的计算只能采用不同的方法进行模拟. 如果能够找到磁有序能函数的表达式, 使其从交换关联能中独立出来, 作为能量泛函表达式的一项, 将推动磁性材料的模拟取得显著进展. 本研究组提出的WEP模型和对典型磁性材料居里温度的解释为此提供了一定的线索[6,7,50], 但还需要深入研究.此外, 目前利用密度泛函进行材料模拟, 把每个原子的所有价电子都在程序中计算, 计算量非常大, 对于稍复杂的体系, 计算量大到无法完成. 然而, 基于上述实验结果和初步改进的价电子结构模型, 大量的价电子都是局域电子, 对于晶体的结合能并无影响. 如果对软件进行改进, 把能够确定的局域电子都当做内层电子, 只计算自由电子和巡游电子, 可大幅度减少计算工作量.
2
4.2.基于氧化物中存在负一价氧离子, 对XPS分析方法进行系统研究
目前关于氧化物的XPS分析, 多数作者还没有注意到负一价氧离子的存在, 对于其中阳离子谱峰的分析还建立在所有氧离子都是负二价离子的基础上. 例如, 对钙钛矿结构锰氧化物La1–xSrxMnO3中Mn离子的XPS主峰进行分峰, 从中得到正三价和四价锰离子的含量比, 而实际上其主峰并没有双峰的特点, 这样的分峰具有非常大的任意性, 只有谱峰本身具有比较明显的双峰特点时才适于做分峰处理, 例如O 1s峰, 二、三价Ti离子峰[6,21]. 因此, 应组织团队, 在这方面开展系统研究, 建立权威的数据库.2
4.3.多系列钙钛矿结构锰氧化物单晶薄膜的原位磁性、电输运和XPS系统研究
以La1–xSrxMnO3为代表的ABO3型钙钛矿结构锰氧化物是一类磁性和电输运性质可多因素调节的材料[6,9,46-49]: 调节A位的Sr含量, 可以使Mn离子的磁矩从反铁磁耦合变为铁磁耦合[46]; 把A位的La换成Pr, 可在A位引入弱的磁矩[47]; 以Cr替代部分Mn, 可引起倾角铁磁耦合[47]; 以Fe, Co, Ni替代部分Mn, 可引起倾角反铁磁耦合[9,48]. 对于这些替代, 在样品磁性发生变化的同时, 电输运性质也发生巨大变化[9,47,48]. 基于改进的价电子结构模型, 对这类材料磁性和电输运性质的精细而系统的分析, 必将加深对于氧化物磁性和电输运物理机制的理解.在这方面, 虽然过去三十多年中有大量研究工作[56-58], 但是, 绝大部分工作都是基于其中氧离子全部为负二价离子, 用双交换和超交换作用模型分析其磁性和电输运性质, 对于存在的问题又用多种不同的模型进行修补, 形成许多不同的观点, 实际上造成了对这类材料物理机制理解的混乱现象.
本研究组在这方面做了多个系列样品的研究工作, 用O 2p巡游电子模型给出了系统的解释[6,8,9,46-49]. 但都是建立在假设其中Mn离子和其他3d过渡金属离子都不存在正四价离子的基础上, 没有对其进行精细的XPS分析. 这是因为所用的样品都是粉末样品, 由于XPS探测深度较小, 表面效应影响较大, 对于其中的负一价和负二价氧离子含量比不能给出精细的分析结果.
所以, 组织力量对这类材料进行单晶薄膜的原位磁性、电输运和XPS系统研究, 澄清这类氧化物磁性和电输运物理机制, 将推动对于其他种类晶体结构氧化物磁耦合机制的理解.
2
4.4.多系列金属、合金磁性和电输运性质的XPS系统研究
利用提出的磁性金属巡游电子模型(IEM模型)和磁有序能来源的外斯电子对模型(WEP模型), 可拟合方俊鑫和陆栋所著《固体物理学》[59]中给出的NiCu合金在不同Cu含量下电阻率随测试温度变化的曲线, 并能成功解释Co, Fe, Ni和这些NiCu合金为什么具有不同的磁矩和居里温度[7]. 迄今为止, 还没有发现其他研究组关于同时系统解释金属与合金磁性和电输运性质的类似报道.如本文开头所指出, 由于材料的磁性和电输运性质都是由其价电子结构所决定的, 只有能够同时解释磁性和电输运性质的磁有序模型, 并且有足够的实验依据, 才能真正反映材料的实际价电子结构.
然而, 对于磁性金属和合金, 由于其电阻率非常小, 一般的设备难于测量. 可以查到一些早期的电阻率数据, 缺乏近年来的系统研究数据, 更难于找到同时给出磁性和电性的研究报告. 所以, 组织力量开展这方面的系统研究, 非常必要.
